

Abstract
Due to their biostability, d-peptides are emerging as an important molecular platform for biomedical applications. Being proteolytically resistant, d-peptides lack interactions with endogenous transporters and hardly enter cells. Here we show that taurine, a natural amino acid, drastically boosts the cellular uptake of small d-peptides in mammalian cells by >10-fold, from 118 μM (without conjugating taurine) to >1.6 mM (after conjugating taurine). The uptake of a large amount of the ester conjugate of taurine and d-peptide allows intracellular esterase to trigger intracellular self-assembly of the d-peptide derivative, further enhancing their cellular accumulation. The study on the mechanism of the uptake reveals that the conjugates enter cells via both dynamin-dependent endocytosis and macropinocytosis, but likely not relying on taurine transporters. Differing fundamentally from the positively charged cell-penetrating peptides, the biocompatibility, stability, and simplicity of the enzyme-cleavable taurine motif promise new ways to promote the uptake of bioactive molecules for countering the action of efflux pump and contributing to intracellular molecular self-assembly.
This Communication reports that incorporating taurine into small d-peptides drastically boosts the cellular uptake of d-peptides for intracellular enzyme-instructed self-assembly and accumulation. Since few pathways are readily available for the biological utilization of d-amino acids, nutritionally important amino acids rarely exist as d-isomers, and naturally occurring proteins consist exclusively of l-amino acids. Though microorganisms, marine invertebrates, and a few other animals do synthesize d-amino acids and, during food processing, racemization occurs to produce d-amino acids, these examples remain exceptions.1 This unique feature of d-amino acids endows d-peptides with enduring stability due to their resistance against endogenous proteases in vitro and in vivo. Kim et al. developed an elegant approach (i.e., mirror-image phage display) to identify d-peptide ligand/inhibitors for proteins.2d-Peptides thus are gaining increased attention and readily find applications in a variety of areas of biology and biomedicine.3,ref4 For example, d-peptides can be used to trace the lineage of cells4 and the growth of axons,5 disrupt protein interactions,6 reduce adverse drug reactions of anti-inflammatory drugs,7 and serve as a medium for drug delivery.8
These advances in the use of d-amino-acid-based materials have mainly occured for extracellular9 applications, because cellular uptake of d-peptides is ineffective and less explored.10 A recent result also reveals that d-peptides exclusively enrich in cell membrane.11 Although it is feasible to inject d-peptides directly into cells,4,5 microinjection is unsuitable for common mammalian cells and is impractical for application to large numbers of cells. To further explore the merits of d-peptides inside cells, it is essential to develop an effective strategy to enhance the cellular uptake of d-peptides. Although there are reports of cell-penetrating peptides (CPPs, e.g., poly(lysine), poly(arginine), or poly(d-arginine)),12 these CPPs still have limitations, such as their susceptibility to metabolic degradation, dependence on cell lines and cellular differentiation state, or poor cellular compatibility (especially of those cationic CPPs).13 Thus, there is an unmet need for new molecular promoters to enhance cellular uptake of d-peptides and other bioactive molecules.
We unexpectedly observed that taurine, a non-proteinogenic but essential amino sulfonic acid,14 apparently enhances cellular uptake. Our results show that the attachment of taurine to a d-peptidic derivative significantly boosts the cellular uptake of the d-peptidic derivative, achieving >10-fold increase. Enzyme-catalyzed removal of the taurine group results in the self-assembly of the d-peptidic derivatives and further enhances the intracellular accumulation of the d-peptide derivative (Figure 1B). Our preliminary study on the mechanism of the uptake reveals that the uptake of the d-peptide derivative relies on both dynamin-dependent endocytosis and macropinocytosis but, surprisingly, little on taurine transporters. As the first example of taurine (an amino sulfonic acid widely distributed in animal tissues)-promoted cellular uptake, this work illustrates a fundamentally new strategy to deliver d-peptides into live cells. This approach ultimately may lead to taurine-based intracellular delivery of other bioactive molecular and therapeutic agents for intracellular self-assembly.15
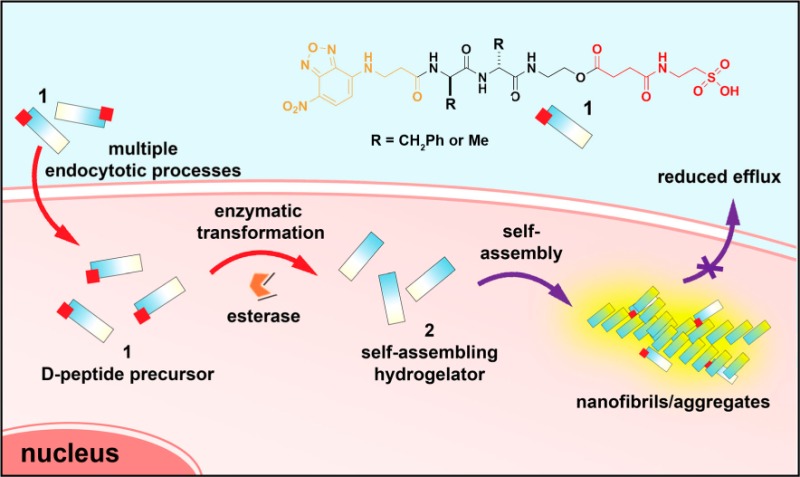
Figure 1.

(A) Molecular structures of precursors 1a and 1b, and the corresponding hydrogelators 2a and 2b after enzymatic transformation. (B) Taurine conjugation boosts cellular uptake of d-peptide precursors and subsequent enzyme-instructed self-assembly to form nanofibers, accumulating inside cells.
Molecule 1a consists of a fluorophore (i.e., 4-nitro-2,1,3-benzoxadiazole, NBD), a dipeptide residue (i.e., d-Phe-d-Phe), an enzyme (i.e., esterase) cleavage site (i.e., an ester bond), and a taurine residue (Figure 1A). The NBD motif, exhibiting enhanced fluorescence in a hydrophobic environment, can efficiently indicate nanofibrils formed by molecular self-assembly in cells.16 The d-diphenylalanine peptide, besides serving as the self-assembly motif,7,17 ensures the proteolytic resistance of the molecules. The ester bond allows esterase to catalyze the conversion of 1a into 2a. To verify the roles of taurine and the ester bond, we design two control molecules, 3a and 4. 3a lacks the taurine residue, and 4 has an amide bond replacing the ester bond in 1a. Based on the design in Figures 1, S1, and S2, we combine solid- and liquid-phase peptide synthesis (Figure S2) to make these molecules in fair yields (50–70%) and reasonable scales (0.1–0.5 g).
We first examine the enzymatic transformation of 1a (and 3a) by analytical HPLC and the subsequent self-assembly of 2a by transmission electron microscopy (TEM). At pH 7.4 and 1.0 wt%, 1a forms a transparent solution (Figure S3A). After treatment with esterase (1 U/mL) for 1 min, the orange solution remains unchanged, though analytical HPLC shows that the esterase has already converted 2.5% of 1a into 2a (1a:2a = 40:1). The TEM image of the solution reveals that there are short nanofibrils (d = 8 ± 2 nm) and a few long nanofibrils (d = 12 ± 2 nm) (Figure S3A). After 24 h, the solution of 1a turns into a hydrogel, with ∼11% of 1a (1a:2a = 8:1) being converted to 2a according to HPLC. TEM shows that the self-assembled nanofibrils (d = 8 ± 2 nm) entangle with each other to form the matrices of the hydrogel, within which are sporadic short nanofibrils (d = 12 ± 2 nm) (Figure S3B). These results indicate that 1a exists mainly as a monomer or oligomer (which cannot be detected by TEM and is readily available to esterase) but has a weak tendency to form nanofibrils in solution, even at a high concentration (1.0 wt%), and that 2a and 1a are able to co-self-assemble to form nanofibrils. These features allow the enzyme-instructed self-assembly of 1a, resulting in the nanofibrils and subsequent hydrogelation. The control molecule 3a, without the taurine residue, is less soluble than 1a and forms only a semi-transparent dispersion at high concentration. After treatment with esterase (1 U/mL), the resulting hydrogel formed by the hydrolysis of 3a is rather soft and exhibits yellow fluorescence (Figure S4).
To match the concentrations used for cell experiments, we evaluate the self-assembly of 1a at a lower concentration (i.e., 100 μM). As shown by TEM (Figure 2A), there are hardly any aggregates/nanofibrils in the solution of 1. After addition of esterase (1 U/mL, 24 h), large amounts of nanofibrils (d = 8 ± 2 nm) appear, interwoven with aggregates (Figure 2B). Correspondingly, static light scattering (SLS) of the solution before esterase treatment exhibits a low signal (almost the same intensity as that of PBS buffer, or PBS with esterase in it), but the signal increases dramatically after addition of esterase (1 U/mL) (Figure 2C), suggesting that the conversion of 1a to 2a at 100 μM results in the self-assembly of 2a. HPLC reveals that >50% of 1a has been converted into 2a. The fluorescence associated with NBD may serve as an efficient indicator to report the self-assembly;162a fluoresces with increased quantum yield because the self-assembled nanofibrils or aggregates encapsulate the NBD residues in a highly hydrophobic environment.18 Significant fluorescence appears in the solution of 1a treated with esterase (Figure 2D) compared with the solution prior to addition of enzyme (Figure 2D inset).
Figure 2.
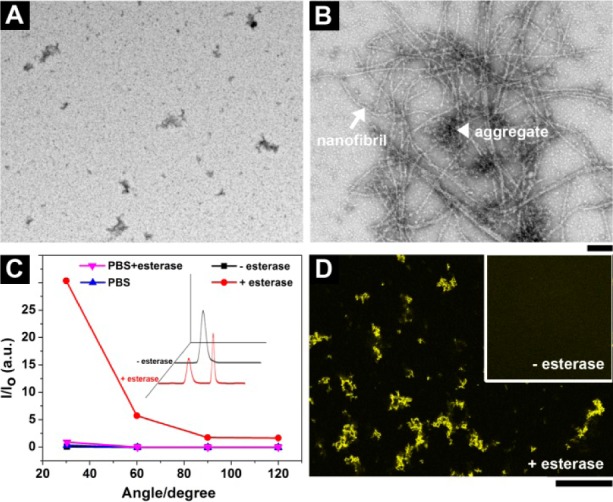
TEM images of the solutions of 1a in PBS buffer (A) before and (B) after addition of esterase. Scale bar = 100 nm. (C) SLS signals of the solution of 1a in PBS buffer without and with addition of esterase. Inset: corresponding analytical HPLC traces. (D) The confocal microscope image shows the appearance of fluorescence in the solution of 1a after esterase treatment (<10 min). Inset: corresponding image before esterase treatment. Scale bar = 100 μm. [1a] = 100 μM in PBS.
The fluorescence contrast before and after formation of nanofibrils allows the evaluation of cellular uptake, as well as intracellular self-assembly, the dynamics, and the localization of the nanofibrils of the small molecules in live cells.16 We incubate HeLa cells with 1a and 3a, respectively, in culture medium (200 μM, 37 °C, 24 h) and take confocal images to examine the cellular distribution of these molecules. We choose 200 μM for a shorter experiment period and better imaging quality. Our results show that HeLa cells treated with 1a exhibit significant fluorescence in cytoplasm (Figures 3 and S5), suggesting that the cells take up a large amount of 1a. Some of 1a turns into 2a upon esterase-instructed hydrolysis, and the 2a then co-self-assembles with 1a to form intracellular nanofibrils that exhibit strong fluorescence. In contrast, images of the HeLa cells treated with 3a show hardly any fluorescence inside the cells (Figure 3). The background outside the cells looks even brighter, indicating that little 3a enters the cells. These results confirm that incorporating taurine boosts cellular uptake of the d-peptide derivatives.
Figure 3.

Fluorescent confocal microscope images showing the fluorescence emission in HeLa cells with the treatment of (upper) 1a and (bottom) 3a at 200 μM concentration in culture medium for 24 h and co-stained with Hoechst 33342 (nuclei). Scale bar = 50 μm.
To prove that cleavage of the ester bond is critical for the significant fluorescence observed in the cytosol of the cells treated by 1, we examine the cellular uptake of 4. As a control of 1a, 4, having an amide bond to link the d-peptide and taurine, is resistant to hydrolysis catalyzed by esterase. There are a few yellow bright spots inside cells after 24-h incubation (Figure S6), validating that self-assembly due to ester bond hydrolysis further facilitates intracellular accumulation of the d-peptide derivatives in the cytosols, probably by reducing efflux.
We develop the procedure in Figure S8 for quantifying the intracellular or uptake concentration, Cu, of the d-peptide derivatives in HeLa cells treated by 1, 3, and 4, respectively. To be clear, we define Cu(1a/b) = [1a/b] + [2a/b], Cu(3a/b) = [3a/b] + [2a/b], and Cu(4) = [4], where [x] is the intracellular concentration of compound x, because both 1a/b and 3a/b would be hydrolyzed to 2a/b by esterase inside the cell, while 4 would not. When treated with 1a (200 μM) for 24 h, HeLa cells take up 1a to reach Cu = 1.6 mM, 8 times the incubating concentration (200 μM) (Figure 4A). In contrast, hardly any 3a (or 2a) is detected inside the HeLa cells. This striking contrast again verifies that covalent conjugation of taurine to the d-peptide derivative significantly increases the cellular uptake. Besides, Cu(4) = 0.8 mM in HeLa cells incubated with 4 (200 μM), half the Cu of 1a at the same condition, suggesting that intracellular esterase-instructed self-assembly of the d-peptide derivatives promotes cellular accumulation based on the taurine-enhanced uptake. The Cu of 4 (800 μM) is 4 times the incubating concentration in culture medium (200 μM). We also find that Cu(1a) gradually rises with increasing incubation time; notably, it keeps rising even after 48-h incubation, confirming that formation of nanofibrils by enzyme-instructed self-assembly maintains the cellular uptake of molecule 1a (Figure 4B).
Figure 4.
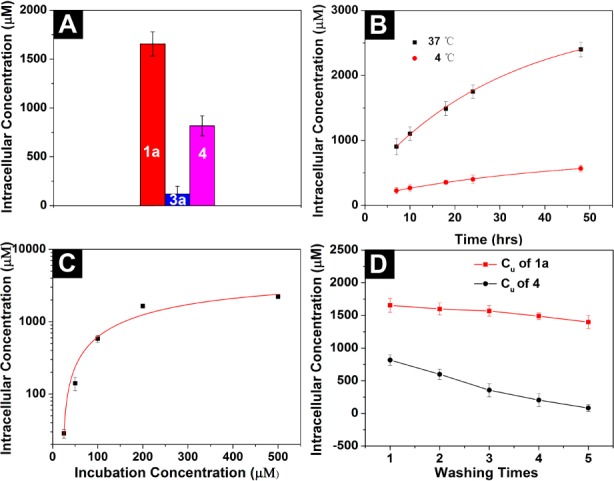
(A) Uptake concentration (Cu) of 1a, 3a, and 4 inside HeLa cells after 24-h incubation. The incubating concentration is 200 μM. (B) Cu of 1a (200 μM) at different time points and temperatures. (C) Cu of 1a at different incubating concentration after 24 h. (D) Cu of 1a and 4 after washing the cells with PBS buffer at each wash.
We also incubate HeLa cells with 1a at different concentrations (i.e., 25, 50, 100, 200, and 500 μM) for 24 h. When the incubating concentration is ≤25 μM, the enhancement of cellular uptake is less pronounced (Figure 4C). It appears that 200 μM is the optimum incubating concentration among those tested, because it reaches an 8-fold enhancement (24 h) compared with the incubating concentration (and 12-fold after 48 h). When the incubating concentrations are 50 and 500 μM, the boosts are about 3- and 4.5-fold, respectively. These results indicate that the activity of enzymes and the minimum aggregation concentration together dictate the enhancement of the cellular uptake. In addition, we find that 1a at high concentration (500 μM) causes intracellular nanofibrils to start disrupting actin filaments (Figure S7). Cell extraction analysis (of the HeLa cells incubated with 1a or 4 (200 μM, 24 h)) by analytical HPLC shows that the intracellular ratio of 2a and 1a is ∼1:4, while 4 remains intact inside cells (Figure S9). This result further confirms the enzymatic transformation of 1a to 2a and the proteolytic resistance of 1a, 4, and 2a.
To evaluate the retention of the d-peptide derivatives inside cells, we rinse the HeLa cells with fresh PBS buffer after 24-h incubation with 1a or 4 (200 μM) and measure the Cu of these d-peptides, respectively. Each wash (i.e., add PBS, mix, centrifuge, remove PBS) only slightly decreases Cu(1a), from 1.6 to 1.4 mM (Figure 4D). In contrast, each wash decreases Cu(4) by ∼25% (e.g., from 0.8 to 0.6 mM after the first wash). After the cells were washed with PBS buffer five times, the Cu(4) dropped significantly (i.e., about an order of magnitude, from 0.8 to 0.08 mM). These results confirm that intracellular cleavage of taurine and formation of the nanofibrils significantly reduce the molecular diffusion, which may open a new way for retaining desired molecules inside cells. Though it is impossible to rule out the contribution of solubility increase resulting from the attachment of taurine, the results in Figure 4D confirm that the increase of the solubility is unable to enhance the retention of the compounds inside cells.
We carry out a preliminary study to elucidate the uptake mechanism. The uptake of 1a reduces significantly at 4 °C, suggesting that the uptake is an energy-dependent process (Figure 4B). Since 1a does not aggregate at 4 °C (Figure S10), the reduced uptake is unlikely due to aggregation outside cells at lower temperature. Surprisingly, cellular uptake of 1a relies little on the assistance of taurine transporter, since addition of neither cyclosporine A (Cs A, 5 μM, taurine transporter inhibitor) nor taurine (3 mM) decreases Cu(1a) (Figure S11). In addition, other competitors (e.g., taurocholic acid (1 mM) or heparin (0.33 mM)) hardly affect the uptake of 1a (Figure S11 and S12). However, the use of dynamin 1, 2, and 3 knockout mouse fibroblast (TKO) cells19 results in reduced uptake of 1a, as there are fewer fluorescent puncta in treated TKO cells than in control (wild-type) cells (Figure S11 and S12). We also use inhibitors of different endocytotic processes to determine the possible pathways of cellular uptake—filipin III for caveolae-mediated endocytosis, chlorpromazine for clathrin-mediated endocytosis, and ethylisopropylamiloride (EIPA20) for macropinocytosis—and find that only the addition of EIPA significantly reduces the uptake of 1a. When we treat TKO cells with EIPA, cellular uptake of 1a is further reduced. These results suggest that this taurine-assisted uptake of 1a involves both dynamin-dependent endocytosis and macropinocytosis.21 Though how molecules escape from endosomes has yet to be determined, these d-peptide derivatives can remain intact in cells for a longer time compared with regular therapeutic molecules due to the proteolytic resistance of d-peptide, even when they end up in lysosomes.
To prove the generality of this approach, we synthesize another pair of molecules, 1b and 3b, with and without a taurine motif, by simply replacing the d-phenylalanine in 1a and 3a with d-alanine (Figure S13A). Though the self-assembly ability of the hydrogelator 2b (formed by esterase-catalyzed hydrolysis of 1b or 3b) is weaker than that of 2a, we still observe enhanced cellular uptake of 1b (with taurine motif) compared with 3b (without taurine) (Figure S13B,C).
In conclusion, we present here a novel strategy to boost the cellular uptake of d-peptide derivatives. This approach relies on covalent conjugation of a cleavable taurine motif with a d-peptide derivative. The use of taurine for enzyme-instructed self-assembly to enrich the molecules inside the cells may provide a new way to generate higher-order molecular assemblies that utilize small peptides instead of proteins for signaling transduction.22 Due to the accessibility of taurine and easiness of its molecular modification, this approach will be useful to facilitate the transport of functional d-peptidesref4,23 and other bioactive molecules through the cell membrane into live cells for controlling the fate of cells.24 For example, intracellular nanofibers formed by enzyme-instructed self-assembly can disrupt actin filaments and enhance the activity of cisplatin against drug-resistant ovarian cancer cells (Figure S14).
Acknowledgments
This work was partially supported by NIH (R01CA142746). J.Z. is a HHMI International Research Fellow. We thank Prof. De Camilli of Yale University for providing dynamin 1, 2, and 3 TKO mouse fibroblast cells.
Supporting Information Available
Synthetic procedures, NMR and LC-MS characterization, and cellular uptake measurement. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b06181.
The authors declare no competing financial interest.
Supplementary Material
References
- Friedman M.; Levin C. E. Amino Acids 2012, 42, 1553. 10.1007/s00726-011-0915-1. [DOI] [PubMed] [Google Scholar]
- Schumacher T. N. M.; Mayr L. M.; Minor D. L. Jr.; Milhollen M. A.; Burgess M. W.; Kim P. S. Science 1996, 271, 1854. 10.1126/science.271.5257.1854. [DOI] [PubMed] [Google Scholar]
- Reich Z.; Schramm O.; Brumfeld V.; Minsky A. J. Am. Chem. Soc. 1996, 118, 6345. 10.1021/ja9600237. [DOI] [Google Scholar]; Morii T.; Tanaka T.; Sato S.-i.; Hagihara M.; Aizawa Y.; Makino K. J. Am. Chem. Soc. 2002, 124, 180. 10.1021/ja017078f. [DOI] [PubMed] [Google Scholar]; Michaud M.; Jourdan E.; Villet A.; Ravel A.; Grosset C.; Peyrin E. J. Am. Chem. Soc. 2003, 125, 8672. 10.1021/ja034483t. [DOI] [PubMed] [Google Scholar]
- Eckert D. M.; Malashkevich V. N.; Hong L. H.; Carr P. A.; Kim P. S. Cell 1999, 99, 103. 10.1016/S0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]; Schumacher T. N. M.; Mayr L. M.; Minor D. L.; Milhollen M. A.; Burgess M. W.; Kim P. S. Science 1996, 271, 1854. 10.1126/science.271.5257.1854. [DOI] [PubMed] [Google Scholar]; Fitzgerald M. C.; Chernushevich I.; Standing K. G.; Kent S. B. H.; Whitman C. P. J. Am. Chem. Soc. 1995, 117, 11075. 10.1021/ja00150a006. [DOI] [Google Scholar]
- Weisblat D. A.; Zackson S. L.; Blair S. S.; Young J. D. Science 1980, 209, 1538. 10.1126/science.6159680. [DOI] [PubMed] [Google Scholar]
- Mason A.; Muller K. J. Nature 1982, 296, 655. 10.1038/296655a0. [DOI] [PubMed] [Google Scholar]
- Liu M.; Li C.; Pazgier M.; Li C. Q.; Mao Y. B.; Lv Y. F.; Gu B.; Wei G.; Yuan W. R.; Zhan C. Y.; Lu W. Y.; Lu W. Y. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 14321. 10.1073/pnas.1008930107. [DOI] [PMC free article] [PubMed] [Google Scholar]; McDonnell J. M.; Beavil A. J.; Mackay G. A.; Jameson B. A.; Korngold R.; Gould H. J.; Sutton B. J. Nat. Struct. Biol. 1996, 3, 419. 10.1038/nsb0596-419. [DOI] [PubMed] [Google Scholar]; Merrifield R. B.; Juvvadi P.; Andreu D.; Ubach J.; Boman A.; Boman H. G. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 3449. 10.1073/pnas.92.8.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. Y.; Kuang Y.; Gao Y.; Du X. W.; Shi J. F.; Xu B. J. Am. Chem. Soc. 2013, 135, 542. 10.1021/ja310019x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G. L.; Yang Z. M.; Zhang R. J.; Li L. H.; Fan Y. J.; Kuang Y.; Gao Y.; Wang T.; Lu W. W.; Xu B. Langmuir 2009, 25, 8419. 10.1021/la804271d. [DOI] [PubMed] [Google Scholar]
- Kuang Y.; Shi J.; Li J.; Yuan D.; Alberti K. A.; Xu Q.; Xu B. Angew. Chem., Int. Ed. 2014, 53, 8104. 10.1002/anie.201402216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida M.; Watanabe H.; Takigawa K.; Kurishita Y.; Oki C.; Nakamura A.; Hamachi I.; Tsukiji S. J. Am. Chem. Soc. 2013, 135, 12684. 10.1021/ja4046907. [DOI] [PubMed] [Google Scholar]; Hayashi T.; Sun Y.; Tamura T.; Kuwata K.; Song Z.; Takaoka Y.; Hamachi I. J. Am. Chem. Soc. 2013, 135, 12252. 10.1021/ja4043214. [DOI] [PubMed] [Google Scholar]
- Wang H.; Wang Y.; Han A.; Cai Y.; Xiao N.; Wang L.; Ding D.; Yang Z. ACS Appl. Mater. Interfaces 2014, 6, 9815. 10.1021/am502250r. [DOI] [PubMed] [Google Scholar]
- Mitchell D. J.; Kim D. T.; Steinman L.; Fathman C. G.; Rothbard J. B. J. Pept. Res. 2000, 56, 318. 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]; Wender P. A.; Galliher W. C.; Goun E. A.; Jones L. R.; Pillow T. H. Adv. Drug Delivery Rev. 2008, 60, 452. 10.1016/j.addr.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rothbard J. B.; Jessop T. C.; Lewis R. S.; Murray B. A.; Wender P. A. J. Am. Chem. Soc. 2004, 126, 9506. 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Melikov K.; Vives E.; Ramos C.; Verbeure B.; Gait M. J.; Chernomordik L. V.; Lebleu B. J. Biol. Chem. 2002, 278, 585. 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]; Sakai N.; Matile S. J. Am. Chem. Soc. 2003, 125, 14348. 10.1021/ja037601l. [DOI] [PubMed] [Google Scholar]
- Huxtable R. J. Physiol. Rev. 1992, 72, 101. [DOI] [PubMed] [Google Scholar]; Price J. C.; Barr E. W.; Glass T. E.; Krebs C.; Bollinger J. M. J. Am. Chem. Soc. 2003, 125, 13008. 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]; Proshlyakov D. A.; Henshaw T. F.; Monterosso G. R.; Ryle M. J.; Hausinger R. P. J. Am. Chem. Soc. 2004, 126, 1022. 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]; Riggs-Gelasco P. J.; Price J. C.; Guyer R. B.; Brehm J. H.; Barr E. W.; Bollinger J. M.; Krebs C. J. Am. Chem. Soc. 2004, 126, 8108. 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- Ye D.; Shuhendler A. J.; Cui L.; Tong L.; Tee S. S.; Tikhomirov G.; Felsher D. W.; Rao J. Nat. Chem. 2014, 6, 519. 10.1038/nchem.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yuan Y.; Sun H.; Ge S.; Wang M.; Zhao H.; Wang L.; An L.; Zhang J.; Zhang H.; Hu B.; Wang J.; Liang G. ACS Nano 2015, 9, 761. 10.1021/nn5062657. [DOI] [PubMed] [Google Scholar]; Tanaka A.; Fukuoka Y.; Morimoto Y.; Honjo T.; Koda D.; Goto M.; Maruyama T. J. Am. Chem. Soc. 2015, 137, 770. 10.1021/ja510156v. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Shi J. F.; Yuan D.; Xu B. Nat. Commun. 2012, 3, 1033. 10.1038/ncomms2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Du X.; Yuan D.; Zhou J.; Zhou N.; Huang Y.; Xu B. Biomacromolecules 2014, 15, 3559. 10.1021/bm5010355. [DOI] [PMC free article] [PubMed] [Google Scholar]; Li J. Y.; Gao Y.; Kuang Y.; Shi J. F.; Du X. W.; Zhou J.; Wang H. M.; Yang Z. M.; Xu B. J. Am. Chem. Soc. 2013, 135, 9907. 10.1021/ja404215g. [DOI] [PMC free article] [PubMed] [Google Scholar]; Reches M.; Gazit E. Science 2003, 300, 625. 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- Soltani C. E.; Hotze E. M.; Johnson A. E.; Tweten R. K. J. Biol. Chem. 2007, 282, 15709. 10.1074/jbc.M701173200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S. M.; Raimondi A.; Paradise S.; Shen H.; Mesaki K.; Ferguson A.; Destaing O.; Ko G.; Takasaki J.; Cremona O.; O’Toole E.; De Camilli P. Dev. Cell 2010, 18, 332. 10.1016/j.devcel.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; Park R. J.; Shen H.; Liu L.; Liu X.; Ferguson S. M.; De Camilli P. J. Cell Sci. 2013, 126, 5305. 10.1242/jcs.138578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakase I.; Niwa M.; Takeuchi T.; Sonomura K.; Kawabata N.; Koike Y.; Takehashi M.; Tanaka S.; Ueda K.; Simpson J. C.; Jones A. T.; Sugiura Y.; Futaki S. Mol. Ther. 2004, 10, 1011. 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Takei K.; Slepnev V. I.; Haucke V.; De Camilli P. Nat. Cell Biol. 1999, 1, 33. 10.1038/9004. [DOI] [PubMed] [Google Scholar]
- Cai X.; Chen J.; Xu H.; Liu S.; Jiang Q.-X.; Halfmann R.; Chen Z. J. Cell 2014, 156, 1207. 10.1016/j.cell.2014.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhou J.; Xu B. Bioconjugate Chem. 2015, 26, 987. 10.1021/acs.bioconjchem.5b00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Pazgier M.; Li J.; Li C.; Liu M.; Zou G.; Li Z.; Chen J.; Tarasov S. G.; Lu W.-Y.; Lu W. J. Biol. Chem. 2010, 285, 19572. 10.1074/jbc.M110.116814. [DOI] [PMC free article] [PubMed] [Google Scholar]; Milton R. C. D.; Milton S. C. F.; Kent S. B. H. Science 1992, 256, 1445. 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- Kuang Y.; Long M. J. C.; Zhou J.; Shi J.; Gao Y.; Xu C.; Hedstrom L.; Xu B. J. Biol. Chem. 2014, 289, 29208. 10.1074/jbc.M114.600288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.