

Abstract
Haemophagocytic lymphohistiocytosis (HLH), also called haemophagocytic syndrome (HPS) is characterised by a dysregulated activation and proliferation of macrophages, leading to uncontrolled phagocytosis of platelets, erythrocytes, lymphocytes and their haematopoietic precursors throughout the reticuloendothelial system. Mycobacterium tuberculosis-associated HPS is a rare and underdiagnosed association. We report a 34-year-old male patient diagnosed with tubercular pleural effusion responding poorly to antitubercular treatment. Patient later developed generalised lymphadenopathy, pancytopaenia and liver dysfunction and was eventually diagnosed as HLH. Despite being treated as per HLH protocol 2004 he could not be saved.
Background
Haemophagocytic lymphohistiocytosis (HLH) is a rare disease associated with various immunocompromised states, malignancies and infections. Association with tuberculosis has high death rate. Early diagnosis and prompt treatment of HLH can prevent mortality. This case highlights a rare secondary outcome of a common disease like tuberculosis leading to fatal outcome.
Case presentation
A 34-year-old man presented with complaints of fever and breathlessness for 1 month diagnosed as tubercular pleural effusion based on pleural fluid aspirate examination showing a total leucocyte count of 512 cells with 95% lymphocytes, sugar 32 mg/dl, protein 5.2 g/dl, adenosine deaminase level 113 IU/ml and PCR positive for Mycobacterium tuberculosis however acid-fast bacilli could not be demonstrated with Ziehl-Neelsen (ZN) stain. The patient was put on antitubercular treatment but had no response to fever; however, pleural effusion subsided with treatment. After a period of 1 month the patient developed yellowish discolouration of eyes and antitubercular treatment was modified and hepatotoxic drugs removed. But after 3 months patient also developed bilateral parotid swelling, periorbital swelling and progressive increase in jaundice. On examination the patient's general condition was poor. The patient had icterus and generalised lymphadenopathy, that is, cervical, axillary and inguinal, ranging from 1×1 cm to 2.5×1 cm. The patient had swelling present around both the eyes, more on the right side than the left side and bilateral parotid gland enlargement with only superficial lobe palpable (figure 1). Abdominal examination revealed hepatosplenomegaly. Liver was palpable 3 cm below the mid-costal line and spleen was palpable 4 cm below the mid costal line.
Figure 1.
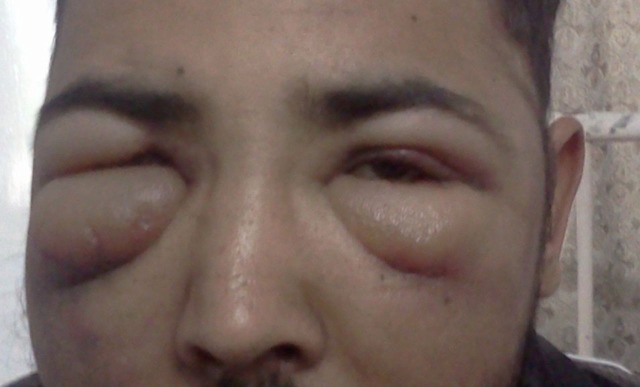
Shows bilateral eye swelling of the patient.
Investigations
Haematological investigations revealed pancytopaenia with a haemoglobin of 7.6 g/dl, total leucocyte count of 1200/mm3, differential count with 70% polymorphs, 30% lymphocytes and platelet count of 60 000/mm3. Reticulocyte count was 1.2% and peripheral smear showed mild anisocytosis, normocytic normochromic red blood cells, leucopaenia and reduced platelets.
Work-up for any other source of fever including malarial antigen test, widal test, leptospira antigen test, blood and urine culture were negative. Serology for HIV, hepatitis A, B, C and E was negative.
Random blood sugar was 93 mg/dl, bilirubin was 13.1 mg/dl with 6.1 mg/dl as direct and 7 mg/dl as indirect fraction, liver enzymes aspartate aminotransferase was 325 U/l, alanine aminotransferase was 309 U/l and alkaline phosphatase level was 1773 U/l, plasma protein was reduced with albumin only 2.4 g/dl, serum calcium was decreased to 6.7 mg/dl, international normalised ratio was raised to 3.7. C reactive protein was raised while antinuclear antibody was negative. ACE inhibitors level was 132 mcg/l.
Whole body contrast-enhanced CT revealed enlarged submandibular glands, bulky parotids, multiple subcentrimetric pretracheal, precarinal, subcarinal lymph nodes, hepatosplenomegaly, bilateral nephrolithiasis and mild ascites (figure 2).
Figure 2.
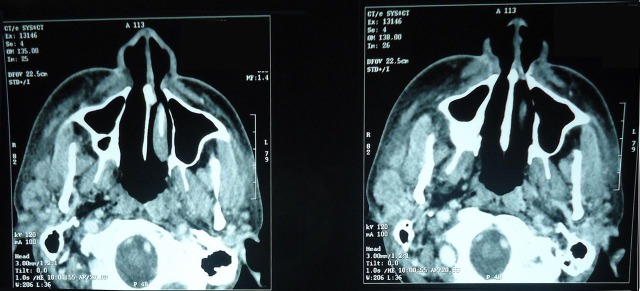
CT showing bilateral parotid swelling with surrounding subcutaneous oedema.
Whole body positron emission tomography (PET) revealed non-fluoro deoxyglucose avid heterogeneous ring-enhancing lesion in high frontal lobe of the brain, ground glass haziness in both lungs with bilateral pleural effusion, mild pericardial effusion and cervical, axillary, mediastinal and inguinal lymph nodes (figure 3).
Figure 3.

Positron emission tomography showing heterogeneous ring-enhancing lesion in high frontal region (arrow), ground glass haziness in both lungs with bilateral pleural effusion, mild pericardial effusion
Keeping in view pancytopaenia and strong suspicion for lymphoreticular malignancy, bone marrow aspiration was done which was found to be haemodilute, aparticulate with no abnormal cells.
Fine needle aspiration cytology from parotids revealed acellular necrotic particles.
Lymph node biopsy was done which revealed numerous large clusters of histiocytes forming ill-defined granulomas surrounded by lymphocytes, no necrosis. Many of histiocytes stained positive for CD68 on immunohistochemistry. ZN stain for acid-fast bacilli was negative and KOH (potassium hydroxide) stain for fungus was also negative.
Bone marrow biopsy revealed significant histiocytosis with phagocytosis of erythroid and myeloid cells. CD68 staining revealed innumerable histiocytes and macrophages throughout the bone marrow interstitium, many of which contained intact haematopoietic cells.
So with diagnosis of haemophagoctic lymphohistiocytosis in mind immunoglobulin (Ig) M and IgG for Epstein-Barr virus (EBV) was done which was negative. Serum ferritin level was 4164 mg/l, fibrinogen level was 1.16 g/l and triglyceride level was 466 mg/dl.
In our case, six of the eight criteria of revised HLH diagnostic criteria were fulfilled. Hence, the case was diagnosed as haemophagocytic syndrome (HPS) associated with tuberculosis.
Treatment
The patient was treated as per HLH 2004 protocol with steroids, etoposide and cyclosporine. Modified antitubercular treatment with no hepatotoxic drug was continued.
Outcome and follow-up
Unfortunately, the patient succumbed to complications of HLH and expired within 2 weeks of diagnosis.
Discussion
HLH, also called HPS, is characterised by a dysregulated activation and proliferation of macrophages, leading to uncontrolled phagocytosis of platelets, erythrocytes, lymphocytes and their haematopoietic precursors throughout the reticuloendothelial system.1 HLH was first described in 1952 using the name familial haemophagocytic reticulosis where an overgrowth of histiocytes and phagocytosis was described in two related cases, with multisystem involvement.2
HLH can be primary or familial due to genetic mutations or secondary or acquired HLH. Primary HLH is observed mostly in infants with an estimated incidence of 0.12/100 000 children per year.3 Secondary HLH is associated with various stimuli-like infections,4 5 T-cell lymphoma and immunocompromised states. Among the infections, EBV is the most common. Others are cytomegalovirus, adenovirus, parvovirus B19, HIV, bacteria like M tuberculosis, some Gram-positive and Gram-negative bacteria, fungi and protozoa like Babesia microti. Few cases of HLH are associated with miliary tuberculosis, HIV and mycoplasma infection, respectively.6–8 Our patient also developed secondary HLH after M tuberculosis infection.
In a study done by Tseng et al9 M tuberculosis was associated with 23% of cases of infection-induced HLH. M tuberculosis-related HLH was associated with higher death rate and longer duration of symptoms (more than 2 weeks) before diagnosis than HLH caused by other infections. M tuberculosis-related HLH in our patient also had a fatal outcome.
HLH is a disease with major diagnostic and therapeutic difficulties. The Revised Diagnostic Guidelines4 for HLH is as shown in box 1. Diagnosis in our patient was made by fulfilling six of eight features of the diagnostic criteria.
Box1. Revised Diagnostic Guidelines for haemophagocytic lymphohistiocytosis (HLH)4.
A molecular diagnosis consistent with HLH
-
Diagnostic criteria for HLH fulfilled (five of the eight criteria below)
Initial diagnostic criteria (to be evaluated in all patients with HLH)
Fever
Splenomegaly
Cytopaenias (affecting ≥2 of 3 lineages in the peripheral blood): haemoglobin <90 g/l (in infants <4 weeks: haemoglobin <100 g/l) platelets <100×109/litre neutrophils <1.0×109/litre
Hypertriglyceridaemia and/or hypofibrinogenaemia
Fasting triglycerides ≥3 mmol/l (ie, ≥265 mg/dl)
Fibrinogen ≤1.5 g/l
Haemophagocytosis in bone marrow or spleen or lymph nodes
No evidence of malignancy
-
New diagnostic criteria
Low or absent natural killer-cell activity (according to the local laboratory reference)
Ferritin ≥500 mg/l
Soluble CD25 (ie, soluble IL-2 receptor) ≥2400 U/ml
The diagnosis of HLH can be established if one of either 1 or 2 below is fulfilled.
Bhattacharyya et al10 reported severe fulminant liver failure and coagulopathy can sometimes predominate the clinical picture of HLH and hence delay the diagnosis. Our patient also developed liver dysfunction contributing to coagulopathy.
Koç et al11 reported that PET can be helpful in directing towards an inflammatory disease with multisystem involvement and suspecting the diagnosis of HLH. PET scan of our patient was also contributory to diagnosis with increased uptake of fluorodeoxyglucose in various organs including brain.
HLH can be rapidly fatal with death occurring in the first 4–8 weeks often from multiorgan failure, bleeding and/or sepsis. These patients need to be diagnosed early and treated according to the HLH 2004 treatment protocol (figure 4)4 with steroids, itoposide, cyclosporine with or without intrathecal methotrexate and treatment of primary cause. Early diagnosis of HPS as well as primary disease and treatment are essential to avoid a fatal outcome. Our patient was also treated as per HLH treatment 2004 protocol but had a fatal outcome.
Figure 4.

Treatment protocol overview for haemophagocytic lymphohistiocytosis 2004 protocol.
Learning points.
Haemophagocytic lymphohistiocyosis is a rare disease.
Association with tuberculosis can be seen.
High degree of clinical suspicion is required for diagnosis.
Early diagnosis can prevent fatal outcome.
Footnotes
Competing interests: None.
Patient consent: Obtained.
References
- 1.Brastianos PK, Swanson JW, Tobenson M, et al. Tuberculosis-associated hemophagocytic syndrome. Lancet Infect Dis 2006;447–54. [DOI] [PubMed] [Google Scholar]
- 2.Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child 1952;27:519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henter JI, Elinder G, Söder O, et al. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand 1991;80:428–35. [DOI] [PubMed] [Google Scholar]
- 4.Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124–31. [DOI] [PubMed] [Google Scholar]
- 5.Chan JK, Ng CS, Law Ck, et al. Reactive hemophagocytic syndrome: a study of seven fatal cases. Pathology 1987;19:43–50. [DOI] [PubMed] [Google Scholar]
- 6.Browett PJ, Varcoe AR, Fraser AG, et al. Disseminated tuberculosis complicated by the hemophagocytic syndrome. Aust N Z J Med 1988;18:79–80. [DOI] [PubMed] [Google Scholar]
- 7.Campo E, Condom E, Mario MJ, et al. Tuberculosis-associated hemophagocytic syndrome: a systemic process. Cancer 1986;58:2640–5. [DOI] [PubMed] [Google Scholar]
- 8.Subhash HS, Sowmya S, Sitaram U, et al. Tuberculosis-associated haemophagocytic syndrome. J Postgrad Med 2001;47:220. [PubMed] [Google Scholar]
- 9.Tseng YT, Sheng WH, Lin BH, et al. Causes, clinical symptoms, and outcomes of infectious diseases associated with hemophagocytic lymphohistiocytosis in Taiwanese adults. J Microbiol Immunol Infect 2011;44:191–7. [DOI] [PubMed] [Google Scholar]
- 10.Bhattacharyya M, Ghosh MK. Review article: hemophagoctic lymphohistiocytosis—recent concept. J Assoc Physicians India 2008;56:453–7. [PubMed] [Google Scholar]
- 11.Koç ZP, Akarsu S, Balci T, et al. PET/CT images of a patient with haemophagocytic lymphohistiocytosis. BMJ Case Rep Published 22 June 2012;10.1136/bcr-03-2012-6026. [DOI] [PMC free article] [PubMed] [Google Scholar]