

Abstract
Immune-mediated drug-induced liver injury (IMDILI) can be devastating, irreversible, and fatal in the absence of successful transplantation surgery. We present a novel approach that combines the methods of pharmacoepidemiology with in silico molecular modeling to identify specific features in toxic ligands that are associated with clinical features of IMDILI. Specifically, from pharmacovigilance data multivariate logistic regression identified 18 drugs associated with IMDILI (P < 0.00015). Eleven of these drugs, along with their known and proposed metabolites, constituted a training set used to develop a four-point pharmacophore model (sensitivity 75%; specificity 85%). Subsequently, this information was combined with information from immune-pathway reviews and genetic-association studies and complemented with ligand-protein docking simulations to support a hypothesis implicating two putative targets within separate, possibly interacting, immune-system pathways: the major histocompatibility complex within the adaptive immune system and Toll-like receptors (TLRs), in particular TLR-7, which represent pattern recognition receptors of the innate immune system.
Study Highlights.
• WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ Idiosyncratic drug-induced liver injuries (iDILI) are rare, but are serious when they occur, often leading to the withdrawal of the offending drug from the market. While it is thought that the immune system is involved in many of these reactions, the mechanisms or the proteins that may be mediating the toxicity remain elusive. • WHAT QUESTION DID THIS STUDY ADDRESS? ☑ How can a multidisciplinary approach combining pharmacovigilance data with structural chemistry be used in the study of complex diseases? • HOW THIS STUDY ADDS TO OUR KNOWLEDGE ☑ Molecules implicated in immune-mediated DILI share 3D patterns of molecular features. These patterns reveal evidence to support the involvement of HLA-B*5701 and TLR-7 as potential toxicity targets for immune-mediated DILI. • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ Understanding the mechanisms behind DILI may enable the development of screening methods to identify at-risk patients, allowing for individualized therapy. Knowledge of toxicity targets will facilitate the development of in vitro and in vivo assays to aid in identifying toxic drugs early in drug development.
Of the barriers that need to be overcome in bringing a new drug to market, idiosyncratic drug reactions rank among the most dreaded due to their unpredictable and rare, but often serious nature. Traditional approaches involving exposure to cells, animals, and humans during drug development have proven incapable of identifying rare idiosyncratic reactions or the characteristics or circumstances of individuals that place them at risk of these reactions. Although such approaches remain in use for predicting and understanding common reactions to drugs, they are beginning to be complemented by data-driven computational methods that draw on and link knowledge across a number of chemical, biological (including genetic), and clinical disciplines to predict the potential for idiosyncratic reactions. A communication by Sara Reardon, a reporter for the journal Nature, in 20131 described an exciting effort in the field of computational toxicology linking drug molecular structure with various biological receptor sites, enabling researchers to predict potential unintended adverse drug effects that can be detrimental to both patients and the prospect for a drug's further development as a pharmacotherapeutic agent (www.drugable.com, presented by Timothy Cardozo at the US National Institutes of Health's High Risk-High Reward Symposium held in November 2013). Similarly, Liebler and Guengerich identified the importance of moving beyond the reductionist science of monodisciplinary methods towards multidisciplinary and systems biology approaches in the study of drug toxicity.2 A number of studies have also proposed network pharmacology as a way of understanding the on- and off-target effects of drugs.3,4
We present a novel approach that combines the methods of pharmacoepidemiology with in silico molecular modeling to identify specific features in toxic ligands that are associated with the clinical features of immune-mediated drug-induced liver injury (IMDILI).
We then investigate whether the structural features of identified toxic molecules are shared with ligands for which putative immunological mechanisms and targets have been proposed, and provide evidence in support of coincident pharmacological interactions in IMDILI.
Drug-structure and intrinsic features such as particular molecular functional groups and substructures that serve as precursors to reactive intermediate metabolites have been identified as factors associated with idiosyncratic hepatotoxic reactions.5
Genetic studies have also revealed a number of host-related factors that are associated with increased risk of idiosyncratic drug-induced liver injuries (iDILI) from certain medicines. Ann Daly (2012)6 summarizes the major associations that have emerged from Genome-Wide Association Studies (GWAS). For example, iDILI due to ximelagatran, flucloxacillin, lumiracoxib, and amoxicillin/clavulanate is associated with specific human leukocyte antigen (HLA) class I and II genotypes.6 The association with HLA genotypes serves to classify and provide a mechanistic understanding of other IMDILI. The correlation is not perfect, however, with only 1 in every 500–1,000 people who possess the genetic predisposition to flucloxacillin DILI (HLA-B*5701) developing liver toxicity upon exposure to the drug.7
Endeavors in unraveling the mechanisms that lead to iDILI have focused on identifying the manifestation patterns of toxicity features in individuals and the drugs that are associated with these reactions. It has been suggested that the low incidence of iDILI may be explained by a requirement for a number of factors to be present coincidentally for toxicity to develop. The rare "perfect storm" that manifests as a serious hepatic reaction is most likely the result of the coexistence of a number of critical drug- and host-related and environmental factors.8 Coincidental factors may involve the generation of a second signal required for immune (T cell) activation from interaction of a drug and/or its metabolites with a second immune-pathway target or from other exogenous factors (e.g., infection or another drug) that primes or augments the immune response. An example in support of this is the observation that lipopolysaccharides from cell walls of Gram-negative bacteria, through interaction with Toll-like receptor 4 (TLR-4), activate immune reactivity and reduce the exposure threshold required for a number of compounds to induce liver injury.9–11
Due to the infrequent nature of iDILI, clinical information regarding drug exposure and the clinical and pathological features of toxicity is limited and is largely derived from case reports and from postmarketing pharmacovigilance data. The quality of such data and levels of suspicion attributed to particular medicine exposure in patients are dependent on the clinical expertise of case reporters, which can vary from specialist physicians to consumers themselves. Nonetheless, postmarketing pharmacovigilance databases can provide sufficient clinical data to signal drug-event associations for rare adverse drug reactions.12
Drug-event association information from analysis of pharmacovigilance data is now being integrated with chemical and biological data in silico to reveal features of drugs and biological targets and infer toxicity mechanisms.13–17 New computational technologies are transforming the field of regulatory science, with the US Food and Drug Administration (FDA) working towards replacing animal studies with a combination of in silico and in vitro approaches.18
We present an approach to expose relationships between IMDILI and the 3D structural features of toxic drug molecules and their metabolites. We hypothesize that drugs (or their metabolites) that produce similar patterns of toxicity interact with targets within common toxicological pathways and that activation of the underlying mechanisms depends on structural features that are shared among toxic molecules. We chose to focus on immune-mediated DILI since these reactions have defined clinical characteristics that allow us to identify cases from population adverse drug event data. Briefly, drugs with the potential to cause IMDILI were identified from population adverse drug reaction (ADR) data. We then identified similarities in molecular structures between toxic drugs using in silico pharmacophore modeling. Subsequently, we test the hypotheses that these similarities are important in molecular interactions between toxic drugs and the immune system proteins HLA and TLR-7 (Figure 1).
Figure 1.

Workflow diagram outlining the approach taken in this study.
METHODS
Identification of drugs that share signals for IMDILI
Pharmacovigilance data were obtained from Australia's Database of Adverse Event Notifications (DAEN) (http://www.tga.gov.au/safety/daen.htm#.UzAVgc4VXjs). This database is curated by the Australian Therapeutic Goods Administration (TGA) and includes information on patient demographics; drug exposure (generic name, dose, frequency, dose form); and adverse reaction terms recorded using the Medical Dictionary for Regulatory Activities (MedDRA) (www.meddra.org) coded as preferred terms (PT) and lower-level terms (LLT). The subset of data used from the DAEN contained 240,137 ADR reports voluntarily reported to the TGA from 1972 to December 2008. Reports were excluded when data were absent for patient age, gender, drug name, or adverse reaction term. This led to 37,293 reports being excluded, leaving 204,844 records for analysis.
Training set
Cases of IMDILI were defined as reports that included a combination of at least one MedDRA preferred term indicative of liver injury (e.g., hepatic failure) and at least one MedDRA preferred term that indicated an immunological reaction (e.g., drug allergy) (Supplementary Table 1). In all, 780 cases of IMDILI were identified. Noncases were defined as all reports that do not meet the case definition.
All drugs from a therapeutic class were selected for inclusion in disproportionality analyses where literature evidence of IMDILI exists for at least one member of the drug class, defined according to the Australian Medicines Handbook (2010). A total of 328 drugs were included (Supplementary Table 2).
Patient gender and age (as a continuous variable) were included as covariates to account for the increased incidence of DILI in females and the increased risk of the elderly due to a greater number of concomitant medications. Antiviral drugs indicated for viral hepatitis were also included. These were used as surrogate markers of underlying liver disease to account for cases where liver injury may not be due to an administered drug. Covariates were included as variables in the multiple logistic regression model.
We investigated a set of candidate drugs for the possession of signals for IMDILI using a case/noncase method, exposing disproportionality of drug-event combinations in pharmacovigilance data.
Univariate analyses were carried out to measure the association of each drug with IMDILI. Those drugs that were significantly associated with IMDILI were then included in multivariate logistic regression analysis. This was carried out using the forward stepwise inclusion method based on the likelihood ratio statistic to determine the association of each drug with IMDILI (expressed as a reporting odds ratio (ROR) and 95% confidence interval (95% CI)).
The overall alpha level was set at 0.00015 following the use of the Bonferroni adjustment to account for the 328 drugs investigated simultaneously.19 Drugs for which the lower limit of the 95% CI of the ROR exceeds 1 were included in the training set for pharmacophore analysis. Drugs with fewer than four ADR reports in the database were excluded. Statistical analyses were conducted using the SPSS 20.0.0 software (SPSS, Chicago, IL).
Test set
We defined the activity of a drug as its IMDILI potential, i.e., active drugs are drugs that have known IMDILI potential and inactive drugs are drugs that are not known to cause IMDILI.
Drugs withdrawn from the market because of DILI concerns as identified by Guengerich (2011)20 were included in the active test set.
We searched through Australia's DAEN database for drugs with >200 case reports, none of which contain ADR descriptors indicative of DILI. Six drugs met these criteria and were included in the inactive test set.
Since the number of inactive drugs was significantly smaller than the number of active drugs (in the training and test sets), we decided to identify a second set of inactives from the literature. Chen et al.21 list two datasets of drugs and the DILI potential of each drug as identified by two different sources. The seven drugs identified in both data sources as without DILI potential were added into the inactive test set.
Literature confirmation of toxicity classification
The IMDILI potential (or lack thereof) of all the drugs included in the training and test sets was confirmed by reviewing published literature. A drug was only included in the training or active test set if there was published evidence of DILI with immune features (e.g., fever, rash, or eosinophilia), or if there are genetic associations with proteins known to activate the adaptive immune system. Drugs were excluded from the inactive test set if there was any published evidence of hepatotoxicity. Drugs with molecular weight >500 were excluded from all in silico analyses.
Identifying similarities between probe drugs
The published literature was reviewed to identify known and proposed metabolites of all drugs in the training and test sets (Supplementary Table 3).
All molecules investigated in this study were built with the molecular modeling and graphical user interface package Maestro (v. 9.3, Schrödinger, New York, NY, 2012). Parent drug structures were downloaded in Simple Data Format (sdf) from the DrugBank database (www.drugbank.ca). Metabolites were drawn in using Maestro's 2D sketcher tool.
The global minimal energy state of all molecules was determined with MacroModel (v. 9.9, Schrödinger). The OPLS_2005 forcefield was used with a constant dielectric of 1.0.
Conformers were generated with Confgen (v. 2.3, Schrödinger).22 The OPLS_2005 forcefield was used with a distance-dependent electrostatic treatment and a dielectric constant of 1.0. Redundant conformers were eliminated using a root mean square deviation cutoff of 0.5Å.
Generation of pharmacophore hypothesis
Pharmacophore development was conducted using Phase (v. 3.4, Schrödinger)23 in the Maestro modeling environment, in accordance with the Phase 3.4 user manual.
Each drug of the training set was grouped with its known and proposed metabolites as an active ligand group. Pharmacophore sites were identified using both point and vector geometries for the following features: H-bond donor, H-bond acceptor, ring, ionic, and hydrophobe. Phase was then asked to identify common four-point pharmacophore hypotheses that match at least 9 of 11 active groups. The maximum tree depth was set to 7. Hypotheses were scored using the default survival formula.
The analysis was repeated on four different computers across Linux (Ubuntu 12.04), UNIX (iOS), and Windows (Windows 7) platforms.
The primary pharmacophore hypothesis was chosen on the basis of reliability across the different platforms, the Phase survival score, and visual inspection.
Validation of pharmacophore hypothesis
The Phase (v. 3.4, Schrödinger) Advanced Pharmacophore Screening tool was used to screen the active and inactive test sets for matches. The intersite distance matching tolerance was set to 2.0Å. The predictive performance measures sensitivity, specificity, and Matthews Correlation Coefficient were calculated based on the number of drug/metabolite groups containing at least one molecule matching the pharmacophore hypothesis.24
Computational modeling of the interaction of abacavir and flucloxacillin metabolites
The crystal structure of HLA-B*5701 complexed with peptide V and abacavir was obtained from the protein data bank (PDB: 3UPR Biological Assembly 1). Similarly, HLA-B*5703 (PDB: 2BVP) was also obtained. All waters were removed and the proteins were optimized and minimized using Maestro's Protein Preparation Wizard.25
Glide (v. 5.8, Schrödinger)25 was used to generate a 14Å cubic receptor grid centered on the crystallized abacavir in HLA-B*5701. In the case of HLA-B*5703, the grid was centered around the F-pocket (residues: Y24, N77, A81, Y123, I95, Y11626). The structures of flucloxacillin and its metabolites were energy minimized to their most stable conformations. Conformers were generated using Confgen (v. 2.3, Schrödinger).22 Extra precision flexible docking of flucloxacillin and its metabolites into both proteins was performed with a 0.8 scaling factor of van der Waals radii and a partial charge cutoff of 0.15. Ligand interaction diagrams were generated using Maestro's Ligand Interaction Diagram tool.25
Pharmacophore screening of TLR-7 agonists
Structures of 36 compounds, including 23 TLR-7 agonists and 13 without activity on TLR-7, were identified from Yoo et al.27 and drawn into Masetro using the 2D sketcher tool.
The Phase (v. 3.4, Schrödinger) Advanced Pharmacophore Screening tool was used to screen the compounds for matches. The intersite distance matching tolerance was set to 2.0Å. The predictive performance measures, sensitivity, specificity, accuracy, Matthews Correlation Coefficient, positive predictive value, and negative predictive value were calculated.24
RESULTS
Identifying structural similarities among IMDILI drugs
Multivariate logistic regression identified 18 drugs (representing 12 drug classes) that were significantly associated with IMDILI (P < 0.00015, Bonferroni-adjusted limit for significance) (Figure 2). Seven of these drugs had fewer than four reports in the database or had molecular weight >500 Da and were excluded from subsequent analyses.
Figure 2.
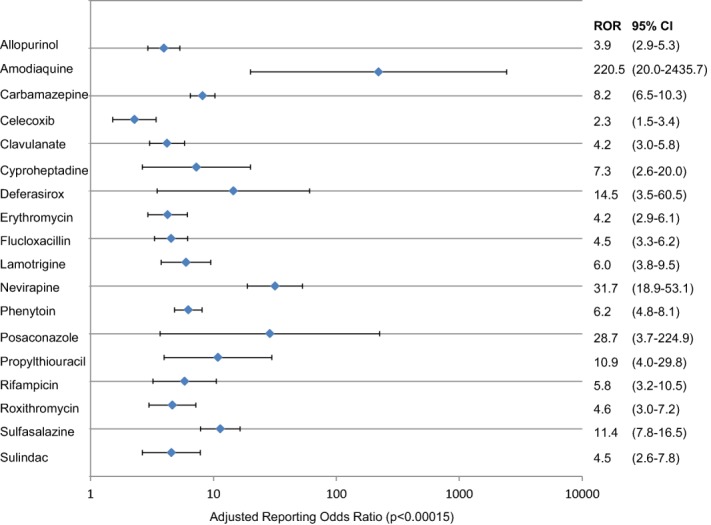
Adjusted reporting odds ratio (with 95% CI) for drugs significantly associated with immune-mediated drug induced liver injury as identified from Australia's postmarketing adverse drug reaction surveillance system (P < 0.00015, Bonferroni adjusted).
The remaining 11 drugs (Table 1) formed the basis of a training set for the in silico development of pharmacophore models. Schrödinger's Phase program (v. 3.4)23 identified a number of four-point pharmacophore hypotheses that were common to 9 out of 11 drug/metabolite groups of the training set. Given the diverse pharmacological actions of these compounds, these structural similarities are unlikely to relate to primary pharmacological targets and effects and we propose that these pharmacophore hypotheses may mirror as-yet unidentified target sites within the toxicity pathways.
Table 1.
List of drug/metabolite groups in the training and test sets and whether or not members of the group contain the pharmacophore hypothesis
| Drug/metabolite group | Pharmacophore | |
|---|---|---|
| Training set | Allopurinol | Yes |
| Carbamazepine | Yes | |
| Celecoxib | Yes | |
| Clavulanate | No | |
| Flucloxacillin | Yes | |
| Lamotrigine | Yes | |
| Nevirapine | Yes | |
| Phenytoin | Yes | |
| Propylthiouracil | Yes | |
| Sulfasalazine | Yes | |
| Sulindac | No | |
| Active test set | Bromfenac | Yes |
| Chlormezanone | No | |
| Lumiracoxib | Yes | |
| Nomifensine | Yes | |
| Oxyphenisation | Yes | |
| Pemoline | No | |
| Tienilic acid | Yes | |
| Ximelagatran | Yes | |
| Inactive test set | Alprazolam | No |
| Aripiprazole | No | |
| Baclofen | No | |
| Fluvoxamine | No | |
| Ketorolac | Yes | |
| Pregabalin | No | |
| Diphenhydramine | No | |
| Betaine hydrochloride | No | |
| Clemastine | No | |
| Isoproterenol | Yes | |
| Methysergide | No | |
| Oxybutynin | No | |
| Phenoxybenzamine | No |
Using a combination of the Phase survival score and visual inspection, we selected a primary pharmacophore hypothesis for validation and further study. This hypothesis consisted of two hydrogen bond acceptors, a hydrogen bond donor, and a ring (Figure 3).
Figure 3.
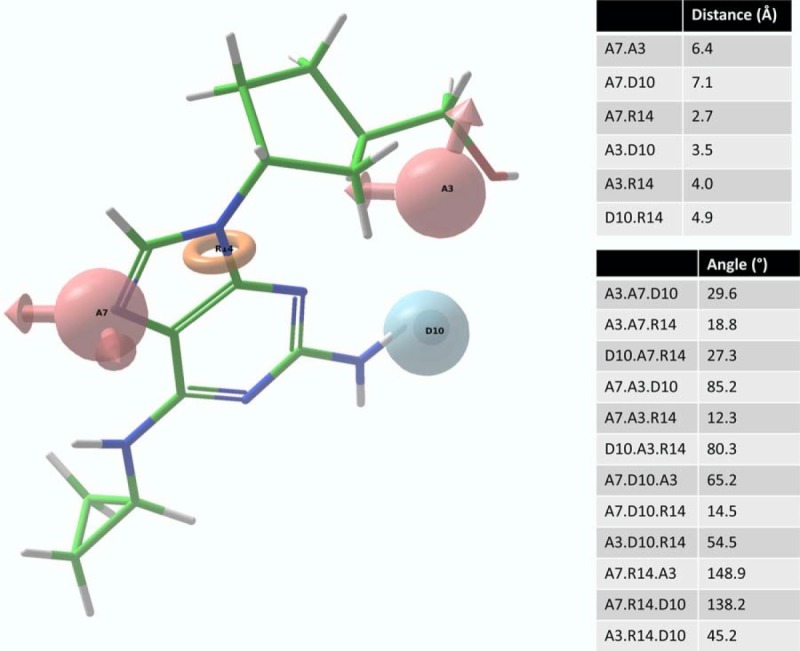
Abacavir superimposed on our pharmacophore hypothesis which consists of two hydrogen bond acceptor sites (A3, A7), a hydrogen bond donor site (D10), and a ring moiety (R14).
External validation of pharmacophore model
Validation of the pharmacophore using an external test set of drugs showed that our model exhibits predictive performance characterized by a Matthews Correlation Coefficient of 0.60 and sensitivity and specificity of 75% and 85%, respectively. The full list of drugs and metabolites possessing the pharmacophore hypothesis is presented in Table 2.
Table 2.
List of molecules that contain the pharmacophore hypothesis
| Training set | Active test set | Inactive test set |
|---|---|---|
| Allopurinol | Bromfenac | Ketorolac |
| Allopurinol M1 | Bromfenac M2 | Ketorolac M1 |
| Allopurinol M2 | Bromfenac M3 | Isoproterenol |
| Allopurinol M3 | Lumiracoxib | Isoproterenol M1 |
| Allopurinol M4 | Lumiracoxib M2 | |
| Carbamazepine M1 | Lumiracoxib M3 | |
| Carbamazepine M2 | Lumiracoxib M5 | |
| Celecoxib | Lumiracoxib M6 | |
| Celecoxib M1 | Lumiracoxib M7 | |
| Celecoxib M2 | Lumiracoxib M8 | |
| Flucloxacillin | Lumiracoxib M9 | |
| Flucloxacillin M1 | Lumiracoxib M10 | |
| Flucloxacillin M2 | Lumiracoxib M11 | |
| Flucloxacillin M3 | Lumiracoxib M12 | |
| Lamotrigine M1 | Lumiracoxib M13 | |
| Nevirapine | Lumiracoxib M15 | |
| Nevirapine M1 | Lumiracoxib M16 | |
| Nevirapine M2 | Lumiracoxib M17 | |
| Nevirapine M3 | Lumiracoxib M18 | |
| Nevirapine M4 | Nomifensine M4 | |
| Nevirapine M5 | Oxyphenisatin | |
| Phenytoin M3 | Tienilic acid | |
| Phenytoin M6 | Tienilic acid M1 | |
| Propylthiouracil M6 | Tienilic acid M2 | |
| Propylthiouracil M7 | Tienilic acid M3 | |
| Sulfasalazine | Tienilic acid M4 | |
| Sulfasalazine M1 | Tienilic acid M5 | |
| Sulfasalazine M2 | Ximelagatran | |
| Sulfasalazine M3 | Ximelagatran M1 | |
| Sulfasalazine M4 | Ximelagatran M2 | |
| Sulfasalazine M5 | Ximelagatran M3 | |
| Sulfasalazine M6 | Ximelagatran M4 | |
| Ximelagatran M5 | ||
| Ximelagatran M6 |
Computational modeling of abacavir and flucloxacillin metabolites
Extra precision flexible docking showed that the metabolites of flucloxacillin were able to dock into abacavir's binding pocket. The pharmacophoric features are positioned to interact with complementary amino acid residues within the HLA binding groove. Furthermore, we demonstrated in silico that the penicilloic acid metabolites of flucloxacillin and of its 5-hydroxymethyl derivative contain structural features positioned to interact with all of the amino acid residues that were associated with abacavir binding as identified by Peters and co-workers28; specifically Tyr9, Tyr-74, Ile-95, Val97, Tyr99, Tyr123, Ile-124, Trp147, Ile3, Leu7, and Val9 (Figure 4).
Figure 4.

Comparison between the docking of abacavir and penicilloic acid, a metabolite of flucloxacillin. (a,b) Abacavir and penicilloic acid respectively docked into HLA:B*5701 (3UPR). (c,d) Interaction of abacavir and penicilloic acid, respectively, with the amino acids in the binding pocket.
The binding affinity of abacavir and the metabolites of flucloxacillin in HLA-B*5703 was significantly lower than that for HLA-B*5701 (Supplementary Table 4).25
TLR-7 as a potential toxicity target
We noted that loxoribine, a model TLR-7 agonist, possesses an arrangement of structural features that is consistent with our pharmacophore hypothesis.
Our primary pharmacophore hypothesis was able to differentiate between active ligands and ligands without activity on TLR-7 with sensitivity and specificity of 74% and 70%, respectively (Table 3).
Table 3.
List of compounds active and inactive on Toll-like receptor 7 and whether or not the compounds contain the pharmacophore hypothesis
| Compound | Structure | Activity on TLR7 | Pharmacophore |
|---|---|---|---|
| 5 |  |
Yes | No |
| 6a |  |
No | No |
| 6b |  |
No | No |
| 11a |  |
No | No |
| 11b |  |
No | No |
| 17 |  |
No | No |
| 19a | 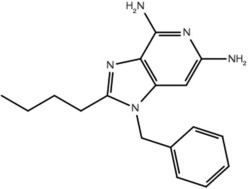 |
Yes | Yes |
| 19b | 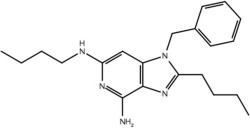 |
Yes | Yes |
| 19c | 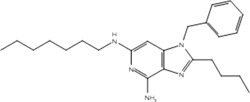 |
Yes | Yes |
| 19d | 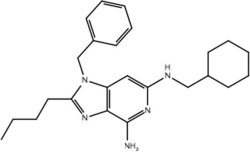 |
Yes | Yes |
| 19e | 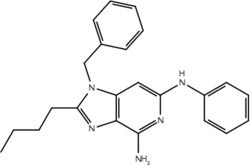 |
Yes | Yes |
| 19f | 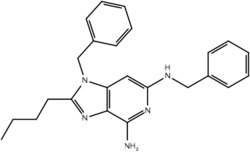 |
Yes | Yes |
| 19g | 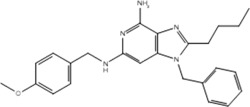 |
Yes | Yes |
| 19h | 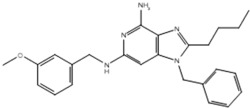 |
Yes | Yes |
| 19i | 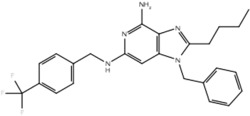 |
Yes | Yes |
| 19j | 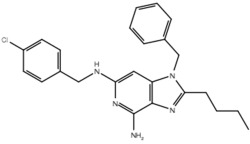 |
Yes | Yes |
| 19k | 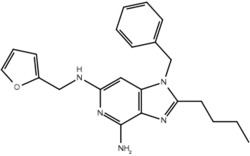 |
Yes | Yes |
| 19l | 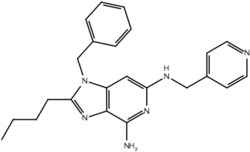 |
Yes | Yes |
| 19m | 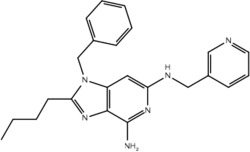 |
Yes | Yes |
| 19n | 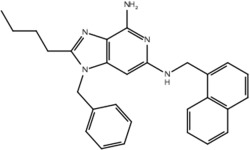 |
No | Yes |
| 19o | 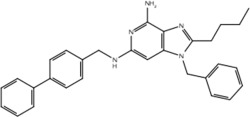 |
Yes | Yes |
| 19p | 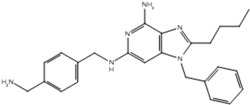 |
Yes | Yes |
| 19q | 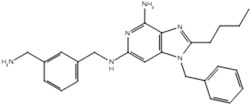 |
Yes | Yes |
| 19r | 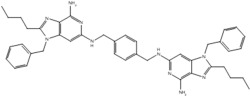 |
Yes | Yes |
| 19s | 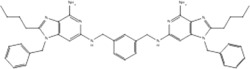 |
No | Yes |
| 23a | 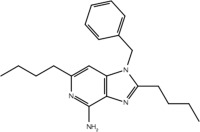 |
Yes | No |
| 23b |  |
No | No |
| 23c |  |
No | Yes |
| 23d |  |
No | No |
| 23e |  |
No | No |
| 23f | 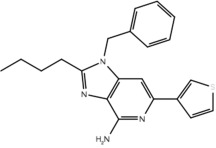 |
No | No |
| 23g | 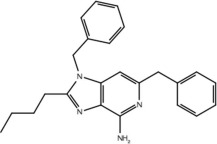 |
Yes | No |
| 23h | 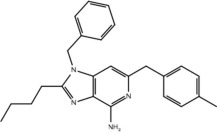 |
Yes | No |
| 23i | 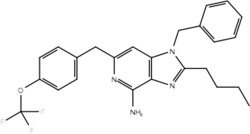 |
No | Yes |
| 23j | 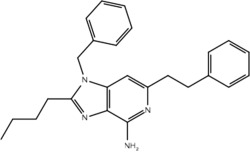 |
Yes | No |
| 30 | 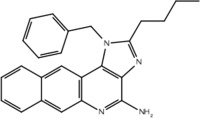 |
Yes | No |
True positive = 17; false positive = 4; true negative = 9; false negative = 6.
DISCUSSION
In this study we integrated the methods of pharmacoepidemiology and computational chemistry to reveal a structure–activity relationship in the form of a four-point pharmacophore associated with IMDILI. Since it is unknown whether the clinical toxicity is due to the parent compound or a metabolite, each drug was grouped with its known and proposed metabolites into a drug/metabolite group. A potential limitation of this approach is that by increasing the number of molecules in each drug/metabolite group, we increase the risk of identifying structural features from molecules not involved in the toxicity. In addition, given the limited size of the training and test sets our pharmacophore hypothesis may not be generalizable. However, the strength of this approach is the ability to identify new potential toxicity targets. In essence, the patterns of exposure and toxicity in the pharmacovigilance data have revealed part of a suspicious molecular fingerprint that may be interacting with immune system targets in vulnerable individuals.
This observation provided a probe set of molecules sharing a unique structural pattern that we used to explore two putative targets within immune pathways that may precipitate tissue injury in vulnerable individuals; namely, the major histocompatability complex (HLA) that is involved in activating T cells as part of the adaptive immune response and TLR-7, a pattern-recognition receptor involved in the innate immune system, which may be involved in priming or augmenting the adaptive immune response.
The hapten hypothesis is the most longstanding theory of IMDILI, and is based on the premise that small molecules (<1,000D) are too small to stimulate an immune response. Reactive metabolites formed from parent drugs act as haptens, binding covalently to cellular proteins that are subsequently processed and presented to T cells by the HLA on antigen-presenting cells (APCs) in order to initiate the adaptive immune response.29 This is supported by the increased likelihood of hepatotoxicity from drugs with greater fractional clearance via hepatic metabolism,30 suggesting that it is metabolites, rather than the parent drug, which are responsible for toxicity.31
However, hapten formation ability alone may be insufficient to induce an immune response. Rather, a secondary signal may be required for the upregulation of costimulatory molecules on APCs before the immune response is initiated.32 This has been termed the "danger hypothesis" since the secondary signal is thought to consist of cytokines released as a result of cellular damage, indicating there is "danger" to the tissue.33
A modification of the hapten hypothesis proposes that rather than T-cell activation via APC presentation of haptenated proteins, the small drug molecules themselves (or their metabolites) can interact with HLA and/or T-cell receptors in a noncovalent manner.34 This has been termed the pharmacological-interaction (p-i) hypothesis, and may explain the IMDILI of some drugs (e.g., ximelagatran35) that do not generate reactive metabolites. Drug molecules and metabolites are thought to reversibly bind to peptides complexed with HLA molecules. The drug then acts like a bridge, linking the T cell and the APC via HLA in order to initiate an immune response.36
We explored this hypothesis in silico by comparing the interaction of HLA-B*5701 with both abacavir and flucloxacillin metabolites. The recently published article on abacavir hypersensitivity28 provides evidence for a modified p-i hypothesis, which the authors termed the altered self-peptide repertoire model. In this model, abacavir binds in the F-pocket at the base of the HLA-B*5701 groove. The presence of abacavir alters the set of endogenous peptides presented to T cells by HLA-B*5701, and this is thought to be the mechanism behind abacavir's observed HLA-B*5701 restricted hypersensitivity.28
We noted with interest that abacavir shares the same four-point pharmacophore as the hepatotoxic molecules identified in our analysis (Figure 3). Hence, we hypothesized that other molecules of our training set may interact with HLA in similar ways to abacavir.
Metabolites of flucloxacillin and abacavir share common interactions with HLA-B*5701 and the cocrystallized peptide, inferring that T-cell activation via HLA-B*5701 may be a common toxicity pathway for both abacavir and flucloxacillin (Figure 4). Indeed, Wuillemin et al.37 recently demonstrated the importance of cytotoxic T cells in mediating flucloxacillin induced DILI in HLA-B*5701+ patients.
In contrast, although both abacavir and the metabolites of flucloxacillin are able to bind to abacavir-insensitive HLA-B*5703, the binding affinities are lower as indicated by their greater GlideScores (Supplementary Table 4). This provides further evidence that abacavir and flucloxacillin may share the same toxicity pathway.
It is increasingly recognized that the innate immune system also plays a vital role in the pathogenesis of IMDILI.38,39 TLRs are a class of pattern recognition receptors that form an integral part of the innate immune system, as they recognize and respond to pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs).
TLRs have a role in liver injury, with recent research linking TLR-9 and liver fibrosis40 and TLR-2 with neutrophil recruitment in acute and chronic liver injury.41 Lipopolysaccharides from Gram-negative bacterial cell walls, TLR-4 ligands, have also been known to potentiate DILI of trovafloxacin in animal models.42 TLR-7, a cytoplasmic endosomal TLR, the subtype investigated in this study, is similar to TLR-9 and is one of the few TLRs for which there are known small molecule modulators. All TLRs are known to activate the MyD88 pathway, the downstream effects of which include the production of inflammatory cytokines and the expression of costimulatory molecules required for the activation of the adaptive immune system.43
Our pharmacophore hypothesis, generated from molecules implicated in IMDILI, was able to accurately differentiate between TLR-7 agonists and antagonists (Table 3). This implies that molecules of our training set share structural features with TLR-7 agonists, suggesting TLR-7 as a potential target that is common to the toxicity pathways of these drugs. It should be noted that TLR-7 is not usually detected in the liver,44 and that our study does not account for relative liver specificity. Nevertheless, we propose that this pathway may be commonly activated in all individuals exposed to these drugs and the factors differentiating toxic manifestations lie in the HLA genotype, which may explain why evidence has not emerged implicating toxicity from all of the "active" drugs in our test and training sets with the same HLA genotype. In other words, the identified structural features of our pharmacophore may be involved in activating the innate immune response but that this only results in toxicity in individuals with vulnerable HLA genotypes.
Given the degree of overlap in TLR signaling pathways, it is not unreasonable to assume that other receptors may also play a part in IMDILI. Although there was insufficient data from our study to determine whether other TLR subtypes are involved in the toxicity pathway, it is certainly worthy of further investigation.
Accurate development and validation of pharmacophore models relies on the correct assignment of activity. As such, our results are reliant on the accuracy of DILI case data. Hence, literature confirmation and refinement of toxicity classification was an important step in our method and acted as a validation step to filter out potentially false signals generated from the pharmacovigilance data. In some cases genetic associations with HLA were taken as evidence of immune involvement even if DILI does not present with prominent clinical hypersensitivity features. In the identification of noncases, it is possible that drugs with very rare incidences of DILI may have been inadvertently misclassified as nontoxic due to a lack of evidence to the contrary. In this study we demonstrated that postmarketing surveillance data can be effectively used to study rare and unpredictable ADRs. In particular, we report a set of probe molecules for IMDILI that can be utilized with confidence. We also identified a relationship between drug structure and hepatotoxicity, and were able to use this relationship to identify and investigate potential immune-system targets: HLA-B*5701 and TLR-7. These in silico results provide promising leads into the mechanisms behind IMDILI and merit further investigation and validation using in vitro and in vivo studies. Our results highlight the utility of linking information from clinical and chemical databases with knowledge from biological pathway and gene-association datasets to reveal potential mechanisms behind idiosyncratic drug reactions. This approach may assist in developing targeted experimental methods; for example, assays to confirm whether our training set drugs are able to activate TLR-7. To this end, we intend to use those molecules identified as sharing a common four-point pharmacophore, implicated in IMDILI in controlled in vitro experiments involving a number of putative receptors, including TLR-7 and HLA-B*5701.
Acknowledgments
The authors thank Australia's Therapeutic Goods Administration for provision of adverse reaction data. S. Ho is supported by the Australian Postgraduate Award scholarship.
Conflict of Interest
The authors declared no conflict of interest.
Author Contributions
S.S.H. wrote the article; D.E.H., S.S.H., A.M., T.F.C., and R.A.F. designed the research; S.S.H performed the research; D.E.H., S.S.H., and R.A.F. analyzed the data.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
References
- Reardon S. Project ranks billions of drug interactions. Nature. 2013;503:449–450. doi: 10.1038/503449a. [DOI] [PubMed] [Google Scholar]
- Liebler DC. Guengerich FP. Elucidating mechanisms of drug-induced toxicity. Nat. Rev. Drug. Discov. 2005;4:410–420. doi: 10.1038/nrd1720. [DOI] [PubMed] [Google Scholar]
- Campillos M, Kuhn M, Gavin A-C, Jensen LJ. Bork P. Drug target identification using side-effect similarity. Science. 2008;321:263–266. doi: 10.1126/science.1158140. [DOI] [PubMed] [Google Scholar]
- Scheiber J, et al. Gaining insight into off-target mediated effects of drug candidates with a comprehensive systems chemical biology analysis. J. Chem. Inf. Model. 2009;49:308–317. doi: 10.1021/ci800344p. [DOI] [PubMed] [Google Scholar]
- Low Y, et al. Predicting drug-induced hepatotoxicity using QSAR and toxicogenomics approaches. Chem. Res. Toxicol. 2011;24:1251–1262. doi: 10.1021/tx200148a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly AK. Using genome-wide association studies to identify genes important in serious adverse drug reactions. Annu. Rev. Pharmacol. Toxicol. 2012;52:21–35. doi: 10.1146/annurev-pharmtox-010611-134743. [DOI] [PubMed] [Google Scholar]
- Daly AK, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009;41:816–819. doi: 10.1038/ng.379. [DOI] [PubMed] [Google Scholar]
- Russmann S, Jetter A. Kullak-Ublick GA. Pharmacogenetics of drug-induced liver injury. Hepatology. 2010;52:748–761. doi: 10.1002/hep.23720. [DOI] [PubMed] [Google Scholar]
- Wu Y, et al. Inhibiting the TLR4-MyD88 signalling cascade by genetic or pharmacological strategies reduces acute alcohol-induced sedation and motor impairment in mice. Br. J. Pharmacol. 2012;165:1319–1329. doi: 10.1111/j.1476-5381.2011.01572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C, et al. Curcumin attenuates Concanavalin A-induced liver injury in mice by inhibition of Toll-like receptor (TLR) 2, TLR4 and TLR9 expression. Int. Immunopharmacol. 2012;12:151–157. doi: 10.1016/j.intimp.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Su Y, et al. Lipopolysaccharide exposure augments isoniazide-induced liver injury. J. Appl. Toxicol. 2014;34:1436–1442. doi: 10.1002/jat.2979. [DOI] [PubMed] [Google Scholar]
- Vilar S, et al. Facilitating adverse drug event detection in pharmacovigilance databases using molecular structure similarity: application to rhabdomyolysis. J. Am. Med. Inf. Assoc. 2011;18(Suppl 1):i73–80. doi: 10.1136/amiajnl-2011-000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursem CJ, et al. Identification of structure-activity relationships for adverse effects of pharmaceuticals in humans. Part A: use of FDA post-market reports to create a database of hepatobiliary and urinary tract toxicities. Regul. Toxicol. Pharmacol. 2009;54:1–22. doi: 10.1016/j.yrtph.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Matthews EJ, et al. Identification of structure-activity relationships for adverse effects of pharmaceuticals in humans: Part B. Use of (Q)SAR systems for early detection of drug-induced hepatobiliary and urinary tract toxicities. Regul. Toxicol. Pharmacol. 2009;54:23–42. doi: 10.1016/j.yrtph.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Matthews EJ, et al. Identification of structure-activity relationships for adverse effects of pharmaceuticals in humans: Part C: use of QSAR and an expert system for the estimation of the mechanism of action of drug-induced hepatobiliary and urinary tract toxicities. Regul. Toxicol. Pharmacol. 2009;54:43–65. doi: 10.1016/j.yrtph.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Matthews EJ. Frid AA. Prediction of drug-related cardiac adverse effects in humans—A: creation of a database of effects and identification of factors affecting their occurrence. Regul. Toxicol. Pharmacol. 2010;56:247–275. doi: 10.1016/j.yrtph.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Frid AA. Matthews EJ. Prediction of drug-related cardiac adverse effects in humans—B: use of QSAR programs for early detection of drug-induced cardiac toxicities. Regul. Toxicol. Pharmacol. 2010;56:276–289. doi: 10.1016/j.yrtph.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Hamburg MA. Advancing regulatory science. Science. 2011;331:987. doi: 10.1126/science.1204432. [DOI] [PubMed] [Google Scholar]
- Bender R. Lange S. Adjusting for multiple testing—when and how? J. Clin. Epidemiol. 2001;54:343–349. doi: 10.1016/s0895-4356(00)00314-0. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug Metab. Pharmacokinet. 2011;26:3–14. doi: 10.2133/dmpk.dmpk-10-rv-062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Borlak J. Tong W. High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology. 2013;58:388–396. doi: 10.1002/hep.26208. [DOI] [PubMed] [Google Scholar]
- Watts KS, et al. ConfGen: a conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 2010;50:534–546. doi: 10.1021/ci100015j. [DOI] [PubMed] [Google Scholar]
- Dixon SL, et al. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006;20:647–671. doi: 10.1007/s10822-006-9087-6. [DOI] [PubMed] [Google Scholar]
- Vihinen M. How to evaluate performance of prediction methods? Measures and their interpretation in variation effect analysis. BMC Genomics. 2012;13:S2. doi: 10.1186/1471-2164-13-S4-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Stewart-Jones GBE, et al. Structures of three HIV-1 HLA-B*5703-peptide complexes and identification of related HLAs potentially associated with long-term nonprogression. J. Immunol. 2005;175:2459–2468. doi: 10.4049/jimmunol.175.4.2459. [DOI] [PubMed] [Google Scholar]
- Yoo E, et al. Structure-activity relationships in Toll-like receptor 7 agonistic 1H-imidazo[4,5-c]pyridines. Org. Biomol. Chem. 2013;11:6526–6545. doi: 10.1039/c3ob40816g. [DOI] [PubMed] [Google Scholar]
- Ostrov DA, et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc. Natl. Acad. Sci. U. S. A. 2012;109:9959–9964. doi: 10.1073/pnas.1207934109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetrecht J. Idiosyncratic drug reactions: current understanding. Annu. Rev. Pharmacol. Toxicol. 2007;47:513–539. doi: 10.1146/annurev.pharmtox.47.120505.105150. [DOI] [PubMed] [Google Scholar]
- Lammert C, Bjornsson E, Niklasson A. Chalasani N. Oral medications with significant hepatic metabolism at higher risk for hepatic adverse events. Hepatology. 2010;51:615–620. doi: 10.1002/hep.23317. [DOI] [PubMed] [Google Scholar]
- Uetrecht J. Idiosyncratic drug reactions: past, present, and future. Chem. Res. Toxicol. 2008;21:84–92. doi: 10.1021/tx700186p. [DOI] [PubMed] [Google Scholar]
- Matzinger P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- Matzinger P. Friendly and dangerous signals: is the tissue in control? Nat. Immunol. 2007;8:11–13. doi: 10.1038/ni0107-11. [DOI] [PubMed] [Google Scholar]
- Adam J, Pichler WJ. Yerly D. Delayed drug hypersensitivity: models of T-cell stimulation. Br. J. Clin. Pharmacol. 2011;71:701–707. doi: 10.1111/j.1365-2125.2010.03764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindmark A, et al. Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenomics J. 2008;8:186–195. doi: 10.1038/sj.tpj.6500458. [DOI] [PubMed] [Google Scholar]
- Pichler WJ. Pharmacological interaction of drugs with antigen-specific immune receptors: the p-i concept. Curr. Opin. Allergy Clin. Immunol. 2002;2:301–305. doi: 10.1097/00130832-200208000-00003. [DOI] [PubMed] [Google Scholar]
- Wuillemin N, et al. T cells infiltrate the liver and kill hepatocytes in HLA-B(*)57:01-associated floxacillin-induced liver injury. Am. J. Pathol. 2014:1–6. doi: 10.1016/j.ajpath.2014.02.018. [DOI] [PubMed] [Google Scholar]
- Uetrecht JP. New concepts in immunology relevant to idiosyncratic drug reactions: the “danger hypothesis” and innate immune system. Chem. Res. Toxicol. 1999;12:387–395. doi: 10.1021/tx980249i. [DOI] [PubMed] [Google Scholar]
- Williams D. Jaeschke H. Role of innate and adaptive immunity during drug-induced liver injury. Toxicol. Res. (Camb) 2012;1:161–170. [Google Scholar]
- Abu-Tair L, et al. Natural killer cell-dependent anti-fibrotic pathway in liver injury via Toll-like receptor-9. PLoS One. 2013;8:e82571. doi: 10.1371/journal.pone.0082571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moles A, et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J Hepatol. 2014;60:782–791. doi: 10.1016/j.jhep.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ, Ganey PE. Roth RA. Idiosyncratic drug-induced liver injury and the role of inflammatory stress with an emphasis on an animal model of trovafloxacin hepatotoxicity. Toxicol. Sci. 2010;118:7–18. doi: 10.1093/toxsci/kfq168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K. Akira S. Toll-like receptors in innate immunity. Int. Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- UniProt. Toll-like receptor 7 (Q9NYK1) [monograph on the Internet]. < www.uniprot.org/uniprot/Q9NYK1 >.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.