

Abstract
We present the case of a 28-year-old gentleman who presented with weakness and wasting in the right arm. He complained about painful cramps in his left leg but there were no sensory, visual or swallowing problems. Neurological examination was significant for asymmetric weakness of both upper and lower limbs and deep tendon reflexes were asymmetrically brisk. Initial laboratory investigations, MRI brain and MRI spine were normal. Electromyography revealed active denervation and chronic neurogenic motor unit action potential. Myokymic discharges were noted in the left deltoid muscle.
Background
Myokymic discharges (MDs) are rarely reported in amyotrophic lateral sclerosis (ALS)1 which is the most frequent form of motor neuron disease (MND).2 MDs are rare, spontaneous, single or few motor potentials that fire in a burst pattern. They may or may not be associated with clinical myokymia and occur as a result of hyperexcitablity of the anterior horn cell or axon membrane.1 The disorders associated with MDs are often classified into two types according to anatomic region, that is limb and facial myokymia.1 3 MDs in cranial muscle have been reported in several diseases and most common of which is multiple sclerosis (MS).1 Limb myokymia is quite rare and is usually found to be associated with radiation-induced plexopathy, neuromyotonia, lead poisoning, thyrotoxicosis, peripheral nerve injury and chronic recurrent polyneuropathy.4 We present the case of MDs in limb muscle in patients with ALS.
Case presentation
A 28-year-old gentleman presented to our hospital with a history of weakness in the right arm and shoulder, with discomfort and difficulty in dressing himself, for the past 5 years. He then noted progressive wasting in the muscles of his right arm and hand with decreased grip strength and developed uncomfortable painful spasms or cramps in his left leg. There were no sensory, visual symptoms or dysphagia. There was no history of trauma and medical history was also insignificant.
On examination he was awake, alert and oriented to time, place and person. Higher mental function, cranial nerves and fundoscopic examination were normal. No tongue fasciculations were noted. Neck was supple and speech was dysarthric. Motor examination showed generalised wasting which was more marked in the right biceps and deltoid and also in the right first dorsal interossei muscle. Fasiculations were noted in the left forearm, left first dorsal interossei and vastus medialis muscle. Strength testing results, using Medical Research Council grades, were as follows: right shoulder abductors 3/5, elbow flexors 4/5 and diminished grip strength 4/5. Left shoulder abductors 4/5, elbow flexors 3/5 and diminished grip strength 4/5. Examination of the lower limbs revealed that bilateral hip flexors and extensors were 3/5, knee flexors were 4/5 on the right side and 3/5 on the left side while knee extensors were 4/5 on the right side and 4/5 on the left side. On dorsiflexion and plantar flexion power was 4/5 bilaterally. Deep tendon reflexes in the right upper limb were 2/4 in brachioradialis and biceps and1/4 in triceps while on the left side it was 3/5 in brachioradialis and 2/5 in biceps and triceps. Knee jerk was 3/4 on the right side while 2/4 on the left side. Ankle jerk was 3/4 bilaterally. His toes were flexors bilaterally. Cerebellar examination was normal. His sensory examination was completely intact. On cardiovascular examination, S1 and S2 were audible with no added sounds or murmur. Respiratory examination revealed chest was clear bilaterally with no viable sounds. The rest of the physical and systemic examination was also unremarkable.
Investigations
His initial laboratory investigations that included complete blood count, electrolytes, serum creatinine phosphokinase, ESR, C-reactive protein, random blood sugar, renal, thyroid and liver functions test were normal. Radiological examinations, such as chest x-ray and ultrasound of abdomen, were normal. Cerebrospinal fluid analysis, MRI brain and MRI cervical and dorsal spine were normal. Electromyography (EMG)/nerve conduction studies were performed according to standard protocols of MND which are used in the clinical neurophysiology laboratory and revealed normal sensory nerve conduction of upper and lower limbs while nerve conduction studies of motor nerves with low (CMAP) amplitudes in upper and lower limbs. F latencies were prolonged while nerve conduction velocities were normal (figures 1 and 2). Needle examination revealed fibrillation, positive sharp waves and fasciculation potentials as well as high-amplitude, broad and polyphasic motor unit action potentials with rapid firing rate and incomplete interference pattern involving cranial, cervical (upper limbs), thoracic paraspinal muscle and lumbosacral regions (lower limbs). Pronounced MDs were noted in the left deltoid muscle (figure 3). On the basis of the available electrical data and clinical findings, these findings are suggestive of pure motor neuron disease.
Figure 1.

Sensory nerve conduction studies.
Figure 2.

Motor nerve conduction studies.
Figure 3.
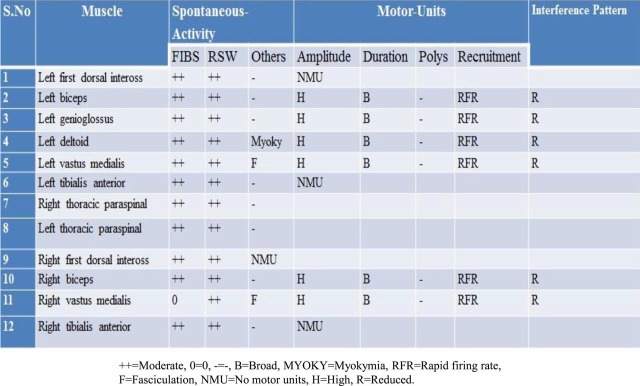
Electromyography.
Treatment
The patient was advised to take Riluzole 50 mg twice daily and was counselled for need of oxygen, nutritional management, physiotherapy, speech therapy and occupational therapy. Supportive management included Quinine, Baclofen and NSAIDs for cramps, discomfort and spasticity, respectively.
Discussion
MND is an adult onset neurodegenerative disease that leads to weakness of limb, bulbar and respiratory muscles.5 Proper clinical studies, electrophysiology and neuroimaging are necessary before reaching a diagnosis of MND5 6 which has been standardised through El Escorial criteria.7 The diagnosis of ALS is considered when signs of upper motor neuron and lower motor neuron (LMN) involvement are found combined with progressive disease course.2 8 Myokymia is typically seen in demyelinating disorders.9 MDs in cranial muscles have been reported in MS, inflammatory demyelinating polyradiculopathies, brain stem neoplasms and following radiation to the head and neck.1
Limb myokymia has been reported previously in association with diffuse disease of the central and peripheral nervous system but is most characteristically seen in radiation-induced nerve damage.10 Although MDs are more commonly found in limb muscles, clinical myokymia is more often observed in facial muscles, probably owing to less subcutaneous tissues than in limb muscles.11 The mechanism of generation of MDs in ALS is unclear.1 12 Multiple mechanisms such as demyelination, radiation, direct neurotoxic effects, ischaemia, hypoxia and oedema are thought to alter the axon membrane microenvironment and thereby form the basis for hyperexcitablity of the axon membrane.12 However, they might be the result of centrally generated rhythmic discharges that electronically spread to abnormally hyperexcitable anterior horn cells in a variety of peripheral and central nervous system disorders.1 The origin of MDs likely involves spontaneous depolarisation or ephaptic transmission along demyelinated segments of nerve.12 The finding of MDs on EMG is very helpful in limiting the differential diagnosis.
ALS is a disorder that begins focally and spreads contiguously and EMG findings will reflect that evolution. Electrodiagnostic studies (nerve conduction studies and EMG) provide supportive evidence of LMN involvement. Sensory nerve conduction studies are normal and motor nerve conduction studies usually reveal only low CMAP amplitude. EMG provides most valuable supporting evidence of LMN injury in the form of widespread active denervation including fasciculation potentials and evidence of reinnervation.13 Management therefore properly focuses on symptom relief and preservation of independence and quality of life.5 14 Riluzole is the only treatment which is currently approved by US Food and Drug Administration and it prolongs life by 3–4 months.13 14
Learning points.
Limb myokymia can be rarely seen in anterior horn cell disorder such as amyotrophic lateral sclerosis.
Footnotes
Competing interests: None.
Patient consent: Obtained.
References
- 1.Whaley NR, Rubin DI. Myokymic discharges in amyotrophic lateral sclerosis (ALS): a rare electrophysiologic finding? Muscle Nerve 2010;41:107–9. [DOI] [PubMed] [Google Scholar]
- 2.Bouche P, Le Forestier N, Maisonobe T, et al. Electrophysiological diagnosis of motor neuron disease and pure motor neuropathy. J Neurol 1999;246:520–5. [DOI] [PubMed] [Google Scholar]
- 3.Kaji M, Shoji H. Myokymia. Nihon Rinsho 1993;51:2866–70. [PubMed] [Google Scholar]
- 4.Hosokawa S, Shinoda H, Sakai T, et al. Electrophysiological study on limb myokymia in three women. J Neurol Neurosurg Psychiatry 1987;50:877–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee CN. Reviewing evidences on the management of patients with motor neuron disease. Hong Kong Med J 2012;18:48–55. [PubMed] [Google Scholar]
- 6.Ross MA, Miller RG, Berchert L, et al. Toward earlier diagnosis of amyotrophic lateral sclerosis: revised criteria. rhCNTF ALS Study Group. Neurology 1998;50:768–72. [DOI] [PubMed] [Google Scholar]
- 7.Arts IM, Overeem S, Pillen S, et al. Muscle ultrasonography: a diagnostic tool for amyotrophic lateral sclerosis. Clin Neurophysiol 2012. 2012;123:1662–7. [DOI] [PubMed] [Google Scholar]
- 8.Léger JM, Salachas F. Diagnosis of motor neuropathy. Eur J Neurol 2001;8:201–8. [DOI] [PubMed] [Google Scholar]
- 9.Kelkar P, Kimura J. Myokymia in axonal disorders. J Clin Neuromuscul Dis 2005;7:55–8. [DOI] [PubMed] [Google Scholar]
- 10.Sinclair A, Davies N. Neurological picture. Conus medullaris lesion causing bilateral lower limb myokymia. J Neurol Neurosurg Psychiatry 2007;78:1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubin DI. Needle electromyography: basic concepts and patterns of abnormalities. Neurol Clin 2012;30:429–56. [DOI] [PubMed] [Google Scholar]
- 12.Glenn SA, Ross MA. Delayed radiation-induced bulbar palsy mimicking ALS. Muscle Nerve 2000;23:814–17. [DOI] [PubMed] [Google Scholar]
- 13.Sorenson EJ. The electrophysiology of the motor neuron diseases. Neurol Clin 2012;30:605–20. [DOI] [PubMed] [Google Scholar]
- 14.Wood-Allum C, Shaw PJ. Motor neurone disease: a practical update on diagnosis and management. Clin Med 2010;10:252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]