

Abstract
We demonstrate that, via controlled anion exchange reactions using a range of different halide precursors, we can finely tune the chemical composition and the optical properties of presynthesized colloidal cesium lead halide perovskite nanocrystals (NCs), from green emitting CsPbBr3 to bright emitters in any other region of the visible spectrum, and back, by displacement of Cl– or I– ions and reinsertion of Br– ions. This approach gives access to perovskite semiconductor NCs with both structural and optical qualities comparable to those of directly synthesized NCs. We also show that anion exchange is a dynamic process that takes place in solution between NCs. Therefore, by mixing solutions containing perovskite NCs emitting in different spectral ranges (due to different halide compositions) their mutual fast exchange dynamics leads to homogenization in their composition, resulting in NCs emitting in a narrow spectral region that is intermediate between those of the parent nanoparticles.
Introduction
Solar cells using hybrid perovskite semiconductors as the active material have recently seen a remarkable rise in performances with device efficiencies exceeding 20%.1,2 The recent renaissance of photovoltaic research has enabled hybrid MAPbX3–xYx (MA = CH3NH3; X, Y = I, Br, Cl) perovskites, in their 3D structural form, to take center stage once more, leading to a plethora of intriguing results. Tunable electroluminescence3 and amplified spontaneous emission4 have been demonstrated by adjusting the band gap of the hybrid semiconductor together with a proper optically pumped laser, both by constructing a simple vertical optical cavity in which the perovskite film was sandwiched5 and by simply pumping perovskite nanowires.6
While the majority of research has focused on thin films, colloidal synthesis routes to hybrid organic–inorganic7−10 or to fully inorganic11 perovskite nanocrystals (NCs) have only been developed very recently. For instance, Protesescu et al. reported a protocol for the synthesis of CsPbX3 NCs (X being either Cl, Br, I or a combination of both), with narrow size distributions as well as narrow emission line widths, and photoluminescence quantum yield (PLQY) up to 90%.11 Methylammonium based NCs (MAPbX3, X = Br, I, Cl) have also been recently reported with PLQY up to 70%, narrow size distributions and wide color gamut,9 with each point in the chromaticity diagram12 corresponding to a different halide composition of the NCs. Bandgap engineering of lead halide perovskites (both in thin film or NCs form) has been so far realized mainly at the synthesis step, for instance by substituting, partially or completely, the cations13 (Sn instead of Pb14 or formamidium instead of MA15,16), or by mixing different ratios of different halide salts in the reaction flask with the aim of preparing mixed-halide perovskites,11,17 or yet by tuning the average crystallite size.18 Halide perovskites are additionally known to exhibit high ionic conductivities (due to anion migration) and CsPbX3 perovskites have been identified as halide-ion conductors since the ‘80s.19 Recently, high ion mobility has been recognized as one of the possible reasons for anomalous hystereses in the current–voltage curves of perovskite based solar cells.20,21 The ability of halide ions to diffuse/migrate in the perovskite lattice explains their ease of replacement with other halide ions in hybrid halide perovskites thin films, using either MA-halides22 or halogen gases23 as sources of the entering halide ions.
Here, we propose a simple method to tune the optical properties of colloidal CsPbX3 (X = Cl, Br, I) NCs via postsynthetic reactions with different compounds capable of delivering halide ions, and demonstrate fast halide exchange with preservation of shape and crystal structure of the initial NCs (Scheme 1). In general, colloidal halide perovskite NCs (for example both the NCs discussed here and the MAPbBr3 NCs reported by Schmidt et al.8) are unstable in polar solvents, which restricts the medium in which postsynthesis treatments can be carried out to nonpolar or moderately polar solvents (toluene in the present case). In our experiments, the amount of added halide precursors regulated the PL of the resulting NCs: depending on the initial NC samples and on the type of exchanging halide ions, such PL could be tuned across the whole visible spectrum. Except when lead halide salts were used as source of anions, the exchanged NCs had optical quality in terms of PLQY and narrow emission linewidth in line with that of directly synthesized NCs. This suggests that the anion exchange process did not deteriorate the structure and the overall stability of the initial NCs. We additionally demonstrate that fast anion exchange can even take place between halide perovskite NCs bearing different halide ions. The inter-NC exchange of anions yielded NCs with homogeneous composition of halide ions in their respective lattices, with halide ratios dictated by the relative amounts of ions in the NCs that were initially mixed.
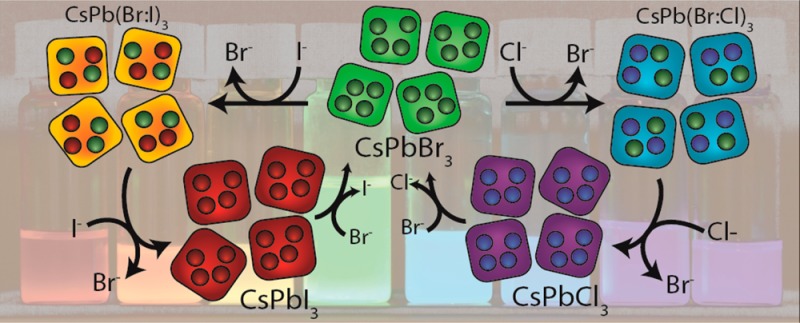
Scheme 1. Overview of the Different Routes and Precursors for the Anion Exchange Reactions on CsPbX3 (X = Cl, Br, I) NCs Reported in This Work.
Experimental Section
Chemicals
Lead(II) chloride (PbCl2, 99.999% trace metals basis), lead(II) bromide (PbBr2, 99.999% trace metals basis), lead(II) iodide (PbI2, 99.999% trace metals basis), cesium carbonate (Cs2CO3, reagentPlus, 99%), tetrabutylammonium iodide (TBAI, reagentPlus, ≥97.0%), tetrabutylammonium bromide (TBABr, reagentPlus, ≥99.0%), tetrabutylammonium chloride (TBACl, ≥97.0%), iodine (I2, 99.99%), methylamine solution (33 wt % in absolute EtOH), absolute ethanol (EtOH, ACS reagent, ≥99.8%), octadecylamine (ODA, 97%), hydrochloric acid (HCl, ACS reagent, ≥37%), hydriodic acid (HI, 57 wt % in H2O), hydrobromic acid (HBr, 48 wt % in H2O, ≥99.99%), octadecene (ODE, technical grade, 90%), oleylamine (OLAM, 70%), trichloroisocyanuric acid (TCICA, technical, ≥95%) and oleic acid (OA, 90%) were purchased from Sigma-Aldrich. Chloroform (CHCl3, anhydrous, 99.95%) and toluene (TOL, anhydrous, 99.8%) were purchased from Carlo Erba reagents. All chemicals were used without any further purification, except for OLAM, OA, and ODE, which were degassed at 100 °C for 2 h under vacuum.
CsPbX3 (X = Cl, Br, I) NCs Synthesis and Purification
CsPbBr3 (and CsPbCl3/CsPbI3) NCs were synthesized as described by Protesescu et al.,11 with some minor adaptations. In a typical synthesis, 69 mg of PbBr2 (0.188 mmol) (or 52/87 mg of PbCl2/PbI2, respectively), 5 mL of ODE, 0.5 mL of OA and 0.5 mL of OLAM were loaded in a 25 mL 3-neck flask and dried under a vacuum for 1 h at 120 °C. After degassing, the temperature was raised to 165 °C and a mixture of 0.6 mL of ODE with 0.4 mL of previously synthesized Cs-oleate (0.4 g of Cs2CO3 degassed in 15 mL of ODE and 1.75 mL of OA at 150 °C) was swiftly injected. Immediately after the injection, the NC solution was quickly cooled down to room temperature with an ice bath, and the NCs were transferred to a glovebox. The NCs as delivered from the synthesis (the “crude” NCs) could be purified via high speed centrifugation (at 12 000 rpm for 30 min), followed by redispersion in TOL.
Synthesis of MA-Br
Methylammonium bromide (MA-Br) used for anion exchange was synthesized following the protocol reported in the literature.3,24 8.5 mL of HBr (48 wt % in H2O) was slowly added to a 250 mL flask containing 24 mL of methylamine solution (33 wt % in absolute EtOH) and 100 mL of EtOH. The solution was kept in ice bath under stirring for 2 h and then the solvent was removed by evaporation, yielding a white powder. The MA-Br salt was then washed three times with diethyl ether and dried on a hot plate.
Synthesis of ODA-X
Octadecylammonium halides (ODA-X) used for anion exchange were synthesized using the same procedure for MA-Br: 0.08 mL of HCl (or 0.5 mL of HI or 0.17 mL of HBr) were slowly added in a flask containing 1 g of ODA and 40 mL of EtOH. The solution was stirred for 2 h and then the solvent was removed by evaporation.
Synthesis of OLAM-X
The OLAM-Cl (most likely a mixture of chloro-oleyalmine and oleylammonium chloride) precursor was synthesized as follows: an excess of Cl2 gas (synthesized in situ by slowly adding 2 mL of 37% HCl to trichloroisocyanuric acid) was bubbled into a flask containing 2 mL of OLAM, under nitrogen flow. From the flask, the N2/Cl2 gaseous mixture was then bubbled in a NaOH solution, to neutralize the unreacted Cl2 gas. After all the trichloroisocyanuric acid had reacted, the oleylamine solution was diluted in 3 mL of TOL. The corresponding OLAM-I precursor was prepared by reacting 0.5 mmol of I2 (1 mmol I–) with 0.250 mL of OLAM overnight. The collected solid was dissolved in 5.75 mL of TOL.
TBA-X Solutions
TBA-X anion precursor solutions were prepared (and stored in a glovebox) by dissolving 1 mmol of halide salt (278 mg of TBA-Cl, 322 mg of TBA-Br or 370 mg of TBA-I) in 1 mL of CHCl3, diluted by 5 mL of TOL.
Anion Exchange Reactions
0.25 mL of crude CsPbX3 NC solution (0.0537 M in X–) were dispersed in 1 mL of TOL, and different quantities (normally ranging from 10 to 400 μL) of 0.17 M halide precursors solution were swiftly injected. The anion exchanges with solid lead halide salts were performed by addition of a 20-fold halide excess (0.14 mmol) of the solid salt to a rapidly stirred NC solution. Similarly, the exchange reaction with ODA-X and MA-Br was performed by adding a 20-fold halide excess (0.14 mmol) of MA-Br salt to 0.25 mL of CsPbBr3 NC solution diluted in 0.75 mL of TOL.
Transmission Electron Microscopy (TEM)
Conventional TEM observations were carried out using a JEOL JEM 1011 microscope equipped with a thermionic gun operating at 100 kV of accelerating voltage. Samples were prepared by dropping washed and diluted (in TOL) NC solutions onto carbon-coated 200 mesh copper grids with subsequent solvent evaporation.
Powder X-ray Diffraction (XRD) Analysis
A Rigaku SmartLab 9 kW diffractometer was used with a Cu source operating at 40 kV and 150 mA. A Göbel mirror was used to obtain a parallel beam and to suppress Cu Kβ radiation (1.392 Å). Samples were prepared by dropcasting a concentrated and washed NC solution on a zero background silicon wafer.
Optical Absorption Spectroscopy
The spectra were recorded on a Varian Cary 5000 UV–vis–NIR spectrophotometer. The NC solutions were prepared by diluting the crude NC solutions in TOL (20 μL in 1 mL), in 1 cm path length quartz cuvettes with airtight screw caps. The sample preparation was performed inside a nitrogen-filled glovebox.
Preparation of Thin Film of CsPbX3 NCs
Thin films of CsPbX3 NCs obtained via anion exchange were prepared as follows: 0.5 mL of crude CsPbX3 NC solution was separated from the organic solvents by centrifugation at 10 000 rpm for 30 min. The NCs were redispersed in 20 μL of TOL and spin coated on a 1.5 by 1.5 cm quartz slide at 2000 rpm for 45 s.
Photoluminescence Measurements
The crude NC solutions were diluted in TOL (∼50 times), to reach an optical density of about 0.1–0.2 at the excitation wavelength. The steady-state PL spectra were collected with an Edinburgh Instruments FLS920 spectrofluoremeter, by exciting the samples at 400 nm using a Xe lamp coupled to a monochromator. For PLQY measurements, the spectra were collected using a spectrofluorimeter (Horiba JobinYvon), by exciting the sample with a monochromated xenon lamp source. The central wavelength was set to λexc= 400 nm for all the samples except for CsPbCl3, for which λexc= 350 nm was used. The exciting light was coupled into an optical fiber connected to an integrating sphere where a quartz cuvette containing the sample was placed. The emitted light was then collected from the sphere with a second fiber coupled to the detection system, made of a spectrometer and a PMT detector. For each sample, four measurements were performed: (i) the sample emission (SEM) that collects the photons emitted by the sample; (ii) the blank emission (BEM), which is a measurement performed with the cuvette containing only the solvent (blank) in the same spectral range used for the SEM measurement; (iii) the sample excitation (SEX), which records the photons at the pumping wavelength that are not absorbed by the sample; (iv) the blank excitation (BEX), which records the photons at the pumping wavelength going through the blank. The photoluminescence quantum yield PLQY was then calculated as
Any reabsorption correction factor was neglected in our calculation of the PLQY, since the solutions investigated were diluted to the point that reabsorption of the PL could be neglected.
Results and Discussion
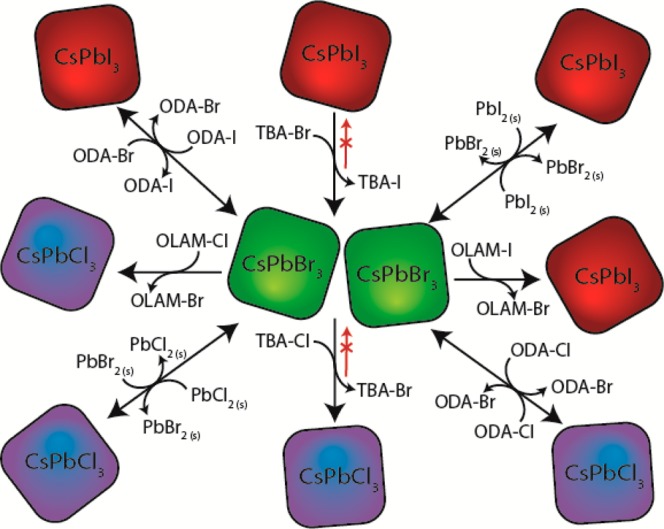
The halide precursors tested in the exchange reactions were: lead halide salts (PbX2), tetrabutylammonium halides (TBA-X), octadecylammonium halides (ODA-X) and oleylammonium halides (OLAM-X). The latter were prepared by reacting oleylamine with halogen molecules (either Cl2 or I2 in the present work, see the Experimental Section). It is known that primary and secondary amines react with halogen molecules to form haloamines (at least for the Cl2 case) and ammonium halides.25 The rationale behind the use of OLAM-X is that the NCs synthesized by us were already coated with oleylamine molecules, therefore OLAM-X should be the reactant that would possibly entail the least interference with the NC ligand shell. In the TBA-X case, exchange reactions worked only for the Br– → Cl– and I– → Br– exchange routes, but not for the reverse cases (Cl– → Br– and Br– → I–). This is understandable in terms of hard/soft acid/base interactions:26 the lyophilic quaternary TBA cation, with its four butyl chains and no possibility of forming hydrogen bonding, is a soft acid that prefers to bind to softer halide ions. Therefore, Cl– ions in CsPbCl3 NCs will not be exchanged with Br– ions coming from TBA-Br because the concurrent transformation from TBA-Br to TBA-Cl is not favored, as Br– ions are softer than Cl– ions and thus prefer to remain associated with TBA. The same reasoning can explain the difficulty in exchanging Br– ions in CsPbBr3 NCs with I– ions coming from TBA-I (this time I– ions are softer than Br– ions). Interestingly, this reasoning does not apply to the much smaller methylammonium halide (MA-X) compounds, which however can be engaged in cation exchange reactions involving the MA cation, as discussed later.
All the anion exchange reactions discussed here led either to a blue shift (for the Br– → Cl– and I– → Br– routes) or to a red shift (for the Br– → I– and Cl– → Br– routes) of the optical features (Figure 1A and 1B), corroborating the incorporation of the new anions. Highly soluble precursors such as OLAM-X and TBA-X led to anion exchange within a few seconds (see Supporting Movies) at room temperature, and could be easily tracked by monitoring changes in the optical absorption and PL spectra of the NCs (later discussed in Figure S1 and Figure 2A, respectively). The exchange with the less soluble ODA-X and lead halides (PbX2) salts was generally slower and required instead at least 1 day under stirring to record a full shift in the PL. Furthermore, exchange was often incomplete in such cases (broadened PL spectra were recorded). This could be easily explained by the low solubility of these compounds in the relatively nonpolar TOL environment. Attempts to improve their solubility, by working at 90 °C, resulted in quick degradation of the NCs (see also later).
Figure 1.
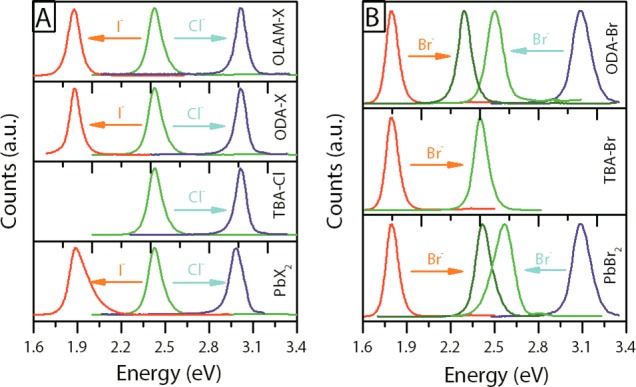
(A) PL of the exchanged NCs obtained from CsPbBr3 by adding: (from top to bottom) OLAM-X, ODA-X, TBA-Cl and PbX2 (X = I, Cl). (B) PL of the CsPbBr3 NCs obtained starting from CsPbCl3 and CsPbI3 NCs using (from top to bottom) ODA-Br, TBA-Br and PbBr2.
Figure 2.
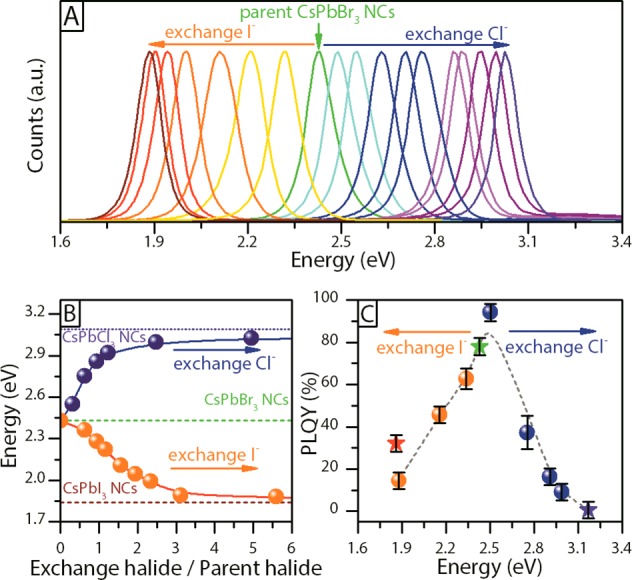
(A) PL spectra of the CsPb(Br:X)3 (X = Cl, I) NCs prepared by anion exchange from CsPbBr3 NCs. (B) PL calibration curves: a targeted emission energy could be obtained by adding a precise amount of halide precursor to a crude solution of CsPbBr3 NCs. The curves are reported as a function of the molar ratio between the added halide (or exchange halide) and the Br amount in the starting NCs. (C) PLQY recorded on the exchanged NCs (dots) as well as on the directly synthesized NCs (stars).
Without loss of generality, we henceforth focus the discussion on the exchange routes starting from CsPbBr3 NCs, due to their stability and strong fluorescence. The NCs, as synthesized, were characterized by a narrow emission band centered at 2.43 eV with a fwhm of 0.12 eV (26 nm) and a PLQY of 78% (Figure 2C, green star shaped marker). After one month from the synthesis, their PLQY dropped only slightly (to 70%). On the other hand, the Cl- or I-based NCs prepared by direct synthesis exhibited a PLQY of ∼1% and 30%, respectively (Figure 2C, violet and red star shaped markers, respectively). Moreover the CsPbI3 NCs were extremely sensitive to moisture, and were degraded in a couple of days if exposed to air. As for the halide precursors, we focus the discussion here on OLAM-I and TBA-Cl, although similar results were found when working with the other precursors, as reported in Figure S1. Also, we focused on room temperature exchange, since the NCs were generally unstable at higher temperatures. For example, the PL intensity of samples of CsPbBr3 NCs dispersed in TOL decreased considerably upon prolonged exposure to heat (90 °C for 2 h), as shown in Figure S2.
As shown in Figure 2A, the PL of the pristine CsPbBr3 NCs could be tuned within an energy interval comprised between 1.88 eV (lowest value attainable by reaction with OLAM-I) and 3.03 eV (highest value attainable by reaction with TBA-Cl) following the titration curves of Figure 2B. The corresponding absorption spectra are reported in Figure S3. Similar tunabilities of the PL using OLAM-Cl and TBA-Br are reported in Figure S1. The upper and lower PL positions of the corresponding samples prepared via anion exchange differed only slightly (by less than 0.1 eV, see Figure S4) from those of the CsPbI3 and CsPbCl3 NCs prepared by direct synthesis, signifying an almost full exchange. The PL emission lines for the CsPbI3 and CsPbCl3 NCs prepared by anion exchange remained narrow (0.11 and 0.10 eV, respectively). Figure 2C shows the measured PLQY, as a function of the energy of the emitted photons, for each of the studied samples. Notably, upon a slight addition of TBA-Cl the PLQY increased from 78% of the initial CsPbBr3 (green star shaped markers in Figure 2C) up to 95%. This is in line with the trend already reported by Pellet et al.22 on CH3NH3PbBr3 thin films prepared via halide substitution from CH3NH3PbCl3 films (a marked PLQY increase was recorded in that case). Apart from the above-mentioned exception, the PLQY of the investigated samples generally dropped from the starting value of 78% when replacing Br– ions with either Cl– or I– ions (Figure 2C). Then, upon full halide exchange, the PLQY settled to a value comparable to the one measured on the directly synthesized CsPbI3 and CsPbCl3 NCs. The lower PLQYs of CsPbI3 and CsPbCl3 with respect to CsPbBr3 are most likely ascribable to intrinsic properties of halide perovskites.4,22,27
Overall, our results indicate that the anion exchange process does not induce any remarkable formation of lattice/surface defects that would lead to deterioration of the optical properties of the NCs. For all NC samples (regardless of whether the NCs were purified from the excess halide precursors or they were stored as crude solutions, see Figure S5), the PL spectral position, its narrow line width and the PLQY remained stable over days. The exchanged NCs remained stable even if deposited as a thin film. These films exhibited PLQYs that were lower than the initial values of the NCs in solution (Figure S6), but were comparable to those recorded from polycrystalline films grown by direct crystallization of chemical precursors on a substrate.4,22,27 Also, it is worthy of note that the exchange worked only for the Cl–Br and Br–I couples (in both directions), but never from CsPbCl3 directly to CsPbI3 nor in the reverse direction. Exchange attempts ended up in complete dissolution of the particles, with loss of PL from solution. One possible explanation here is that the direct exchange between Cl– and I– anions would involve a structural stress on the perovskite lattice that cannot be tolerated without serious degradation of the NCs.
Anion exchange did not alter the cubic shape of the initial CsPbBr3 NCs (Figure 3D), although after exchange with Cl– their size decreased slightly, from (8.4 ± 1.0) nm to (8.0 ± 1.4) nm (Figure 3A), whereas the exchange with I– led to a slight increase in size, to (9.1 ± 1.3) nm (Figure 3C; see also Figure S7 and S8). The XRD pattern collected on the pristine CsPbBr3 NCs (Figure 3D) could be indexed as cubic CsPbBr3 (a = 5.874 Å, space group Pm3̅m, ICSD 29073) as detailed in Figure S9, in agreement with what reported by Protesescu et al.11 Anion exchange reactions did not alter the crystal phase of the NCs (see Figures 3D, S4B and S9) and the patterns collected on the almost fully exchanged NCs were in good agreement with those recorded on directly synthesized CsPbI3 (a = 6.18 Å, space group Pm3̅m, ICSD 181288) and CsPbCl3 (a = 5.605 Å, space group Pm3̅m, ICSD 29072) NCs. The XRD patterns of partially exchanged NCs too could be ascribed to the same cubic phase: as expected, upon incorporation of Cl–, the cell shrunk and all the peaks shifted to higher angles, while the incorporation of I– expanded the cell and the peaks shifted to lower angles (Figure 3D).
Figure 3.
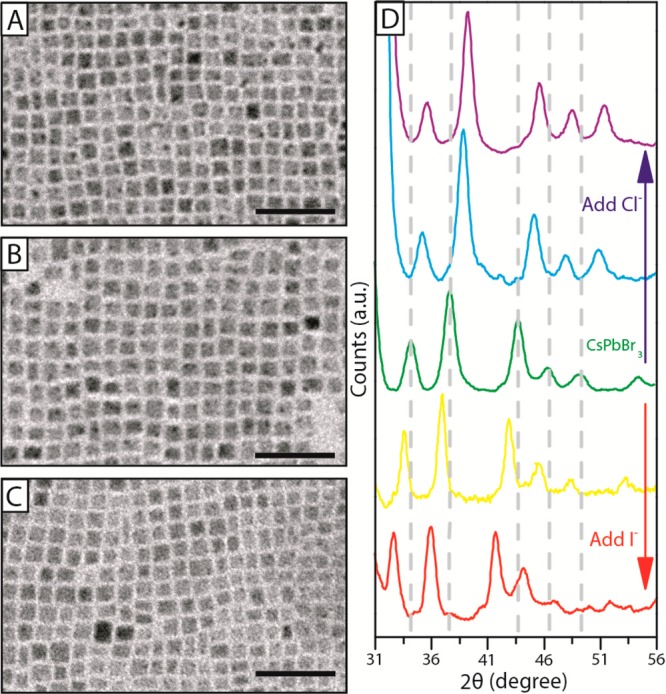
(A–C) TEM images of pristine CsPbBr3 NCs (B) and of fully exchanged CsPbCl3 (A) and CsPbI3 NCs (C), indicating overall size and size preservation upon anion exchange. Scale bars correspond to 50 nm. (D) Zoom of the XRD patterns of pristine CsPbBr3 NCs (middle pattern in green), of the anion exchanged CsPbCl3 (top, violet), CsPbI3 (bottom, red) and of two intermediate Cl– (middle-top, light blue) and I– (middle-bottom, yellow) exchanged samples. Full XRD patterns are reported in Figure S9.
Table 1 lists the results of compositional analyses of the various samples on which XRD patterns were collected. These were performed via energy dispersive X-ray spectroscopy (EDX) in the scanning electron microscope (SEM). The table also lists the molar ratios between the halide ions added to the NC crude solutions and the Br– ions in the starting NCs, and the corresponding ratios in the exchanged NCs. From the table, it can be seen that the yield of exchange differed in the two cases (Cl– and I–), the one with the larger I– ions being less efficient than the one with the smaller Cl– ions (see also the titration curves of Figure 2B): while a 1.5 ratio of added Cl– ions to Br– ions initially present in the NCs yielded NCs with a Cl:Br ratio over 3, the same reaction conditions, in the case of I– ions, led to NCs with a I:Br ratio of only 0.5.
Table 1. Chemical Composition of the Exchanged NCs (by SEM-EDX).
| PL (eV) | X (reactant):Br (NCs) | EDX composition | X:Br (in the NCs) |
|---|---|---|---|
| 2.91 | 1.5:1 | Cs0.9PbBr0.7Cl2.3 | 3.3:1 |
| 2.77 | 0.9:1 | Cs0.9PbBr1.6Cl1.7 | 1.1:1 |
| 2.43 | – | Cs0.9PbBr3.01 | – |
| 2.21 | 1.5:1 | Cs0.9PbBr2.1I1 | 0.5:1 |
| 1.87 | 4:1 | Cs0.9PbBr1.1I2.1 | 1.9:1 |
The reagents listed above were obviously not the only ones capable of triggering anion exchange. For example, methylammonium halides (MA-X) worked as well. However, the use of salts with small cations such as MA+ adds the further complication that the MA+ cation also may engage in exchange reactions, this time with the Cs+ ions in the NCs. When for example CsPbBr3 NCs were exposed to MA-Br their photoluminescence (PL) peak shifted from 2.43 to 2.36 eV (see Figure S10), and indeed the latter value is comparable to the one reported for MAPbBr3 NCs.8,10 At the same time, a slight expansion of the lattice parameters could be inferred from the analysis of the XRD patterns (Figure S10), further supporting the replacement of Cs+ ions with the larger MA+ ions. Similar results were found when CsPbCl3 NCs were exposed to MA-Br (Figure S11).
An extreme case of halide precursor is represented by the NCs themselves: anion exchange could be achieved even by mixing solutions containing perovskite NCs of different halide compositions and therefore emitting in different spectral ranges (Figure 4A). The resulting NCs emitted in a narrow spectral region that was intermediate between those of the parent particles. This suggests a fast partition dynamics of halide ions between the NCs and the solution phase, likely meditated by the residual excess organic stabilizers present in solution. A few examples are reported in Figure 4C and Figure S12. Pure CsPbI3 NCs from a direct synthesis, mixed with CsPbBr3 NCs, yielded different emitting samples, with PL energies depending on the ratios of the CsPbX3 NCs that were mixed, and PLQYs in line with those observed when performing exchanges with the other halide precursors, as shown in Table 2. Furthermore, interparticle exchange did not affect size and shape of the NCs: all NC samples, before mixing, had comparable average sizes and shapes. These remained practically the same after mixing (see TEM images of Figure 4D,E).
Figure 4.

(A) Sketch of interparticle anion exchange. (B) XRD patterns of the pristine CsPbCl3, CsPbBr3 and CsPbCl3 NCs and of the samples after mixing. (C) Optical absorption and PL spectra of various CsPb(Br:Cl)3 and CsPb(Br:I)3 NCs prepared via interparticle exchange. (D) and (E) TEM images of CsPbBr3:CsPbCl3 1:1 (8.5 ± 1.2 nm) and CsPbBr3:CsPbCl3 1:1 (8.9 ± 1.4 nm), respectively. Scale bars correspond to 50 nm.
Table 2. PL of Different CsPb(Br:X)3 NC Mixtures.
| ratio CsPbBr3:CsPbX3 (X = I or Cl) | PL (eV) | PLQY (%) |
|---|---|---|
| CsPbCl3 NCs | 3.18 | 1 |
| 2:3 | 2.91 | – |
| 1:1 | 2.72 | 24 |
| 3:2 | 2.65 | – |
| CsPbBr3 NCs | 2.43 | 78 |
| 3:2 | 2.25 | – |
| 1:1 | 2.14 | 50 |
| 2:3 | 2.08 | – |
| CsPbI3 NCs | 1.87 | 36 |
Anion exchange between NCs took up to several minutes (under stirring) to reach steady state, most likely because the halides ions had to shuttle from NC to NC through the solution phase, a process that in these exploratory experiments was not optimized, for example by the addition of proper stabilizing agents. Anion exchange was further supported by XRD patterns (Figure 4B) of the CsPb(Br:Cl)3 and CsPb(Br:I)3 samples prepared by mixing pure CsPbBr3 and CsPbI3 NCs solutions, which indicated that the halides ions redistributed homogeneously throughout the NCs: XRD patterns were indeed compatible with single phase NCs with average unit cells spacings that were intermediate between those of the parent particles. Finally, in analogy with the failed Cl ↔ I exchange attempts discusses earlier, mixing CsPbCl3 and CsPbI3 NCs resulted in a transparent clear sample and with a quenched PL (see Figure S13), indicating dissolution of the NCs.
In summary, we have demonstrated a fast and simple postsynthesis anion exchange approach to access a wide variety of CsPbX3 perovskite NCs, with preservation of shape and crystal structure of the starting CsPbBr3 sample. The exchanged NCs span the whole visible region of the electromagnetic spectrum, with the PL easily controllable by the amount of added halide precursors, and in general with optical and structural properties in line with those of the same NCs prepared by direct synthesis. We have also shown that the ability of CsPbX3 perovskite NCs to mutually exchange halide ions can be exploited as an additional tool to tune the optical and structural feature of the resulting materials, by simply mixing solutions of NCs having different halide compositions.
Acknowledgments
The research leading to these results has received funding from the 7th European Community Framework Programme under grant agreement no. 614897 (ERC Consolidator Grant “TRANS-NANO”) and from the Cariplo Foundation through grant agreement no. 2013-0656 (“Green nanomaterials for next-generation photovoltaics, GREENS”). We thank Duilio Farina for useful discussions, and Marina Gandini for help with the PLQY experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b05602.
Titration curves with TBA-Br and OLAM-Cl; comparison of optical properties and diffraction patterns of CsPbCl3 and CsPbI3 NCs obtained by direct synthesis and by anion exchange; TEM images of exchanged NCs; full XRD patterns of the NCs obtained by anion exchange; more results on mixed CsPbX3 NCs and results of ion exchange reactions with MA-Br. (PDF)
Movie clip of exchange of CsPbBr3 NCs with I− and Cl−, with stirring. (AVI)
Movie clip of exchange of CsPbBr3 NCs with I− and Cl−, without stirring. (AVI)
The authors declare no competing financial interest.
Supplementary Material
References
- Jeon N. J.; Noh J. H.; Yang W. S.; Kim Y. C.; Ryu S.; Seo J.; Seok S. I. Nature 2015, 517, 476–480 10.1038/nature14133. [DOI] [PubMed] [Google Scholar]
- Yang W. S.; Noh J. H.; Jeon N. J.; Kim Y. C.; Ryu S.; Seo J.; Seok S. I. Science 2015, 348, 1234–1237 10.1126/science.aaa9272. [DOI] [PubMed] [Google Scholar]
- Tan Z. K.; Moghaddam R. S.; Lai M. L.; Docampo P.; Higler R.; Deschler F.; Price M.; Sadhanala A.; Pazos L. M.; Credgington D.; Hanusch F.; Bein T.; Snaith H. J.; Friend R. H. Nat. Nanotechnol. 2014, 9, 687–692 10.1038/nnano.2014.149. [DOI] [PubMed] [Google Scholar]
- Xing G.; Mathews N.; Lim S. S.; Yantara N.; Liu X.; Sabba D.; Grätzel M.; Mhaisalkar S.; Sum T. C. Nat. Mater. 2014, 13, 476–480 10.1038/nmat3911. [DOI] [PubMed] [Google Scholar]
- Deschler F.; Price M.; Pathak S.; Klintberg L. E.; Jarausch D.-D.; Higler R.; Hüttner S.; Leijtens T.; Stranks S. D.; Snaith H. J.; Atatüre M.; Phillips R. T.; Friend R. H. J. Phys. Chem. Lett. 2014, 5, 1421–1426 10.1021/jz5005285. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Fu Y.; Meng F.; Wu X.; Gong Z.; Ding Q.; Gustafsson M. V.; Trinh M. T.; Jin S.; Zhu X. Y. Nat. Mater. 2015, 14, 636–642 10.1038/nmat4271. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Carrero S.; Galian R. E.; Perez-Prieto J. J. Mater. Chem. A 2015, 3, 9187–9193 10.1039/C4TA05878J. [DOI] [Google Scholar]
- Schmidt L. C.; Pertegas A.; Gonzalez-Carrero S.; Malinkiewicz O.; Agouram S.; Espallargas G. M.; Bolink H. J.; Galian R. E.; Perez-Prieto J. J. Am. Chem. Soc. 2014, 136, 850–853 10.1021/ja4109209. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Zhong H.; Chen C.; Wu X.-g.; Hu X.; Huang H.; Han J.; Zou B.; Dong Y. ACS Nano 2015, 9, 4533–4542 10.1021/acsnano.5b01154. [DOI] [PubMed] [Google Scholar]
- Tyagi P.; Arveson S. M.; Tisdale W. A. J. Phys. Chem. Lett. 2015, 6, 1911–1916 10.1021/acs.jpclett.5b00664. [DOI] [PubMed] [Google Scholar]
- Protesescu L.; Yakunin S.; Bodnarchuk M. I.; Krieg F.; Caputo R.; Hendon C. H.; Yang R. X.; Walsh A.; Kovalenko M. V. Nano Lett. 2015, 15, 3692–3696 10.1021/nl5048779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S.; Xiao F.; Pan Y. X.; Ma Y. Y.; Zhang Q. Y. Mater. Sci. Eng., R 2010, 71, 1–34 10.1016/j.mser.2010.07.001. [DOI] [Google Scholar]
- Filip M. R.; Eperon G. E.; Snaith H. J.; Giustino F. Nat. Commun. 2014, 5, 5757. 10.1038/ncomms6757. [DOI] [PubMed] [Google Scholar]
- Hao F.; Stoumpos C. C.; Cao D. H.; Chang R. P. H.; Kanatzidis M. G. Nat. Photonics 2014, 8, 489–494 10.1038/nphoton.2014.82. [DOI] [Google Scholar]
- Amat A.; Mosconi E.; Ronca E.; Quarti C.; Umari P.; Nazeeruddin M. K.; Grätzel M.; De Angelis F. Nano Lett. 2014, 14, 3608–3616 10.1021/nl5012992. [DOI] [PubMed] [Google Scholar]
- Eperon G. E.; Stranks S. D.; Menelaou C.; Johnston M. B.; Herz L. M.; Snaith H. J. Energy Environ. Sci. 2014, 7, 982–988 10.1039/c3ee43822h. [DOI] [Google Scholar]
- Noh J. H.; Im S. H.; Heo J. H.; Mandal T. N.; Seok S. I. Nano Lett. 2013, 13, 1764–1769 10.1021/nl400349b. [DOI] [PubMed] [Google Scholar]
- D’Innocenzo V.; Srimath Kandada A. R.; De Bastiani M.; Gandini M.; Petrozza A. J. Am. Chem. Soc. 2014, 136, 17730–17733 10.1021/ja511198f. [DOI] [PubMed] [Google Scholar]
- Mizusaki J.; Arai K.; Fueki K. Solid State Ionics 1983, 11, 203–211 10.1016/0167-2738(83)90025-5. [DOI] [Google Scholar]
- Tress W.; Marinova N.; Moehl T.; Zakeeruddin S. M.; Nazeeruddin M. K.; Gratzel M. Energy Environ. Sci. 2015, 8, 995–1004 10.1039/C4EE03664F. [DOI] [Google Scholar]
- Stranks S. D.; Snaith H. J. Nat. Nanotechnol. 2015, 10, 391–402 10.1038/nnano.2015.90. [DOI] [PubMed] [Google Scholar]
- Pellet N.; Teuscher J.; Maier J.; Grätzel M. Chem. Mater. 2015, 27, 2181–2188 10.1021/acs.chemmater.5b00281. [DOI] [Google Scholar]
- Karunadasa H. I.; Solis-Ibarra D.; Smith I. C. Chem. Sci. 2015, 6, 4054–4059 10.1039/C5SC01135C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edri E.; Kirmayer S.; Kulbak M.; Hodes G.; Cahen D. J. Phys. Chem. Lett. 2014, 5, 429–433 10.1021/jz402706q. [DOI] [PubMed] [Google Scholar]
- Wayne T. B. A.; Veltmau P. L. US patent US2808439A, 1957.
- Pearson R. G. J. Am. Chem. Soc. 1963, 85, 3533–3539 10.1021/ja00905a001. [DOI] [Google Scholar]
- Stranks S. D.; Burlakov V. M.; Leijtens T.; Ball J. M.; Goriely A.; Snaith H. J. Phys. Rev. Appl. 2014, 2, 034007. 10.1103/PhysRevApplied.2.034007. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.