

Abstract
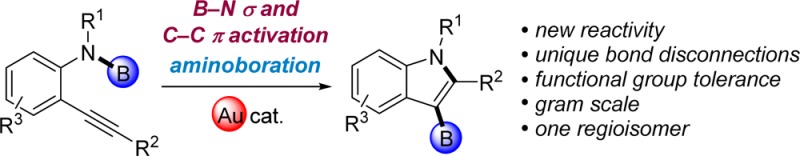
This communication demonstrates the first catalytic aminoboration of C–C π bonds by B–N σ bonds and its application to the synthesis of 3-borylated indoles. The regiochemistry and broad functional group compatibility of this addition reaction enable substitution patterns that are incompatible with major competing technologies. This aminoboration reaction effects the formation of C–B and C–N bonds in a single step from aminoboronic esters, which are simple starting materials available on the gram scale. This reaction generates synthetically valuable N-heterocyclic organoboron compounds as potential building blocks for drug discovery. The working mechanistic hypothesis involves a bifunctional Lewis acid/base catalysis strategy involving the combination of a carbophilic gold cation and a trifluoroacetate anion that activate the C–C π bond and the B–N σ bond simultaneously.
Boron–element additions to unsaturated compounds have played a pivotal role in organic synthesis since the discovery of hydroboration by Hurd1 and Brown.2 These transformations provide a route to synthetically valuable organoboron compounds that are widely used as versatile reagents in C–C and C–heteroatom bond forming reactions, such as the Suzuki–Miyaura reaction3 and Chan–Lam cross-coupling.4 Thus, transition-metal-catalyzed 1,2-addition of boron–element bonds (B–E, where E = H,5,6 B,6,7 C,8 Si,6,9,10 Sn,6,11 S,12 Cl,13 Br,14 I14) to C–C multiple bonds have received significant attention (Figure 1a). In most of these methods, the B–E single bonds are activated via oxidative addition to a low-valent late transition metal center (e.g., Rh, Ni, Pd, Pt) or via σ-bond metathesis with a late-metal catalyst (e.g., Cu15) prior to insertion across alkynes. Given that amines are present in 85% of all pharmaceutical compounds, it is striking that the corresponding addition chemistry with B–N bonds has not been developed for synthetic applications; possibly because the relatively high strength of the B–N σ bond prevented application of existing mechanistic strategies. We herein realize this aminoboration reaction by employing a different mechanistic strategy: activation of the C–C π bond—the other partner in the reaction—with a carbophilic Lewis acid (Figure 1c). This reaction employs starting materials containing B–N σ bonds that are readily available on the gram scale from their corresponding amines and commercially available B-chlorocatecholborane. It generates 3-borylated indoles via unique bond disconnections as potential building blocks for drug discovery, and it is orthogonal to major competing technologies, as it tolerates aryl halides, nitriles, and esters.
Figure 1.
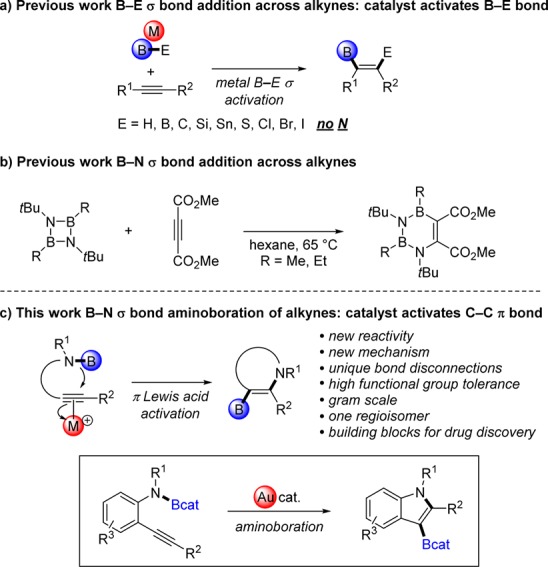
Addition of B–E σ bonds across C–C π bonds.
The resistance of B–N σ bonds to react with unactivated C–C π bonds is not surprising given that aminoboranes (R2B–NR2) are known to have an isostructural and isoelectronic relationship to alkenes due to the π-interaction between nitrogen’s lone pair and boron’s empty p orbital.16 Resultantly, prior work on aminoboration was mostly limited to the addition of B–N bonds to polarized C–heteroatom π bonds that included isocyanate,17 isothiocyanate,17a and carbodiimide.18 Such an approach is less synthetically useful, however, as the resulting B–heteroatom bond formed in those transformations is usually hydrolyzed and the boryl component is lost at the end of the reaction.17,18 To our knowledge, the only reported well-characterized example of aminoboration of a C–C multiple bond with a B–N σ bond is the [4 + 2] cycloaddition of a strained diazadiboretidine with dimethylacetylenedicarboxylate, which leads to a heterocycle of limited synthetic utility for downstream functionalization (Figure 1b).19 More recently, a formal aminoboration of alkenes was reported using a Cu-catalyzed borylation with bis(pinacolato)diboron (pinB–Bpin) followed by an electrophilic amination using O-benzoyl-N,N-dialkylhydroxylamine (R2N–OBz).20 Furthermore, despite a recent development on the addition chemistry involving the analogous B–P bond,21 the catalytic aminoboration of alkynes with B–N σ bond addition was unknown prior to this work (Figure 1c).
Indoles are privileged scaffolds found in numerous biologically active molecules and employed in medicinal chemistry,22 including recent therapeutic leads.23 Thus, the construction of an indole ring system was targeted (initial development, Table 1), which would give access to 3-borylated indoles as potential building blocks for drug discovery. During initial reaction identification, the requisite B–N bond of aminoboronic ester 2 was formed from the reaction of 2-alkynylaniline 1 and commercially available catecholborane in d8-toluene with heating and the release of H2. The formation of this B–N bond was monitored and confirmed by 11B NMR spectroscopy, at δ ≈ 26 ppm24 with concurrent disappearance of the signal from catecholborane at δ 28.7 ppm.
Table 1. Initial Development of Aminoboration.
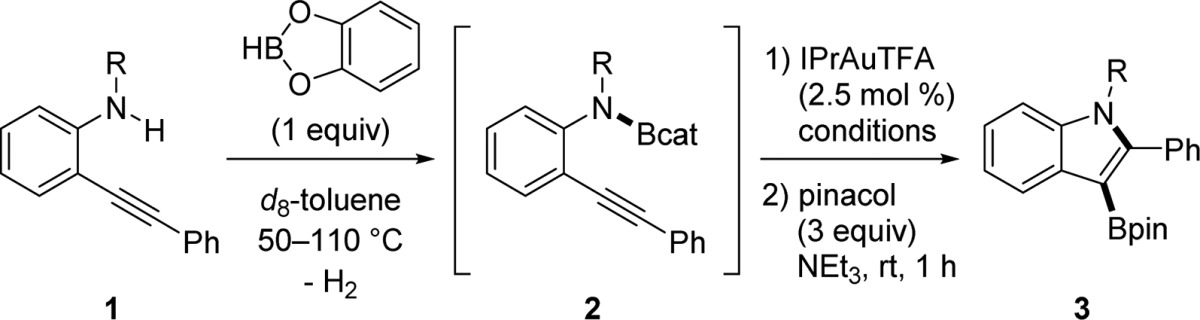
| entry | substrate | R | conditions | yield (%)a of 3 |
|---|---|---|---|---|
| 1 | 1a | H | 50 °C, 15.5 h | 0b |
| 2 | 1b | CH2Ph | 80 °C, 5 h | n.r.c |
| 3 | 1b | CH2Ph | 110 °C, 17 h | 55 |
| 4 | 1c | Ts | 50 °C, 4 h | n.r.c |
| 5 | 1c | Ts | 80 °C, 20 h | 64d |
| 6 | 1d | Mbs | 80 °C, 20 h | 66 |
Isolated yield of the Bpin product.
Only 2-phenyl-1H-indole was obtained in 69% yield.
No reaction observed as monitored by 1H NMR spectroscopy.
Average of two runs.
When the aminoboronic ester 2a (Table 1, entry 1), derived from primary aniline 1a, was treated with the IPrAuTFA catalyst25 and heated at 50 °C, the desired 3-borylated indole 3 was not obtained. Instead, only 2-phenyl-1H-indole was isolated in 69% yield, suggesting that protodemetalation occurs in the presence of a N–H bond. To circumvent this problem, secondary anilines 1b–1d (entries 2–6) were examined for aminoboration. Gratifyingly, the use of a N-benzyl substituent provided the first aminoboration reactivity in 55% yield (entry 3), but high temperatures of 110 °C were required. Examination of N-sulfonyl substituents, tosyl (entries 4 and 5) and 4-methoxybenzenesulfonyl (Mbs, entry 6), showed that the reaction can be accomplished at the lower temperature of 80 °C. The moderate yields obtained under the conditions of initial reaction development are attributed to the degradation of catecholborane (HBcat) into B2cat3 via catechol ligand redistribution,26 observed by a signal at δ 23.1 ppm in the 11B spectrum, as well as unreacted 1 remaining from the first aminolysis step with HBcat.
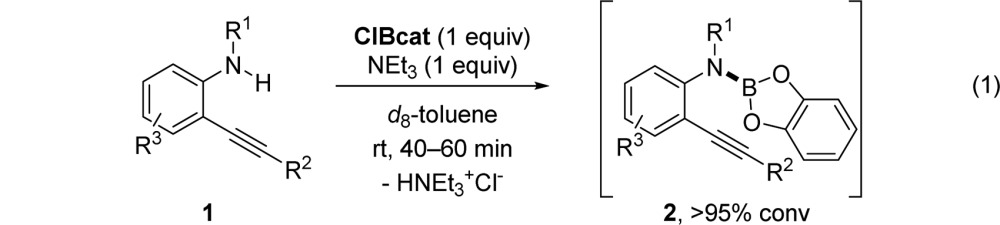
Switching to aminolysis of commercially available B-chlorocatecholborane (ClBcat) by 2-alkynlaniline 1 provided a solution for easily assembling the requisite B–N bond at rt within 1 h using triethylamine as a base to trap the released HCl byproduct (eq 1).27,28 In situ formed 2 does not undergo cyclization in the absence of the IPrAuTFA catalyst; for example, substrate 2d remains unreacted with the B–N bond intact when heated to 110 °C in d8-toluene for 20 h in the absence of a catalyst.
 |
1 |
This reaction displays a notable dependence on the presence of the NaTFA additive. Optimal conditions occurred with 5 mol % IPrAuTFA catalyst and 20 mol % NaTFA additive (using eq 1, in Chart 1) and not simply with the IPrAuTFA catalyst alone (in Table 1). No reaction occurred when aminoboronic ester 2, generated from the route (eq 1) using ClBcat and NEt3, was heated to 110 °C in d8-toluene for 20 h using 2.5 mol % IPrAuTFA catalyst in the absence of added NaTFA. A control reaction showed that 20 mol % NaTFA alone did not catalyze the cyclization of 2 under the conditions reported in Chart 1 (80 °C, 20 h), thereby supporting cationic gold as the Lewis acid catalyst for the reaction. The added NaTFA may overcome catalyst inhibition from trace HNEt3Cl leftover during the preparation of 2 via the optimized chlorocatecholborate route.29
Chart 1. Aminoboration Scope of 3-Borylated Indolesa with Functional Groups Incompatible with Competing Metalation or Pd(0)-Catalyzed Technologies Shown in Blue.
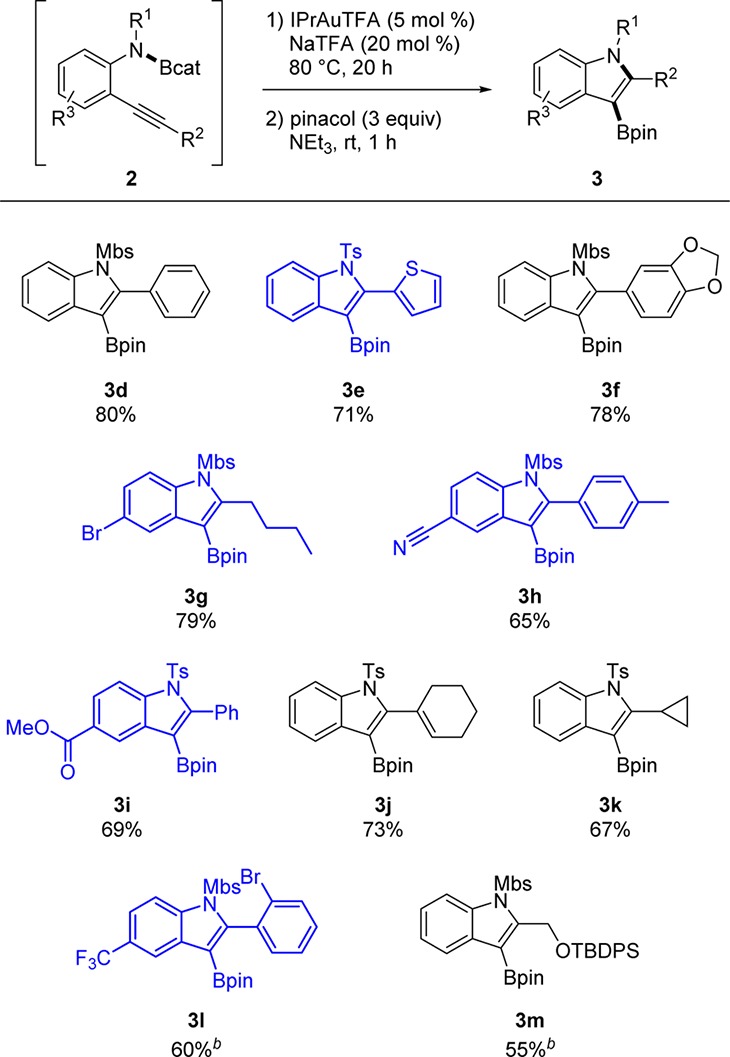
a Isolated yield of the Bpin product.
b 110 °C.
Our aminoboration strategy allows access to 3-borylated indoles with complementary regiochemistry and with functional groups that are incompatible with conventional routes or recent synthetic efforts (Chart 1). Alternative Ir-catalyzed C–H activation routes are predominantly regioselective for borylation at the 2-30 or 7-positions31 of indoles. The 3-position32 of indoles can be borylated using Ir catalysts with certain nitrogen protecting groups under limited conditions. The traditional metal–halogen exchange/borylation strategy33 of aryl halides with magnesium or o-lithiation/borylation34 of arenes with organolithium reagents are unable to tolerate aryl bromides, nitriles, esters, and other metalation-sensitive heterocycles. Pd-catalyzed Miyaura borylation35 of aryl halides and borylative cyclization36 of 2-alkynylanilines are also incompatible for the synthesis of brominated indole 3-boronic esters due to the potential oxidative addition of Pd into the Ar–Br bond.
The aminoboration reaction is compatible with a variety of these potentially sensitive functional groups. Compounds with these functional groups are shown in blue in Chart 1. Pharmaceutically relevant thiophenes (3e), aryl bromides (3g and 3l), nitriles (3h), and esters (3i) are tolerated by this method, as are alkenes (3j) and silyl-protected alcohols (3m). For bulkier substituents at R2, o-substituted aryl (3l) and OTBDPS (3m), increased temperatures of 110 °C were required to induce the cyclization. Pharmaceutically relevant37 1,3-benzodioxoles (3f) also can be tolerated in this transformation.
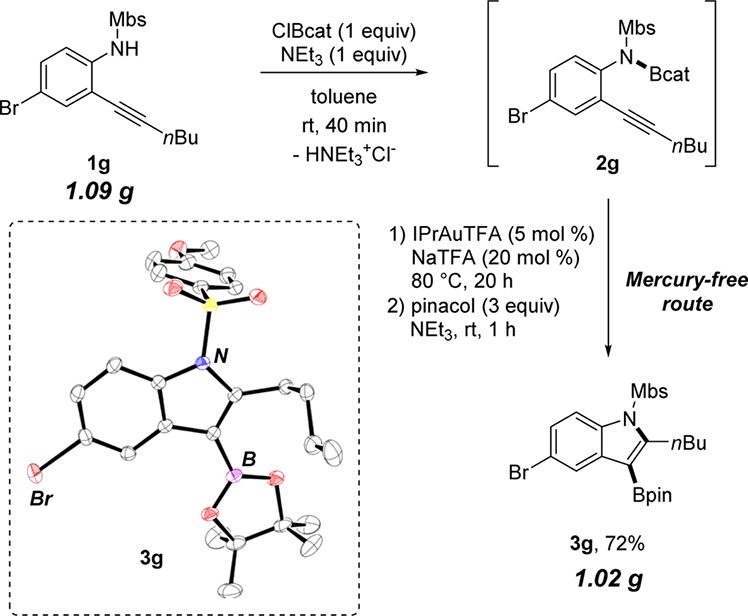
Due to the challenge of achieving the required functional group tolerance with alternative metalation and Pd(0)-catalyzed methods, borylated bromoindoles previously were made via routes with highly toxic mercury acetate.23,38 For example, in the total synthesis of dragmacidin D by Stoltz et al., a tosylated bromoindole was borylated at the 3-position using mercuration with Hg(OAc)2 followed by Hg/B exchange with BH3.38 Recent industrial pharmaceutical leads have also been synthesized by mercuration routes in order generate these borylated bromoindoles.23 In contrast, the functional group tolerance of the aminoboration method provides a mercury-free route to the synthesis of borylated bromoindoles (e.g., 3g).
The aminoboration reaction of 2-alkynylanilines can be easily run on the gram scale (Scheme 1). The scalability of this halide-tolerant aminoboration transformation demonstrates its synthetic utility and allows for generation of quantities suitable for multistep synthesis for downstream manipulation of the C–Br and C–B bonds in cross-coupling reactions.3,4 The solid-state molecular structure of 3g, obtained from single-crystal X-ray diffraction analysis, verifies the structure of the aminoborylated product arising from anti 1,2-addition of B–N across alkynes.
Scheme 1. Hg-Free Synthesis of 3-Borylated Bromoindole 3g on 1 g Scale and Characterization by X-ray Crystallography.
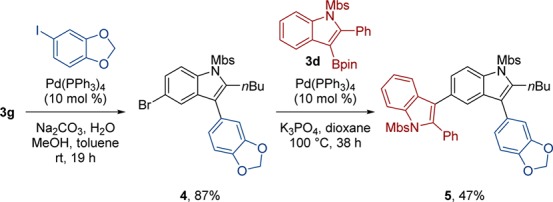
The employment of bromine-containing 3-borylated indole 3g in sequential Suzuki cross-coupling reactions highlights this downstream synthetic utility (Scheme 2). The C–B bond of 3g accesses a selective cross-coupling with an aryl iodide at rt to generate a new C–C bond,38 while maintaining the C–Br bond intact, to afford functionalized indole 4. The Ar–Br bond of 4 is available for a second cross-coupling with organoboronic acid derivatives, and here we further demonstrate the synthetic utility of 3-borylated indole 3d as a building block for C–C bond formation to construct biindole 5. Biindole scaffolds have reported biological activities,39 and this aminoboration facilitates access to such structures.
Scheme 2. Synthetic Versatility of Aminoboration Products.
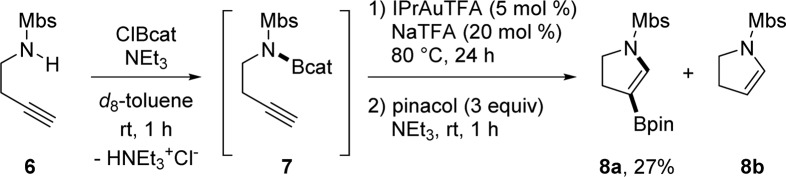
The synthetic utility of aminoboration is not limited to indole scaffolds and has potential for applications in the synthesis of other N-heterocyclic organoboron compounds (Scheme 3). When a simple amine 6, prepared from commercially available homopropargyl amine, is subjected to standard aminoboration conditions, 4-borylated dihydropyrrole 8a can be synthesized. The protodeborylated product 8b is the major byproduct, possibly due to a source of an acidic proton from the terminal alkyne of 7. This preliminary result demonstrates that a rigid backbone to aid cyclization and a gain of product aromaticity are not absolute requirements for the aminoboration reactivity.
Scheme 3. Synthesis of 4-Borylated Dihydropyrrole.
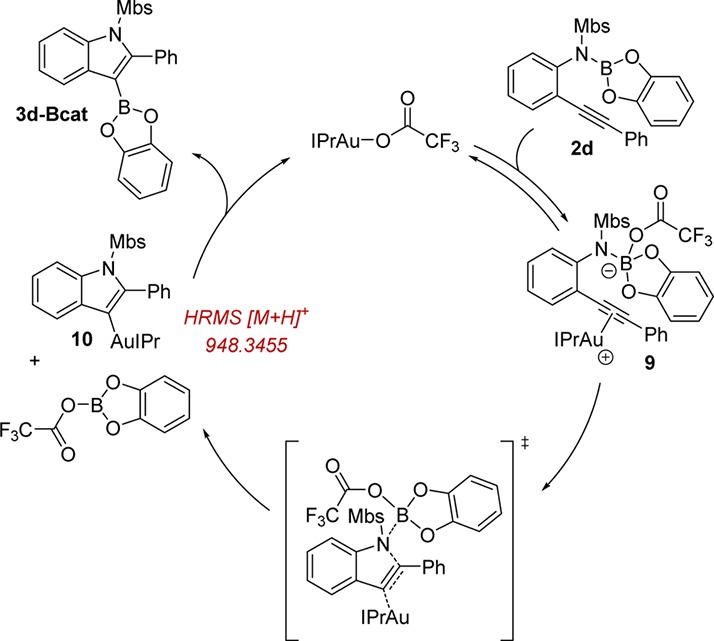
Numerous examples of Lewis acidic gold-mediated cyclization of 2-alkynylanilines exist for the synthesis of indole derivatives,40 but not for the installation of boron on the indole backbone for downstream reactivity. Based on the known carbophilicity of gold40,41 and our previous report on bifunctional catalysis on alkoxyboration of alkynes involving B–O bond activation,42 we propose the catalytic cycle shown in Scheme 4. The potentially bifunctional Lewis acidic/basic catalyst IPrAuTFA and substrate 2d associate to generate intermediate 9 containing a tetracoordinate borate. Simultaneous B–N bond activation and cyclization of 9 releases organogold intermediate 10 and B-(trifluoroacetyl)catecholborane. The presence of proposed intermediates is supported by the detection of neutral organogold 10 in the reaction mixture by HRMS prior to completion of the reaction. Next, organogold species 10 and B-(trifluoroacetyl)catecholborane undergo Au-to-B transmetalation43 to furnish the 3-borylated indole product 3d-Bcat and regenerate the IPrAuTFA catalyst. This reaction manifold is mechanistically distinct from the previous metal-catalyzed boron–element bond addition routes that proceed through metal-mediated breaking of the B–element bond through oxidative addition or σ-bond metathesis.
Scheme 4. Postulated Mechanism of Aminoboration.
In summary, we have developed the first catalytic aminoboration reaction that adds B–N σ bonds across C–C π bonds. We have shown that the easily generated but rather unreactive B–N σ bond can be used for this addition reaction, thereby providing a new bond disconnection strategy for the efficient construction of indole building blocks for potential applications in drug discovery. The use of a carbophilic gold catalyst and mildly basic trifluoroacetate provides tolerance of functional groups that are either incompatible with or difficult to access using existing methods. Ongoing work focuses on the development of aminoboration reactivity for the synthesis of other compound classes.
Acknowledgments
This work was supported by a grant from the NIH (1R01GM098512-01). We thank Dr. Joseph W. Ziller and Dr. Jordan F. Corbey for assistance with X-ray crystallography.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b06678.
The authors declare the following competing financial interest(s): US Pat. App. No. 14/303,684 and US Provisional Pat. App. No. 62/198,410 have been filed by the University of California.
Supplementary Material
References
- Hurd D. T. J. Am. Chem. Soc. 1948, 70, 2053. 10.1021/ja01186a020. [DOI] [Google Scholar]
- Brown H. C.; Rao B. C. S. J. Am. Chem. Soc. 1956, 78, 5694. 10.1021/ja01602a063. [DOI] [Google Scholar]
- Miyaura N.; Suzuki A. Chem. Rev. 1995, 95, 2457. 10.1021/cr00039a007. [DOI] [Google Scholar]
- Qiao J. X.; Lam P. Y. S. Synthesis 2011, 2011, 829. 10.1055/s-0030-1258379. [DOI] [Google Scholar]
- a Männig D.; Nöth H. Angew. Chem., Int. Ed. Engl. 1985, 24, 878. 10.1002/anie.198508781. [DOI] [Google Scholar]; b Crudden C. M.; Edwards D. Eur. J. Org. Chem. 2003, 2003, 4695. 10.1002/ejoc.200300433. [DOI] [Google Scholar]; c Carroll A. M.; O'Sullivan T. P.; Guiry P. J. Adv. Synth. Catal. 2005, 347, 609. 10.1002/adsc.200404232. [DOI] [Google Scholar]
- Miyaura N.Hydroboration, Diboration, Silylboration, and Stannylboration. In Catalytic Heterofunctionalization; Togni A., Grützmacher H., Eds.; Wiley-VCH: Weinheim, Germany, 2001; pp 1–45. [Google Scholar]
- a Marder T. B.; Norman N. C. Top. Catal. 1998, 5, 63. 10.1023/A:1019145818515. [DOI] [Google Scholar]; b Ishiyama T.; Matsuda N.; Miyaura N.; Suzuki A. J. Am. Chem. Soc. 1993, 115, 11018. 10.1021/ja00076a081. [DOI] [Google Scholar]
- a Suginome M.; Yamamoto A.; Murakami M. J. Am. Chem. Soc. 2003, 125, 6358. 10.1021/ja0349195. [DOI] [PubMed] [Google Scholar]; b Suginome M.; Shirakura M.; Yamamoto A. J. Am. Chem. Soc. 2006, 128, 14438. 10.1021/ja064970j. [DOI] [PubMed] [Google Scholar]; c Suginome M. Chem. Rec. 2010, 10, 348. 10.1002/tcr.201000029. [DOI] [PubMed] [Google Scholar]
- Suginome M.; Nakamura H.; Ito Y. Chem. Commun. 1996, 2777. 10.1039/cc9960002777. [DOI] [Google Scholar]
- Suginome M.; Nakamura H.; Ito Y. Angew. Chem., Int. Ed. Engl. 1997, 36, 2516. 10.1002/anie.199725161. [DOI] [Google Scholar]
- Onozawa S.; Hatanaka Y.; Sakakura T.; Shimada S.; Tanaka M. Organometallics 1996, 15, 5450. 10.1021/om9607199. [DOI] [Google Scholar]
- Ishiyama T.; Nishijima K.; Miyaura N.; Suzuki A. J. Am. Chem. Soc. 1993, 115, 7219. 10.1021/ja00069a020. [DOI] [Google Scholar]
- Lappert M. F.; Prokai B. J. Organomet. Chem. 1964, 1, 384. 10.1016/S0022-328X(00)80030-3. [DOI] [Google Scholar]
- Hara S.; Dojo H.; Takinami S.; Suzuki A. Tetrahedron Lett. 1983, 24, 731. 10.1016/S0040-4039(00)81511-7. [DOI] [Google Scholar]
- Semba K.; Fujihara T.; Terao J.; Tsuji Y. Chem. - Eur. J. 2012, 18, 4179. 10.1002/chem.201103612. [DOI] [PubMed] [Google Scholar]
- Bakus R. C.; Atwood D. A.. Boron–Nitrogen Compounds. In Encyclopedia of Inorganic Chemistry; John Wiley & Sons, Ltd: 2006. [Google Scholar]
- a Cragg R. H.; Lappert M. F.; Tilley B. P. J. Chem. Soc. 1964, 2108. 10.1039/jr9640002108. [DOI] [Google Scholar]; b Cragg R. H.; Miller T. J. J. Organomet. Chem. 1983, 255, 143. 10.1016/0022-328X(83)87017-X. [DOI] [Google Scholar]; c Singaram B. Heteroat. Chem. 1992, 3, 245. 10.1002/hc.520030309. [DOI] [Google Scholar]
- Jefferson R.; Lappert M. F.; Prokai B.; Tilley B. P. J. Chem. Soc. A 1966, 1584. 10.1039/j19660001584. [DOI] [Google Scholar]
- a Schreyer P.; Paetzold P.; Boese R. Chem. Ber. 1988, 121, 195. 10.1002/cber.19881210202. [DOI] [Google Scholar]; b For possibly an early example of aminoboration that is not well characterized, see: Chandra G.; George T. A.; Lappert M. F. Chem. Commun. 1967, 116. 10.1039/c19670000116. [DOI] [Google Scholar]; c For an example of adding a B–N triple bond across an alkyne, see: Braunschweig H.; Damme A.; Jimenez-Halla J. O. C.; Pfaffinger B.; Radacki K.; Wolf J. Angew. Chem., Int. Ed. 2012, 51, 10034. 10.1002/anie.201205795. [DOI] [PubMed] [Google Scholar]
- a Matsuda N.; Hirano K.; Satoh T.; Miura M. J. Am. Chem. Soc. 2013, 135, 4934. 10.1021/ja4007645. [DOI] [PubMed] [Google Scholar]; b Hirano K.; Miura M. Pure Appl. Chem. 2014, 86, 291. 10.1515/pac-2014-5004. [DOI] [Google Scholar]; c Sakae R.; Matsuda N.; Hirano K.; Satoh T.; Miura M. Org. Lett. 2014, 16, 1228. 10.1021/ol5001507. [DOI] [PubMed] [Google Scholar]; d Sakae R.; Hirano K.; Satoh T.; Miura M. Angew. Chem., Int. Ed. 2015, 54, 613. 10.1002/anie.201409104. [DOI] [PubMed] [Google Scholar]; e Sakae R.; Hirano K.; Miura M. J. Am. Chem. Soc. 2015, 137, 6460. 10.1021/jacs.5b02775. [DOI] [PubMed] [Google Scholar]
- Daley E. N.; Vogels C. M.; Geier S. J.; Decken A.; Doherty S.; Westcott S. A. Angew. Chem., Int. Ed. 2015, 54, 2121. 10.1002/anie.201410033. [DOI] [PubMed] [Google Scholar]
- a Horton D. A.; Bourne G. T.; Smythe M. L. Chem. Rev. 2003, 103, 893. 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]; b Welsch M. E.; Snyder S. A.; Stockwell B. R. Curr. Opin. Chem. Biol. 2010, 14, 347. 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bandini M.; Eichholzer A. Angew. Chem., Int. Ed. 2009, 48, 9608. 10.1002/anie.200901843. [DOI] [PubMed] [Google Scholar]; d Zhang M.-Z.; Chen Q.; Yang G.-F. Eur. J. Med. Chem. 2015, 89, 421. 10.1016/j.ejmech.2014.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabart M.Indolyldihydroimidazopyrimidinone Derivatives, Preparation Thereof and Therapeutic Use Thereof. International Patent WO2014/053568A1, April 10, 2014.
- Wynberg N. A.; Leger L. J.; Conrad M. L.; Vogels C. M.; Decken A.; Duffy S. J.; Westcott S. A. Can. J. Chem. 2005, 83, 661. 10.1139/v05-078. [DOI] [Google Scholar]
- IPrAuTFA was prepared from the salt metathesis reaction of commercially available IPrAuCl and AgTFA.
- Westcott S. A.; Blom H. P.; Marder T. B.; Baker R. T.; Calabrese J. C. Inorg. Chem. 1993, 32, 2175. 10.1021/ic00062a048. [DOI] [Google Scholar]
- Gerrard W.; Lappert M. F.; Mountfield B. A. J. Chem. Soc. 1959, 1529. 10.1039/jr9590001529. [DOI] [Google Scholar]
- Lappert M. F.; Majumdar M. K.; Tilley B. P. J. Chem. Soc. A 1966, 1590. 10.1039/j19660001590. [DOI] [Google Scholar]
- Presumably trace chloride ions can compete with a trifluoroacetate anion for binding to boron and/or gold.
- Robbins D. W.; Hartwig J. F. Org. Lett. 2012, 14, 4266. 10.1021/ol301570t. [DOI] [PubMed] [Google Scholar]
- a Paul S.; Chotana G. A.; Holmes D.; Reichle R. C.; Maleczka R. E.; Smith M. R. J. Am. Chem. Soc. 2006, 128, 15552. 10.1021/ja0631652. [DOI] [PubMed] [Google Scholar]; b Robbins D. W.; Boebel T. A.; Hartwig J. F. J. Am. Chem. Soc. 2010, 132, 4068. 10.1021/ja1006405. [DOI] [PubMed] [Google Scholar]
- a Takagi J.; Sato K.; Hartwig J. F.; Ishiyama T.; Miyaura N. Tetrahedron Lett. 2002, 43, 5649. 10.1016/S0040-4039(02)01135-8. [DOI] [Google Scholar]; b Kallepalli V. A.; Shi F.; Paul S.; Onyeozili E. N.; Maleczka R. E.; Smith M. R. J. Org. Chem. 2009, 74, 9199. 10.1021/jo901822b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Preshlock S. M.; Plattner D. L.; Maligres P. E.; Krska S. W.; Maleczka R. E.; Smith M. R. Angew. Chem., Int. Ed. 2013, 52, 12915. 10.1002/anie.201306511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K.-T.; Chien Y.-Y.; Liao Y.-L.; Lin C.-C.; Chou M.-Y.; Leung M.-K. J. Org. Chem. 2002, 67, 1041. 10.1021/jo011073j. [DOI] [PubMed] [Google Scholar]
- Snieckus V. Chem. Rev. 1990, 90, 879. 10.1021/cr00104a001. [DOI] [Google Scholar]
- Ishiyama T.; Murata M.; Miyaura N. J. Org. Chem. 1995, 60, 7508. 10.1021/jo00128a024. [DOI] [Google Scholar]
- Huang J.; Macdonald S. J. F.; Harrity J. P. A. Chem. Commun. 2010, 46, 8770. 10.1039/c0cc03577g. [DOI] [PubMed] [Google Scholar]
- Murray M. Curr. Drug Metab. 2000, 1, 67. 10.2174/1389200003339270. [DOI] [PubMed] [Google Scholar]
- Garg N. K.; Sarpong R.; Stoltz B. M. J. Am. Chem. Soc. 2002, 124, 13179. 10.1021/ja027822b. [DOI] [PubMed] [Google Scholar]
- Hu B.Substituted Sulfonamide-Indoles. U.S. Patent 7,442,805B2, October 28, 2008.
- Abbiati G.; Marinelli F.; Rossi E.; Arcadi A. Isr. J. Chem. 2013, 53, 856. 10.1002/ijch.201300040. [DOI] [Google Scholar]
- Fürstner A.; Davies P. W. Angew. Chem., Int. Ed. 2007, 46, 3410. 10.1002/anie.200604335. [DOI] [PubMed] [Google Scholar]
- Hirner J. J.; Faizi D. J.; Blum S. A. J. Am. Chem. Soc. 2014, 136, 4740. 10.1021/ja500463p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirner J. J.; Blum S. A. Tetrahedron 2015, 71, 4445. 10.1016/j.tet.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.