

Abstract
Purpose
To determine the possible antiangiogenic effect of metalloproteinase (MMP) 14 cleavage of vascular endothelial growth factor receptor 1 (VEGFR1) in the cornea.
Methods
Recombinant mouse (rm) VEGFR1 was incubated with various concentrations of recombinant MMP14 to examine proteolysis in vitro. The reaction mixture was analyzed by SDS-PAGE and stained with Coomassie blue. The fragments resulting from rmVEGFR1 cleavage by MMP14 were subjected to Edman degradation, and the amino acid sequences were aligned with rmVEGFR1 sequences. Surface plasmon resonance was used to determine the equilibrium dissociation constant (KD) between MMP14 and rmVEGFR1. The KD value of rmVEGFR1 and the 59.8-kDa cleavage product binding to VEGF-A165 was also determined. Cell proliferation assays were performed in the presence of VEGF-A165 plus the 59.8-kDa VEGFR1 fragment or VEGF-A165 alone.
Results
Matrix metalloproteinase 14 binds and cleaves rmVEGFR1 to produce 59.8-kDa (N-terminal fragment, Ig domains 1–5), 35-kDa (C-terminal fragment containing IgG and His-tag), and 21-kDa (Ig domains 6–7) fragments. The 59.8-kDa fragment showed binding to VEGF-A165 and inhibited VEGF-induced endothelial cell mitogenesis.
Conclusions
Our findings suggest that VEGFR1 cleavage by MMP14 in the cornea leads to a VEGF-trap effect, reducing the proangiogenic effect of VEGF-A165, thereby reducing corneal angiogenesis.
Keywords: matrix metalloproteinase 14, vascular endothelial growth factor receptor, angiogenesis, cleavage product
MMP14 binds and cleaves VEGFR1.
The membrane-bound matrix metalloproteinase 14 (MMP14, MT1-MMP) is one of the most well-studied members of the metalloproteinase family, both in vivo and in vitro, and in both normal physiological and disease models. Matrix metalloproteinase 14 has a variety of roles in processes that involve angiogenesis, including wound healing, inflammation, and cancer invasion and metastasis.1–6 In the MMP14 knockout (KO) mouse,7 corneal angiogenesis fails to occur following normal stimulation techniques, including the application of basic fibroblast growth factor (bFGF).7 Other experiments have shown that angiogenesis and tumor growth are impaired in the absence of MMP14.4,8–10 Matrix metalloproteinase 14 has been implicated in angiogenesis not only as a proangiogenic factor (e.g., MMP-dependent endothelial cell migration8,11) but also as an antiangiogenic factor (e.g., in corneal epithelium6,12), and both up- and downregulation of MMP14 have been implicated in a variety of disease models of angiogenesis.9,13,14
In most of these systems, the most crucial role of MMP14 is thought to be the remodeling of the extracellular matrix (ECM), where it acts to modify essentially all components of the ECM, including collagens, proteoglycans, and fibronectins.5,15,16 The role of MMP14 in the proteolysis of other transmembrane or nonmatrix extracellular proteins remains less well characterized.17,18 In this capacity, it is best known for its ability to cleave, in conjunction with tissue inhibitor of metalloproteinase 2 (TIMP2), its fellow matrix metalloproteinase 2 (MMP2) from its pro-form to its active form.2,19 Previous studies have indicated that the vascular endothelial growth factor receptor 1 (VEGFR1) is cleaved in response to stimulation by its ligand, VEGF.20,21 The agent performing this cleavage has not been identified, but it is suspected to be a member of the metalloproteinase family because cleavage can be blocked by GM6001, a general inhibitor of some metalloproteinases. We hypothesize that MMP14 might be the factor responsible for VEGFR1 cleavage.
Vascular endothelial cells express the VEGF receptors VEGFR1 and VEGFR2, which have overlapping and complementary roles during angiogenesis.22 Both VEGFR1 and VEGFR2 (but not VEGFR3) bind VEGF, but with differing affinities and downstream effects. Vascular endothelial growth factor receptor 1 has a much higher affinity for VEGF (approximately 10× greater) compared to VEGFR2, but VEGFR2 initiates a much higher level of tyrosine kinase activity (approximately 10× greater than VEGFR1).23–26 Vascular endothelial growth factor receptor 1 forms both homo- and heterodimers with family receptors on endothelial cells, in varying proportions depending on conditions.27,28 While much is known about the individual activity of each of these important receptors, less is known about the cellular function of heterodimers with family receptors. It has been suggested that the principal role of VEGFR1 may be to act as a modulator of VEGFR2 activity because VEGFR1 initiates almost no tyrosine kinase activity itself25 and is thought to initiate less tyrosine kinase activity than the VEGFR2.29 Alterations to levels of VEGFR1 on the cell surface could, therefore, have a major impact on VEGFR2-induced kinase activity during the initiation and maintenance of angiogenesis.27 In addition, soluble VEGFR1, formed by alternative splicing, can trap free VEGF and thus serve as a specific antagonist of VEGF function,30 acting as an antiangiogenic factor.
To test our hypothesis that MMP14 cleaves VEGFR1 during angiogenesis, we conducted in vitro proteolytic experiments. Our results provide evidence confirming our hypothesis that VEGFR1 is cleaved by MMP14, and give important insights into the specificity of this activity as well as a potential functional purpose for the products of VEGFR1 cleavage.
Materials and Methods
In Vitro Proteolysis
Recombinant mouse (rm) VEGFR1 (R&D Systems, Minneapolis, MN, USA) was incubated alone or with the catalytic domain of MMP14 (cdMMP14; Calbiochem, Billerica, MA, USA). Recombinant mouse VEGFR1 also was incubated with heat-inactivated (10 minutes at 100°C) MMP14 (MMP14*, Calbiochem) or GM6001 (Calbiochem). Matrix metalloproteinase 2 (Calbiochem) was used as a positive control for these reactions. Reactions were allowed to occur for 4 hours at 37°C. Purification of 59.8-kDa cleaved VEGFR1 fragment at the N-terminus was done by pulldown with Ni-NTA agarose beads (Qiagen, Valencia, CA, USA) to remove 35-kDa C-terminal fragment containing IgG and His-tag. Unbound 59.8-kDa cleaved VEGFR1 fragment was separated from another unbound small 21-kDa (Ig domains 6–7) fragment using Amicon 30 K membrane filter (Millipore, Billerica, MA, USA).
Western Blots
The products of completed reactions were separated by 4% to 20% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) under nonreducing conditions unless stated otherwise and transferred to nitrocellulose (NC) membranes. Reducing conditions, when used, consisted of treatment with 100 mM β-mercaptoethanol solution followed by boiling for 10 minutes. Sections were stained with Coomassie blue and then destained with acetic acid and methanol. Western blot experiments were blocked using 5% milk or 3% bovine serum albumin (BSA). Membranes were incubated overnight with primary antibody (anti-His tag, GenScript, Piscataway, NJ, USA; anti-p-ERK or anti-ERK, Santa Cruz Biotechnology, Santa Cruz, CA, USA; anti-VEGFR1 and anti-MMP14, Abcam, Cambridge, MA, USA; anti-actin, Cell Signaling Technology, Inc., Danvers, MA, USA). The NC membrane was incubated with fluorescence 800CW-conjugated secondary antibody (Li-Cor, Lincoln, NE, USA). Protein bands were detected by Li-Cor Odyssey system.
Wild-type and MMP14 Δexon4 mouse corneal fibroblasts were isolated, cultured, and prepared as described previously.31 Twenty micrograms total lysates from cultured wild-type and MMP14 Δexon4 (exon 4 deleted) mouse corneal fibroblasts was subjected to Western blot to compare VEGFR1 levels.
Edman Degradation
Matrix metalloproteinase 14-cleaved rmVEGFR1 was subjected to SDS gel electrophoresis, transferred to a polyvinylidene difluoride (PVDF) membrane, and stained with Coomassie blue. Bands were excised and sequenced by capillary Edman degradation. The sequences derived from the Edman degradation were compared with mouse VEGFR1 protein sequences.
SPR Analyses
Surface plasmon resonance (SPR) analyses were performed using a BIAcore T200 instrument (GE Healthcare, Marlboro, MA, USA). Recombinant mouse VEGFR1 and the 59.8-kDa N-terminal VEGFR1 fragment (derived from rmMMP14 cleavage of rmVEGFR1) were immobilized on a CM5 sensor chip (Series S Sensor Chip CM5; GE Healthcare) using standard amine coupling. In short, flow channels were activated by 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrocholoride (EDC)/N-hydroxy succinimide (NHS) mixture, and each protein was immobilized followed by ethanolamine blocking of unoccupied surface area at 25°C. Blank immobilization was performed on flow channel 1 as a control. Immobilization levels of flow channels 2 (rmVEGFR1) and 4 (59.8-kDa cleaved VEGFR1) were 3130 and 2736 response units (RU), respectively. Vascular endothelial growth factor-A165 or Placental growth factor (PlGF) binding was measured at a flow rate of 20 μL/min in running buffer PBS-P (0.5% Surfactant P20 [Tween 20], pH 7.4) containing varied concentrations (0–20 nM at 2-fold dilution) of analyte proteins, unless otherwise indicated. Data were normalized with blank (enthanolamine) RU values. BIAcore T200 evaluation software v2.0 and SigmaPlot 12.0 (Systat Software, San Jose, CA, USA) were used to fit the data to a single rectangular hyperbolic curve to determine equilibrium dissociation constant (KD) values. The hyperbola, y = ymax·x/(KD + x), was used to plot RU and corresponding concentration, where y is the response, ymax is the maximum response, and x is the concentration of analyte (VEGF-A165 or PlGF).
VEGF-A165 Binding Affinity for the 59.8-kDa N-Terminal VEGFR1 Fragment of MMP14-Cleaved rmVEGFR1
For the assay of VEGF-A165 binding affinity for the 59.8-kDa N-terminal VEGFR1 fragment of MMP14-cleaved rmVEGFR1, 100 ng of rmVEGFR1 and the N-terminal 59.8-kDa fragment resulting from cleavage of rmVEGFR1 by MMP14 were coated onto separate wells of a 96-well clear-bottom plate. Wells were washed with PBS to remove unbound protein and blocked with 3% nonfat milk/PBS. Vascular endothelial growth factor-A165 (0, 1, or 10 nM; R&D Systems) was added to wells and incubated. Anti-VEGF-A165 (R&D Systems) conjugated to horseradish peroxidase (HRP) was added to capture the receptor/VEGF-A165 complex. Chemiluminescence was read on an enzyme-linked immunosorbent assay (ELISA) reader (Bio-Tek, Winooski, VT, USA).
Cell Proliferation Assays
Calf pulmonary artery endothelial cells (CPAEs) were maintained with endothelial cell growth medium including supplements (EBM-2; Lonza, Allendale, NJ, USA). First, 3 × 103 CPAEs were seeded in 96-well clear-bottom plates and then incubated overnight. Cells were washed with PBS and then incubated in 100 μL EBM-2 containing 0.5% fetal bovine serum for 6 hours for serum starvation. Cells were then treated for 48 hours with various concentrations (2-fold dilution of 1 to 0.015 μg/mL) of purified 59.8-kDa N-terminal VEGFR1 fragment that had been pre-exposed to 10 ng/mL VEGF-A165 for 30 minutes. Cells treated with VEGF-A165 alone were used as a positive control, which is considered the reference of 100% proliferation. Bromodeoxyuridine (10 M; Roche, Mannheim, Germany) was added for measurement of cell proliferation following the manufacturer's instructions.
Results
MMP14 Cleaves VEGFR1, but Not VEGFR2 or VEGFR3, In Vitro
Recombinant VEGFR1 was incubated with MMP14 for 4 hours (Fig. 1A, lane 4). Reactions were allowed to occur for 4 hours at 37°C in order to maximize enzyme activity of the MMP14. Matrix metalloproteinase 14 cleaved VEGFR1 in a concentration-dependent manner (Fig. 1B); complete cleavage was achieved with 200 ng MMP14/1 μg VEGFR1. Under nonreducing conditions, three distinct cleavage products of approximately 70, 59.8, and 21 kDa were produced. Cleavage required only the MMP14 catalytic domain (Fig. 1A, lane 4) and did not occur when the MMP14 catalytic domain was heat inactivated prior to incubation (marked as MMP14* in Fig. 1A, lanes 2 and 5). Cleavage was completely blocked by the general MMP inhibitor GM6001 (Fig. 1A, lanes 6 and 7). These results indicate that MMP14 proteolysis may act to modulate VEGFR1 levels on endothelial cells during angiogenesis.
Figure 1.
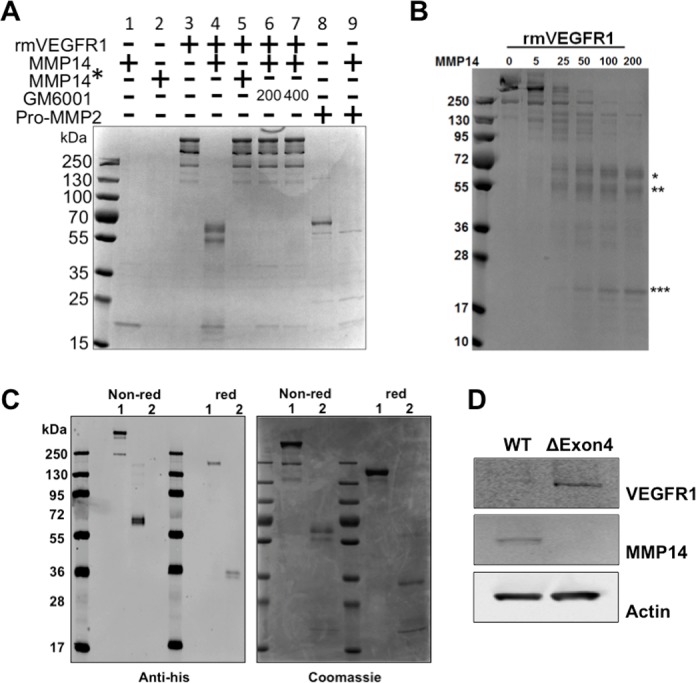
MMP14 cleaves VEGFR1 in vitro. Cleavage of VEGFR1 produces three fragments and requires the MMP14 catalytic domain. Reactions subjected to SDS-PAGE and stained with Coomassie blue. (A) MMP14 (200 ng, lane 1) and heat-inactivated MMP14 (MMP14*, 200 ng, lane 2) alone were run as controls. Mouse recombinant VEGFR1 (rmVEGFR1, 1 μg, lane 3) was run alone, or incubated with 200 ng MMP14 catalytic domain (lane 4) or 200 ng heat-inactivated MMP14* (lane 5). Undigested rmVEGFR1 runs as several bands, all above 130 kDa (lane 3). rmVEGFR1 digestion by MMP14 produced three distinct products at approximately 70, 59.8, and 21 kDa (lane 4). rmVEGFR1 incubated with heat-inactivated MMP14 (MMP14*, 200 ng, lane 5) or MMP14 and the MMP14 catalytic inhibitor GM6001 (200 or 400 ng, lanes 6 and 7) resulted in no cleavage. Pro-MMP2, a known substrate of MMP14, was used as a control (lanes 8 and 9). (B) Cleavage of VEGFR1 by MMP14. One microgram each of rmVEGFR1 was incubated with various concentrations (0–200 ng) of the catalytic domain of MMP14. Digested samples were subjected to SDS-PAGE under nonreducing conditions and stained with Coomassie blue. (C) Western blot analysis using an anti-His antibody shows that uncleaved rmVEGFR1 runs above 130 kDa under either nonreducing or reducing conditions (lane 1, both conditions). MMP14 cleavage of rmVEGFR1 (lane 2, both conditions) produces a 70-kDa His-tag fragment under nonreducing conditions and a 35-kDa His-tag fragment under reducing conditions, indicating that this fragment corresponds to the human IgG1/His-tag domain of recombinant VEGFR1 protein, which forms a dimer under nonreducing conditions. Duplicated samples were subjected to SDS-PAGE and then stained with Coomassie blue. (D) Total protein lysates from wild-type and MMP14 Δexon4 mouse corneal fibroblasts were subjected to Western blotting using anti-VEGFR1 antibodies. Blots reveal that no MMP14 activity is produced by MMP14 Δexon4 cells (row 2) and that VEGFR1 accumulates in MMP14 Δexon4 fibroblasts and is virtually eliminated in wild-type fibroblasts (row 1). Anti-actin was used as an internal control (row 3). *70-kDa fragment; **59.8-kDa fragment; ***21-kDa fragment.
Characterization of VEGFR1 Fragments Produced by MMP14 Cleavage In Vitro
We next explored whether the fragments produced by MMP14 cleavage of VEGFR1 include domains 1 to 7 of VEGFR1. Fragments were exposed to nonreducing and reducing conditions. Analysis revealed that the 70-kDa fragment is a dimer (Fig. 1C), while the 59.8- and 21-kDa fragments are monomers. Western blotting furthermore showed that the 70- and 35-kDa fragments were labeled by an anti-His tag antibody under the nonreducing condition and reducing condition, respectively. This reveals that it contains the C-terminal domain (35-kDa) of rmVEGFR1. This was also confirmed with gels stained by Coomassie blue.
Loss of MMP14 Results in an Accumulation of VEGFR1 in Cornea Fibroblasts
To investigate the effects of MMP14 on the VEGFR1 level in cells, we evaluated VEGFR1 levels in wild-type and MMP14 Δexon4 mouse cornea fibroblasts.31 We used cells derived from MMP14Δexon4 mouse that contain no catalytic domain of MMP14, which disabled the cleavage function of MMP14. Western blotting results with cell lysates showed that a higher level of VEGFR1 remained in MMP14 Δexon4 cells than in wild-type cells (Fig. 1D). These results indicate that MMP14 may be responsible for cleaving VEGFR1 in cornea fibroblast cells, and the loss of MMP14 results in an accumulation of VEGFR1.
MMP14 Binds VEGFR1 to Cleave
We observed that MMP14 cleaved VEGFR1, and we hypothesized that MMP14 may bind to VEGFR1 in order to cleave it. The SPR technique was used to determine binding affinities of MMP14 to VEGFR1 for direct comparison. Sensorgrams of a series of increasing concentrations of MMP14 flown on a VEGFR1-immobilized CM5 sensor surface are shown (Fig. 2A) with the fitting curve (Fig. 2B). The determined KD value for VEGFR1 was 1.26 ± 0.42 (μM), indicating that MMP14 interacts only with VEGFR1. These results support our hypothesis.
Figure 2.

MMP14 binds rmVEGFR1 and cleaves putative VEGFR1 at two points in the extracellular domain. Surface plasmon resonance (SPR) analyses determined the equilibrium dissociation constant (KD) between MMP14 and immobilized rmVEGFR1. Sensorgrams (A) and a fitting curve (B) are shown. (C) Diagram of the products of MMP14 cleavage of rmVEGFR1 based on Edman sequencing results (in parentheses). An N-terminal fragment (SKLKVP) corresponding to aa27 to 560 (Ig domains 1–5) forms a 59.8-kDa monomer that does not dimerize. A middle fragment (FHVSL[E]K) corresponding to aa560 to 744 (Ig domains 6–7) forms a 21-kDa monomer that does not dimerize. A C-terminal fragment (YLTVQGTSDK) corresponding to aa745-C-terminus corresponds to a 70-kDa dimer/35-kDa monomer that contains the recombinant human IgG1/His domains. Proposed sites of VEGFR1 processing by MMP14 are shown relative to known sites of γ-secretase processing and the point at which homology ends between full-length and soluble VEGFR1 proteins (alternative splicing site). (D) MMP14 cleavage sites in the VEGFR1 protein sequence.
Sequencing of MMP14-Cleaved VEGFR1 Fragments
Once we confirmed that MMP14 binds and cleaves VEGFR1, we further analyzed cleaved VEGFR1 fragments by Edman degradation (Fig. 2C). This analysis revealed that MMP14 cleavage of VEGFR1 produces an N-terminal fragment (59.8 kDa) that begins with SKLKVP and contains Ig domains 1 through 5 (amino acids [aa] 27–559), an intermediate fragment (21 kDa) that begins with FHVSL(E)K and contains Ig domains 6 and 7 (aa 560–744), and a C-terminal fragment that begins with YLTVQGTSDK (at aa 745) and contains the human IgG1 and His-tag (Table; Figs. 2C, 2D).
Table.
Cleavage Site of Extracellular Region of VEGF Receptor 1 by MMP14

The N-Terminal Product of MMP14 Cleavage of VEGFR1 Binds VEGF-A165
Cleavage of VEGFR1 by MMP14 produces an extracellular N-terminal fragment that contains the complete VEGF binding domain. We therefore used an ELISA to determine the binding affinity between VEGF-A165 and the N-terminal cleavage fragment. Our investigation of the binding capacity of VEGF-A165 to VEGFR1 and the 59.8-kDa N-terminal VEGFR1 fragment revealed that the N-terminal fragment bound to VEGF-A165 but at a lower affinity than intact VEGFR1 (Fig. 3A). Surface plasmon resonance was used for further quantification of the binding affinities of VEGFR1 and the 59.8-kDa N-terminal VEGFR1 fragment with VEGF-A165. The determined binding affinity (KD) value between VEGF-A165 and VEGFR1 (Fig. 3B) was 0.62 ± 0.37 nM. The KD for the 59.8-kDa N-terminal VEGFR1 fragment (Fig. 3C) was 1.16 ± 0.32 nM. The SPR results suggest that the binding affinity of VEGFR1 is 1.87 stronger than that of the 59.8-kDa fragment. Additionally, we further investigated whether other known ligands of VEGFR1 such as PlGF could recognize and bind to 59.8-kDa cleaved fragments using SPR. We found that PlGF indeed showed binding to both VEGFR1 and 59.8-kDa cleaved VEGFR1 fragments with binding affinities of 6.3 nM and 13.0 nM, respectively (Fig. 3D). The KD value of the 59.8-kDa fragment was two times weaker than that of the VEGFR1, which is similar to that of VEGF-A165.
Figure 3.
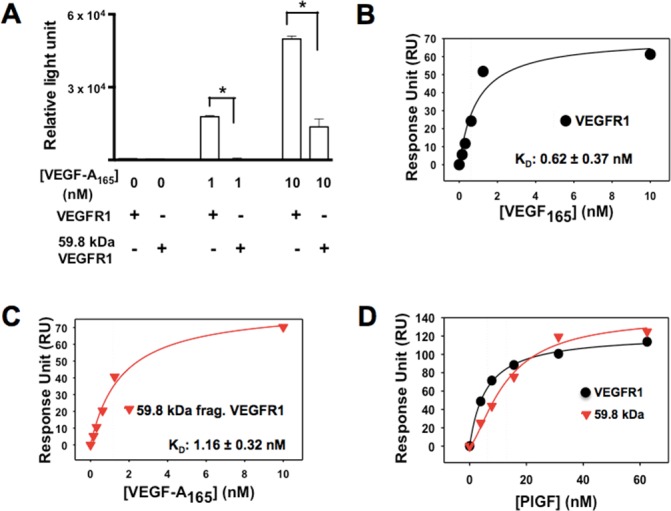
Reduced binding of VEGF-A165 to the N-terminal fragment produced by MMP14 cleavage of rmVEGFR1. (A) rmVEGFR1 and the N-terminal protein fragment produced by MMP14 cleavage of mVEGFR1 were coated onto 96-well plates and incubated with VEGF-A165 (0, 1, or 10 nM) followed by the addition of an anti-VEGF-A165-HRP. Chemiluminescence levels were analyzed with an ELISA reader to determine the binding affinity of each receptor to VEGF-A165. SPR fitting curves of VEGF-A165 with rmVEGFR1 (B) and 59.8-kDa N-terminal fragment (C) VEGF-A165 binding affinities (KD) to the immobilized rmVEGFR1 and the 59.8-kDa N-terminal VEGFR1 fragment were determined by SPR analysis using a CM5 chip. (D) SPR fitting curve of PlGF with VEGFR1 and 59.8-kDa N-terminal VEGFR1 fragment.
N-Terminal VEGFR1 Cleavage Product Inhibits VEGF-A165-Induced Cell Proliferation
Last, we sought to determine whether the 59.8-kDa N-terminal fragment produced by MMP14 cleavage of VEGFR1, which we have shown effectively binds VEGF-A165, might function to inhibit VEGF-A165-induced cell proliferation by binding free VEGF-A165, thus preventing it from binding to VEGFR1 or VEGFR2 on the cell surface. CPAEs were stimulated for 10 minutes with either VEGF-A165 alone (10 ng/mL) or VEGF-A165 that had been preincubated with 1 μg/mL of the 59.8-kDa N-terminal VEGFR1 fragment. We observed that activation of p-ERK protein by VEGF-A165 alone (Fig. 4A, lane 2) was reduced in CPAEs by preincubation of VEGF with the N-terminal fragment (Fig. 4A, lane 3). In addition, CPAEs were incubated with either VEGF-A165 alone (10 ng/mL) or VEGF-A165 that had been preincubated with increasing concentrations of the 59.8-kDa N-terminal VEGFR1 fragment (0–1 μg/mL at 2-fold dilution). After a 48-hour incubation period, we observed that VEGF-A165-induced cell proliferation was reduced and eventually eliminated in a dose-dependent manner (Fig. 4B).
Figure 4.
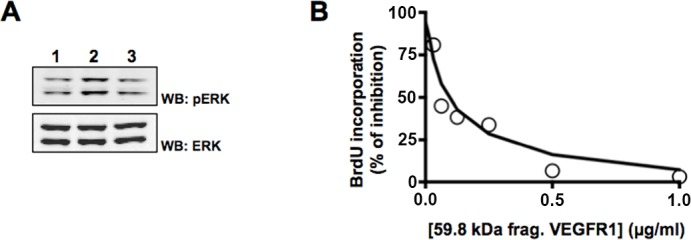
Inhibitory trap effects of the 59.8-kDa N-terminal VEGFR1 fragment of cleaved rmVEGFR1 on VEGF-A165-induced mitogenic activity. (A) Cleaved 59.8-kDa rmVEGFR1 fragments were preincubated with 10 ng/mL VEGF-A165 for 30 minutes and then added onto plated CPAEs for 10 minutes. Total lysates were harvested for Western blot analysis. Greater ERK protein activation (p-ERK) was observed in cells treated with VEGF-A165 only (lane 2) compared to the control (nonstimulated, lane 1). VEGF-induced ERK protein phosphorylation was diminished following the incubation of cells with the cleaved fragment (lane 3). (B) Cleaved 59.8-kDa rmVEGFR1 fragments were preincubated with 10 ng/mL VEGF-A165 for 30 minutes and then added onto plated CPAEs for 48 hours. Bromodeoxyuridine incorporation was used to measure proliferating cells. CPAEs were stimulated with VEGF-A165 only as a positive control.
Discussion
Matrix metalloproteinase 14 is upregulated in many tissues during angiogenesis.13 It is, in particular, upregulated in tip cells that lead angiogenic vessel growth.32 The most well known function for MMP14 at the angiogenic front is ECM remodeling and, specifically, the activation of the gelatinase MMP2.19 Matrix metalloproteinase 14 has been shown, however, to be involved in the canonical VEGF- and bFGF-stimulated angiogenic pathways,33,34 although the specific role of MMP14 in these processes remains unclear. Matrix metalloproteinase 14 is generally regarded as proangiogenic because it is upregulated during angiogenesis in endothelial tissue and loss of MMP14 results in a loss of the ability of endothelial cells to form new vessels in response to bFGF.7,13,33 In the present study, we aimed to determine whether MMP14 interacts with the VEGF-VEGFR signaling axis by directly cleaving the membrane receptors in the VEGF family. We found that MMP14 cleaves VEGFR1 in vitro (Figs. 1A, 1B), that this cleavage produces three distinct fragments (70, 59.8, and 21 kDa), and that it requires the catalytic domain of MMP14 (Fig. 1B).35 We followed these results with experiments aimed at exploring possible antiangiogenic functions for the specificity of MMP14 cleavage of VEGFR1, as well as mechanisms through which the cleavage products might act.
Vascular endothelial growth factor receptor 2 has been previously attributed with the direct activation, via tyrosine kinase activity, of multiple signaling cascades that are required for angiogenesis. It has been proposed that a primary function of VEGFR1 might be to prevent the initiation of these signaling cascades through the maintenance of less functional VEGFR2. We found that loss of MMP14 catalytic function in endothelial cells results in an increase of VEGFR1 in the cell membrane (Fig. 1D). We therefore hypothesized that MMP14 cleavage of VEGFR1 may act to increase N-terminal VEGFR1 fragment levels in the extracellular milieu resulting in trapping free VEGF-A165 to decrease the activation of VEGFR2 signaling on the cell surface and thereby enhance the antiangiogenic potential of the endothelial cell.
We found that cleavage of VEGFR1 by MMP14 occurs at two locations, both within the extracellular domain. The 59.8-kDa N-terminal VEGFR1 fragment is of particular interest because it contains Ig domains 1 through 5, which includes the ligand binding domains (Ig domains 2–3) for VEGF (Fig. 2C; Table). We determined that this N-terminal fragment is capable of binding VEGF-A165 but less than intact VEGFR1 (Fig. 3A). Because this fragment can bind to VEGF-A165, we hypothesized that it may, upon shedding into the extracellular space, act in an antiangiogenic manner by acting as a trap for free VEGF-A165, thereby reducing the level of free VEGF-A165 available to interact with VEGFR 2 and thus decreasing VEGF-driven angiogenesis. Indeed, our results show that the 59.8-kDa N-terminal VEGFR1 fragment reduces and eventually eliminates VEGF-A165-induced proliferation and p-ERK activation in endothelial cells (Fig. 4). This is consistent with previous studies, which have shown that a naturally occurring soluble VEGFR1 (sVEGFR1), generated by alternative splicing that results in a truncated receptor very similar to the N-terminal fragment produced by MMP14 cleavage of VEGFR1, can bind free VEGF-A165 to inhibit VEGF-A165-induced proliferation in human umbilical vein endothelial cells36 and trap free VEGF-A165 for preserving avascularity in the cornea.30 Matrix metalloproteinase 14 cleavage of membrane-bound VEGFR1 may therefore act to enhance the levels of VEGF trap in the extracellular space.
Our results indicate that while MMP14 is required for normal angiogenesis, it may have antiangiogenic functions. We show that MMP14 cleavage of VEGFR1 produces a fragment that is capable of binding the proangiogenic factor VEGF and inhibiting its interaction with endothelial cells.
Acknowledgments
Supported by National Institutes of Health Grants EY10101 (DTA), EY023691, EY021886, I01 BX002386 (J-HC), EY01792, and an unrestricted grant from Research to Prevent Blindness, New York, New York, United States.
Disclosure: K.-Y. Han, None; J. Dugas-Ford, None; H. Lee, None; J.-H. Chang, None; D.T. Azar, None
References
- 1. Rooprai HK,, Van Meter T,, Rucklidge GJ,, Hudson L,, Everall IP,, Pilkington GJ. Comparative analysis of matrix metalloproteinases by immunocytochemistry immunohistochemistry and zymography in human primary brain tumours. Int J Oncol. 1998; 13: 1153–1157. [DOI] [PubMed] [Google Scholar]
- 2. Atkinson SJ,, Patterson ML,, Butler MJ,, Murphy G. Membrane type 1 matrix metalloproteinase and gelatinase A synergistically degrade type 1 collagen in a cell model. FEBS Lett. 2001; 491: 222–226. [DOI] [PubMed] [Google Scholar]
- 3. Koike T,, Vernon RB,, Hamner MA,, Sadoun E,, Reed MJ. MT1-MMP but not secreted MMPs, influences the migration of human microvascular endothelial cells in 3-dimensional collagen gels. J Cell Biol. 2002; 86: 748–758. [DOI] [PubMed] [Google Scholar]
- 4. Chun TH,, Sabeh F,, Ota I,, et al. MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J Cell Biol. 2004; 167: 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mimura T,, Han KY,, Onguchi T,, et al. MT1-MMP-mediated cleavage of decorin in corneal angiogenesis. J Vasc Res. 2009; 46: 541–550. 19571574 [Google Scholar]
- 6. Azar DT,, Casanova FH,, Mimura T,, et al. Corneal epithelial MT1-MMP inhibits vascular endothelial cell proliferation and migration. Cornea. 2010; 29: 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou Z,, Apte SS,, Soininen R,, et al. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc Natl Acad Sci U S A. 2000; 97: 4052–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Malinowski M,, Pietraszek K,, Perreau C,, et al. Effect of lumican on the migration of human mesenchymal stem cells and endothelial progenitor cells: involvement of matrix metalloproteinase-14. PLoS One. 2012; 7: e50709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaimal R,, Aljumaily R,, Tressel SL,, et al. Selective blockade of matrix metalloprotease-14 with a monoclonal antibody abrogates invasion, angiogenesis, and tumor growth in ovarian cancer. Cancer Res. 2013; 73: 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haage A,, Nam DH,, Ge X,, Schneider IC. Matrix metalloproteinase-14 is a mechanically regulated activator of secreted MMPs and invasion. Biochem Biophys Res Commun. 2014; 450: 213–218. [DOI] [PubMed] [Google Scholar]
- 11. Lu C,, Li XY,, Hu Y,, Rowe RG,, Weiss SJ. MT1-MMP controls human mesenchymal stem cell trafficking and differentiation. Blood. 2010; 115: 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang JH,, Javier JA,, Chang GY,, Oliveira HB,, Azar DT. Functional characterization of neostatins the MMP-derived, enzymatic cleavage products of type XVIII collagen. FEBS Lett. 2005; 579: 3601–3606. [DOI] [PubMed] [Google Scholar]
- 13. Ye HQ,, Maeda M,, Yu FS,, Azar DT. Differential expression of MT1-MMP (MMP-14) and collagenase III (MMP-13) genes in normal and wounded rat corneas. Invest Ophthalmol Vis Sci. 2000; 41: 2894–2899. [PubMed] [Google Scholar]
- 14. Littlepage LE,, Sternlicht MD,, Rougier N,, et al. Matrix metalloproteinases contribute distinct roles in neuroendocrine prostate carcinogenesis, metastasis, and angiogenesis progression. Cancer Res. 2010; 70: 2224–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gabison E,, Chang JH,, Hernandez-Quintela E,, et al. Anti-angiogenic role of angiostatin during corneal wound healing. Exp Eye Res. 2004; 78: 579–589. [DOI] [PubMed] [Google Scholar]
- 16. Shi F,, Sottile J. MT1-MMP regulates the turnover and endocytosis of extracellular matrix fibronectin. J Cell Sci. 2011; 124: 4039–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Basile JR,, Holmbeck K,, Bugge TH,, Gutkind JS. MT1-MMP controls tumor-induced angiogenesis through the release of semaphorin 4D. J Biol Chem. 2007; 282: 6899–6905. [DOI] [PubMed] [Google Scholar]
- 18. Sugiyama N,, Gucciardo E,, Tatti O,, et al. EphA2 cleavage by MT1-MMP triggers single cancer cell invasion via homotypic cell repulsion. J Cell Biol. 2013; 201: 467–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Williamson RA,, Hutton M,, Vogt G,, et al. Tyrosine 36 plays a critical role in the interaction of the AB loop of tissue inhibitor of metalloproteinases-2 with matrix metalloproteinase-14. J Biol Chem. 2001; 276: 32966–32970. [DOI] [PubMed] [Google Scholar]
- 20. Cai J,, Jiang WG,, Grant MB,, Boulton M. Pigment epithelium-derived factor inhibits angiogenesis via regulated intracellular proteolysis of vascular endothelial growth factor receptor 1. J Biol Chem. 2006; 281: 3604–3613. [DOI] [PubMed] [Google Scholar]
- 21. Raikwar NS,, Liu KZ,, Thomas CP. Protein kinase C regulates FLT1 abundance and stimulates its cleavage in vascular endothelial cells with the release of a soluble PlGF/VEGF antagonist. Exp Cell Res. 2013; 319: 2578–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Olsson AK,, Dimberg A,, Kreuger J,, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006; 7: 359–371. [DOI] [PubMed] [Google Scholar]
- 23. Waltenberger J,, Claesson-Welsh L,, Siegbahn A,, Shibuya M,, Heldin CH. Different signal transduction properties of KDR and Flt1 two receptors for vascular endothelial growth factor. J Biol Chem. 1994; 269: 26988–26995. [PubMed] [Google Scholar]
- 24. Guo D,, Jia Q,, Song HY,, Warren RS,, Donner DB. Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains. Association with endothelial cell proliferation. J Biol Chem. 1995; 270: 6729–6733. [DOI] [PubMed] [Google Scholar]
- 25. Hiratsuka S,, Minowa O,, Kuno J,, Noda T,, Shibuya M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A. 1998; 95: 9349–9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bruns AF,, Bao L,, Walker JH,, Ponnambalam S. VEGF-A-stimulated signalling in endothelial cells via a dual receptor tyrosine kinase system is dependent on co-ordinated trafficking and proteolysis. Biochem Soc Trans. 2009; 37: 1193–1197. [DOI] [PubMed] [Google Scholar]
- 27. Mac Gabhann F,, Popel AS. Systems biology of vascular endothelial growth factors. Microcirculation. 2008; 15: 715–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ahmadova Z,, Yagublu V,, Forg T,, et al. Fluorescent resonance energy transfer imaging of VEGFR dimerization. Anticancer Res. 2014; 34: 2123–2133. [PubMed] [Google Scholar]
- 29. Cudmore MJ,, Hewett PW,, Ahmad S,, et al. The role of heterodimerization between VEGFR-1 and VEGFR-2 in the regulation of endothelial cell homeostasis. Nature Commun. 2012; 3: 972. [DOI] [PubMed] [Google Scholar]
- 30. Ambati BK,, Nozaki M,, Singh N,, et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature. 2006; 443: 993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Han KY,, Dugas-Ford J,, Seiki M,, Chang JH,, Azar DT. Evidence for the involvement of MMP14 in MMP2 processing and recruitment in exosomes of corneal fibroblasts [published online ahead of print July 11, 2014] Invest Ophthalmol Vis Sci. doi:http://dx.doi.org/10.1167/iovs.14-14417. [DOI] [PMC free article] [PubMed]
- 32. De Smet F,, Segura I,, De Bock K,, Hohensinner PJ,, Carmeliet P. Mechanisms of vessel branching: filopodia on endothelial tip cells lead the way. Arterioscler Thromb Vasc Biol. 2009; 29: 639–649. [DOI] [PubMed] [Google Scholar]
- 33. Onguchi T,, Han KY,, Chang JH,, Azar DT. Membrane type-1 matrix metalloproteinase potentiates basic fibroblast growth factor-induced corneal neovascularization. Am J Pathol. 2009; 174: 1564–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Han KY,, Fahd DC,, Tshionyi M,, et al. MT1-MMP modulates bFGF-induced VEGF-A expression in corneal fibroblasts. Protein Pept Lett. 2012; 19: 1334–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cao J,, Kozarekar P,, Pavlaki M,, Chiarelli C,, Bahou WF,, Zucker S. Distinct roles for the catalytic and hemopexin domains of membrane type 1-matrix metalloproteinase in substrate degradation and cell migration. J Biol Chem. 2004; 279: 14129–14139. [DOI] [PubMed] [Google Scholar]
- 36. Kendall RL,, Thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci U S A. 1993; 90: 10705–10709. [DOI] [PMC free article] [PubMed] [Google Scholar]