

Abstract
A photocatalytic system for CO2 reduction exhibiting visible-light harvesting was developed by preparing a hybrid consisting of a supramolecular metal complex as photocatalyst and periodic mesoporous organosilica (PMO) as light harvester. A RuII–ReI binuclear complex (Ru–Re) with methylphosphonic acid anchor groups was adsorbed on acridone or methylacridone embedded in the walls of PMO mesochannels to yield the hybrid structure. The embedded organic groups absorbed visible light, and the excitation energy was funneled to the Ru units. The energy accumulation was followed by electron transfer and catalytic reduction of CO2 to CO on the Re unit. The light harvesting of these hybrids enhanced the photocatalytic CO evolution rate by a factor of up to ten compared with that of Ru–Re adsorbed on mesoporous silica without a light harvester.
Keywords: artificial photosynthesis, co2 reduction, light harvesting, mesoporous materials, photochemistry
The conversion of CO2 to energy-rich compounds using solar light has recently attracted considerable attention owing to global warming and energy shortage problems. In particular, significant efforts are focused on the development of supramolecular photocatalysts constructed of redox photosensitizer and catalyst units for CO2 reduction because of their high efficiency, durability, speed, and selectivity to products.1 However, these photocatalysts require certain additional functionalities. Specifically, light harvesting (LH) is needed in molecular photocatalysts with a small cross section for absorption because the photon flux of solar light is too low.2 In natural photosynthesis, typical LH systems consisting of regularly arranged chlorophylls collect sunlight and efficiently transfer the excitation energy to a reaction center that initiates redox reactions. Many artificial LH systems have been developed from porphyrin assemblies,3a dendrimers comprising organic chromophores or transition metal complexes,3b metal–organic frameworks,3c and dye/clay hybrids.3d However, despite efficient energy accumulation, utilization of harvested photon energy in catalytic reactions has been very rare, especially for CO2 reduction.
In this study, periodic mesoporous organosilica (PMO)4 acted as LH material in which numerous acridone groups are embedded in the silica framework as visible-light absorbers5 and was combined with a supramolecular photocatalyst comprising a RuII photosensitizer and ReI catalyst for CO2 reduction1c (Scheme 1). Various PMO materials have been proven to be effective artificial LH systems.5, 6 However, only one hybrid material incorporating a ReI complex in biphenyl-embedded PMO mesochannels has served as a CO2 reduction photocatalyst.6a But this material did not harvest visible light because the biphenyl groups absorbed only UV light and the turnover number (TON) of CO2 reduction to CO was unfortunately very low (TONCO=2). In contrast, the new hybrid LH photocatalyst described herein utilizes visible light and accelerates the photocatalytic reduction of CO2 up to ten times compared with the corresponding molecular photocatalyst. Its TONCO reached 635, and CO formation was highly selective (>98 %).
Scheme 1.
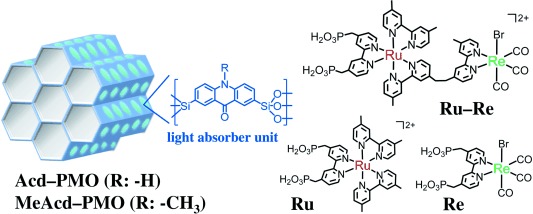
Schematic representation of PMO and metal complex structures
LH materials with acridone (Acd–PMO) or methylacridone (MeAcd–PMO) embedded in PMO mesochannels were synthesized using two types of acridone-bridged organosilane precursors5 (Scheme 1). The X-ray diffraction (XRD) patterns (Figure S1 in the Supporting Information) and N2 adsorption/desorption isotherms (Figure S2) of the synthesized PMOs showed well-defined mesoporous structures. The mesochannels exhibited pore diameters of 4.6 and 3.7 nm for Acd–PMO and MeAcd–PMO, respectively, exceeding the longest molecular length in the RuII−ReI complex estimated from the van der Waals radii (ca. 2.4 nm) and, thus, providing enough space for encapsulation of metal complexes. Only the hybridization and photophysical properties of Acd–PMO are described in detail below because both materials exhibited similar characteristics (Figure S3).
To facilitate the hybridization of Acd–PMO with the supramolecular photocatalyst, a RuII−ReI complex with a 2,2′-bipyridine ligand bearing methylphosphonic acids as anchor groups6b (Ru−Re, Scheme 1) was synthesized. The hybrid material (Ru−Re/Acd–PMO) was easily obtained by adding Acd–PMO powder to a Ru−Re solution in MeCN and stirring the dispersion overnight at an ambient temperature. Next, the dispersed powder was collected by filtration and washed with MeCN several times. All dissolved Ru−Re was adsorbed in Acd–PMO when the amount of Ru−Re in the solution was below 70 μmol per gram of Acd–PMO ([MC]Ads: μmol g−1 is used as the unit for the hybrid materials in the remainder of the manuscript). When the amount of dissolved Ru−Re exceeded this concentration, the adsorbed Ru−Re gradually reached saturation at 95±2 μmol g−1 (Figure S4). No elution of the complex or decomposition of Ru−Re was observed even when the hybrid was dispersed and stirred overnight in the 5:1 N,N-dimethylformamide–triethanolamine (DMF–TEOA, v/v) mixture, which was used as a solvent for CO2 reduction. The XRD pattern of Ru−Re/Acd–PMO (63 μmol g−1) was similar to that of Acd–PMO (Figure S5), suggesting that Acd–PMO retained its original structure upon Ru−Re adsorption. Table 1 summarizes the textural properties of Acd–PMO and Ru−Re/Acd–PMO (63 μmol g−1). Ru−Re/Acd–PMO exhibited roughly 15 % smaller pore volume than Acd–PMO. This decrease in pore volume upon Ru−Re adsorption closely agrees with the total volume of adsorbed Ru−Re, which was estimated from molecular volumes of the mononuclear model complexes [Ru(dmb)2(L1)](PF6)2 (11.5 nm3) and [Re(dmb)(CO)3]Br (4.2 nm3) (dmb=4,4′-dimethyl-2,2′-bipyridine, L1=4,4′-bis(methyl-phosphonate)-2,2′-bipyridine), indicating that Ru−Re mostly adsorbed in the mesochannels of Acd–PMO.
Table 1.
Textural Properties of PMO, MCM-41 and their Ru−Re Hybrid Materials.
| Material | SBET[a] [m2 g−1] | Vt[b] [cm3 g−1] | dDFT[c] [nm] | Particle size [μm] |
|---|---|---|---|---|
| Acd–PMO | 947 | 0.80 | 4.6 | 1.5 |
| Ru−Re/Acd–PMO[d] | 805 | 0.68 | 3.5 | – |
| MCM-41(I) | 1028 | 0.89 | 4.6 | 0.1 |
| Ru−Re/MCM-41(I)[d] | 755 | 0.62 | 3.7 | – |
| MeAcd–PMO | 760 | 0.40 | 3.7 | 0.2 |
| MCM-41(II) | 897 | 0.64 | 3.7 | 0.1 |
[a] Brunauer–Emmett–Teller specific surface area. [b] Pore volume calculated from the t-plot. [c] Pore diameter calculated by density functional theory. [d] Adsorbed Ru−Re amounts were 64 and 63 μmol g−1 in Acd–PMO and MCM-41(I), respectively.
The UV/Vis diffuse reflectance spectra of Acd–PMO and Ru−Re/Acd–PMOs showed a strong absorption band at around 380 nm, which corresponds to the π–π* transition of the Acd units in Acd–PMO (Figure S6). Ru−Re/Acd–PMO showed an additional absorption band at around 460 nm attributable to the metal-to-ligand charge transfer (1MLCT) absorption band of Ru−Re because of its similarity to that of Ru−Re adsorbed in mesoporous silica MCM-41(I),7 which has mesochannels identical to Acd–PMO (Table 1, Figure S7) without absorbing light above 350 nm. The FTIR spectrum of a KBr pellet of Ru−Re/Acd–PMO showed similar peaks to those of Ru−Re (Figure S8).
Figure 1 A shows emission spectra of Acd–PMO and Ru−Re/Acd–PMO dispersions in MeCN for different amounts of adsorbed Ru−Re at an excitation wavelength of 405 nm. Approximately 88 % of these excitation photons were absorbed by Acd units, even for the highest amount of adsorbed Ru−Re (93 μmol g−1) (see the Supporting Information). In the absence of Ru−Re, Acd groups showed a maximum emission at 520 nm with a quantum yield (Φ) of 0.029. An increase in the adsorbed Ru−Re amount quenched the emission of the Acd units and enhanced a new emission band at around 640 nm, which resembled that of Ru−Re/MCM-41(I) upon excitation at 456 nm (Figure S9).
Figure 1.
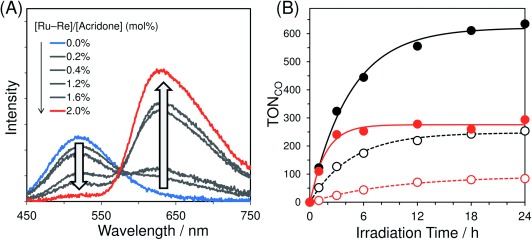
(A) Emission spectra of Acd–PMO and Ru−Re/Acd–PMO dispersions in MeCN at various adsorbed Ru–Re amounts. (B) Photocatalytic CO formation using Ru−Re/Acd–PMO (black solid line), Ru−Re/MCM-41(I) (black dotted line), Ru−Re/MeAcd–PMO (red solid line), and Ru−Re/MCM-41(II) (red dotted line).
The hybrid materials Ru/MCM-41(I) and Re/MCM-41(I), consisting of Ru or Re mononuclear complexes adsorbed in MCM-41(I) (Scheme 1), were used to determine emission quantum yields for each component (Acd–PMO, Ru, and Re units). Ru/MCM-41(I) displayed an emission band at around 640 nm with a quantum yield of 0.102 upon excitation at 460 nm. Re/MCM-41(I) emitted around 610 nm with a quantum yield of 0.015 when excited at 400 nm. Direct excitation of the Ru unit in Ru−Re/Acd–PMO at 460 nm produced an emission band (λmax=635 nm, ΦRu−Re=0.109) closely resembling that of Ru/MCM-41(I). A global fitting analysis was conducted for Ru−Re/Acd–PMO at an excitation wavelength of 405 nm (Figure S10, Table S1) using emission spectra of Acd–PMO, Ru/MCM-41(I), and Re/MCM-41(I). This analysis indicated that the emission maximum detected at 620 nm only resulted from the 3MLCT excited state of the Ru unit and almost no emission originated from the Re unit. Interestingly, the emission from Acd–PMO was largely quenched in Ru/Acd–PMO (80 μmol g−1), but not efficiently quenched in Re/Acd–PMO (80 μmol g−1; Figure S11). These results clearly indicate that the Förster-type energy transfer from Acd–PMO to the Ru unit proceeds with high efficiency but less effectively from Acd–PMO to the Re unit because the spectral overlap is relatively larger between the absorption of the Ru unit and emission of Acd–PMO than between the absorption of the Re unit and emission of Acd–PMO (Figure S12).8
Emission lifetimes measured at 500 nm for Acd units in Acd–PMO and Ru−Re/Acd–PMO (Figure S13 and Table S2) also showed that the Ru−Re complex efficiently quenched the excited state of Acd–PMO. A new emissive species was observed at 700 nm, which was attributed to the emission from the Ru unit by comparison with the lifetime of Ru/MCM-41(I) (Table S2).
The quantum yield of emission from the excited Ru unit produced via energy transfer from the Acd unit to the Ru unit [Φhybrid(Ru)] was calculated using Equation (1).
| 1 |
where nabs(Acd) is the number of photons absorbed by Acd–PMO and nem(Ru) is the number of photons emitted by the Ru unit through energy transfer from the Acd unit (see the Supporting Information). On the other hand, the direct excitation of the Ru unit in Ru−Re/Acd–PMO at 460 nm resulted in an emission quantum yield (ΦRu−Re) of 0.109, as described above. Energy transfer efficiencies (ηET) were obtained for various Ru−Re/Acd–PMO materials at different adsorbed Ru−Re amounts (Figure S14) using Equation (2).
| 2 |
When the ratio between Ru−Re and Acd units increased but remained relatively low (below 2 mol %), ηET increased linearly. For ratios exceeding 2 mol %, ηET reached a saturation point of (80±1) %. At a ratio of 2 mol %, only 10 % of the light emitted by the Acd units remained. Two possible mechanisms explain the “missing” excited state of the Acd units (ca. 10 %): a trivial energy transfer mechanism and/or another deactivation process induced by Ru−Re adsorption. The trivial mechanism involves a radiation (from the Acd units)+re-absorption (by Ru−Re) process (the emission quantum yield of the Acd units only equaled 0.029).
The number of excited Acd units that potentially transfer excitation energy to one Ru unit (N) was calculated using Equation (3) (Table S1).6b
| 3 |
For example, for N=42, photons absorbed by 42 Acd units can be accumulated into one Ru unit at a ratio of 1.9 mol % (64 μmol g−1), which was used for CO2 reduction.
The photocatalytic performance of Ru−Re/Acd–PMO (63 μmol g−1) for CO2 reduction was evaluated in 5:1 DMF–TEOA (v/v) using 1,3-dimethyl-2-phenylbenzimidazoline (BIH, 0.1 M)1d as a sacrificial reductant by irradiation at 405 nm employing a high-pressure mercury lamp with solution filters comprising 1 % NaNO2 in H2O (w/v) and 0.75 % I2 in CCl4 (w/v) [Eq. (4)].
| 4 |
The irradiation of a Ru−Re/Acd–PMO dispersion at 405 nm under CO2 atmosphere caused a catalytic and selective CO formation with a very small amount of H2 and without HCOOH formation (entry 1 in Table 2). For the initial 3 h irradiation, CO was continuously evolved with a turnover frequency (TOFCO) of 109 h−1 and TONCO reached 635 depending on the amount of Ru−Re used after 24 h irradiation. On the other hand, in control experiments using hybrids without Ru−Re (i.e., with Ru or Re instead of Ru−Re), in the dark, or under an Ar atmosphere instead of CO2, no CO or only small amounts of CO were observed (Table S3). Ru−Re/MeAcd–PMO (54 μmol g−1) also showed a catalytic and selective CO formation with TOFCO and TONCO values of 81 and 294, respectively (entry 3).
Table 2.
Photocatalytic CO2 Reduction.
| Entry | Photocatalyst | [MC]Ads [μmol g−1] | TOFCO[a] [h−1] | TONCO[b] | TON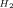 [b]
[b]
|
|---|---|---|---|---|---|
| 1 | Ru−Re/Acd–PMO | 64 | 109 | 635 | 10 |
| 2 | Ru−Re/MCM-41(I) | 63 | 36 | 254 | 29 |
| 3 | Ru−Re/MeAcd–PMO | 54 | 81 | 294 | 4 |
| 4 | Ru−Re/MCM-41(II) | 57 | 8 | 84 | 10 |
| 5 | (Ru+Re)/Acd–PMO[c] | 62 | – | 33 | 21 |
[a] After 3 h irradiation. [b] The turnover number was calculated based on the metal complex used. [c] Both [MC]Ads of Ru and Re were 62 μmol g−1.
To evaluate the effect of LH on the photocatalytic reaction, control experiments were conducted using hybrids combining Ru−Re with mesoporous silica MCM-41(I) and (II), which have mesochannel diameters similar to Acd–PMO and MeAcd–PMO, respectively. MCM-41(I) and (II) did not present any light-harvesting ability as they did not contain any organic light absorber. The Ru−Re/MCM-41(I) (63 μmol g−1) and Ru−Re/MCM-41(II) (57 μmol g−1) dispersions photocatalyzed the CO2 reduction under the same reaction conditions as the PMO hybrids because the adsorbed Ru−Re in the MCM-41 samples was directly excited by irradiation at 405 nm. However, their activities were much lower than those of the corresponding PMO hybrids (Figure 1 B), clearly indicating that the LH ability of PMOs enhanced the photocatalytic activity of Ru−Re for CO2 reduction.
The Ru−Re/Acd–PMO photocatalyst exhibited a threefold larger TOFCO than Ru−Re/MCM-41(I). The TOFCO was tenfold larger for Ru−Re/MeAcd–PMO than for Ru−Re/MCM-41(II). Considering the ratio between the numbers of photons absorbed by each unit in the hybrids and the energy transfer efficiency from PMO to Ru units, the number of excited Ru units in PMO hybrids is expected to be about tenfold larger than in MCM-41 hybrids (see the Supporting Information). The difference in the photocatalytic activity enhancement between Acd–PMO and MeAcd–PMO should be attributed to the difference in their light scattering at 405 nm. The particle sizes of Acd–PMO were on the microscale, which were larger than those of MeAcd–PMO, MCM-41(I), and MCM-41(II) (Table 1, Figure S15). Under photocatalytic conditions, visible-light scattering resulted in the devitrification of the Ru−Re/Acd–PMO suspension and limited its light absorption (405 nm). On the other hand, Ru−Re/MeAcd–PMO, Ru−Re/MCM-41(I), or Ru−Re/MCM-41(II) suspensions looked almost transparent because their sizes were equal to or less than 0.2 μm (Figure S16). This suggests that both Acd–PMO and MeAcd–PMO exhibited effective LH in photocatalytic reactions and that most photons accumulated by the Acd or MeAcd groups were used for CO2 reduction.
Another hybrid [(Ru+Re)/Acd–PMO] combining Acd–PMO with mononuclear Ru and Re complexes (62 μmol g−1 each) was also synthesized, and its photocatalytic activity was investigated under the same reaction conditions. However, its TONCO and selectivity (entry 5) were much lower than for Ru−Re/Acd–PMO (64 μmol g−1, entry 1). This result indicates that the covalent bonding between the Ru and Re units in the supramolecular photocatalyst played a crucial role in the photocatalytic activity of the hybrid material for CO2 reduction. This bonding may enhance the electron transfer efficiency from the one-electron-reduced Ru unit to the Re unit in the supramolecular system compared with the case where Ru and Re adsorb separately in the Acd–PMO mesochannels.
Although Ru−Re/MeAcd–PMO and Ru−Re/Acd–PMO display exceptionally high activities relative to other photocatalytic systems with LH ability,6a the potential ability of the supramolecular photocatalyst Ru−Re may not be fully developed in these hybrid systems, as suggested by a previous homogeneous solution system that has achieved higher photocatalytic ability, for example, TONCO>3000.1d Interestingly, small-pore Ru−Re/MeAcd–PMO (entry 3, Table 2) and Ru−Re/MCM-41(II) (entry 4) presented lower activities than the corresponding large-pore Ru−Re/Acd–PMO (entry 1) and Ru−Re/MCM-41(I) (entry 2), respectively. The slower diffusion of substrates, that is, BIH and CO2, and probably products through the mesochannels may be reflected in the lower activity of the hybrids. Specifically, the Stern–Volmer constant (KSV) of the excited Ru unit in Ru−Re/MeAcd–PMO (KSV=8.2 L mol−1) and Ru−Re/MCM-41(II) (7.5 L mol−1) by the reductant BIH were smaller than those in Ru−Re/Acd–PMO (22 L mol−1) and Ru−Re/MCM-41(I) (24 L mol−1) (Table S4). Consequently, large-pore hybrids (dDFT=4.6 nm) showed higher quenching ratios than their small-pore counterparts (dDFT=3.7 nm; Figure S17). The lifetime of emission from the Ru unit was shortened by the addition of BIH (Figure S19): without BIH τem=787 (92 %) and 382 ns (8 %); with BIH (0.1 M), τem=779 (7 %), 342 (12 %), 94 (27 %), and 7 ns (54 %). Since the longest component (τem≈780 ns) still remained even in the presence of 0.1 M BIH, a small part of Ru−Re (less than 10 %) in relatively narrow mesopores of Acd–PMO might be isolated from BIH. The insufficient quenching phenomena are expected to decelerate photocatalysis and possibly induce a photochemical decomposition of the metal complexes. The slower escape of CO and the oxidation product of BIH may also hinder the photocatalysis.
In conclusion, an efficient photocatalytic system with visible-light harvesting was developed for CO2 reduction. In this system, numerous Acd (MeAcd) units embedded in the walls of PMO absorbed visible light, and an extremely small number of Ru units efficiently accumulated the excitation energy stepwise. This LH function of PMO strongly enhanced the photocatalytic ability of the hybrids. An investigation of the effects of mesochannel pore sizes clearly indicated that wider pores resulted in improved photocatalysis in the hybrids because of the faster substrate and product diffusions in the mesochannels. However, the LH function needs to be maintained even when using hybrids with large-pore mesochannels. Further improvements of the LH function and photocatalysis of the hybrids are under investigation.
Acknowledgments
This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas ‘Artificial photosynthesis (AnApple)’ (Japan Society for the Promotion of Science (JSPS), No. 24107005), and CREST (Japan Science and Technology Agency (JST).
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1. For examples:
- 1a.Schulz M, Karnahl M, Schwalbe M, Vos JG. Coord. Chem. Rev. 2012;256:1682–1705. [Google Scholar]
- 1b.Takeda H, Ishitani O. Coord. Chem. Rev. 2010;254:346–354. [Google Scholar]
- 1c.Koike K, Naito S, Sato S, Tamaki Y, Ishitani O. J. Photochem. Photobiol. A. 2009;207:109–114. [Google Scholar]
- 1d.Tamaki Y, Koike K, Morimoto T, Ishitani O. J. Catal. 2013;304:22–28. [Google Scholar]
- 2.Inoue H, Shimada T, Kou Y, Nabetani Y, Masui D, Takagi S, Tachibana H. ChemSusChem. 2011;4:173–179. doi: 10.1002/cssc.201000385. [DOI] [PubMed] [Google Scholar]
- 3a.Miyatake T, Tamiaki H. Coord. Chem. Rev. 2010;254:2593–2602. [Google Scholar]
- 3b.Balzani V, Bergamini G, Ceroni P, Marchi E. New J. Chem. 2011;35:1944–1954. [Google Scholar]
- 3c.Kent CA, Mehl BP, Ma L, Papanikolas JM, Meyer TJ, Lin W. J. Am. Chem. Soc. 2010;132:12767–12769. doi: 10.1021/ja102804s. [DOI] [PubMed] [Google Scholar]
- 3d.Ishida Y, Shimada T, Masui D, Tachibana H, Inoue H, Takagi S. J. Am. Chem. Soc. 2011;133:14280–14286. doi: 10.1021/ja204425u. [DOI] [PubMed] [Google Scholar]
- 4.Inagaki S, Guan S, Ohsuna T, Terasaki O. Nature. 2002;416:304–307. doi: 10.1038/416304a. [DOI] [PubMed] [Google Scholar]
- 5a.Takeda H, Goto Y, Maegawa Y, Ohsuna T, Tani T, Matsumoto K, Shimada T, Inagaki S. Chem. Commun. 2009:6032–6034. doi: 10.1039/b910528j. [DOI] [PubMed] [Google Scholar]
- 5b.Takeda H, Ohashi M, Goto Y, Ohsuna T, Tani T, Inagaki S. Chem. Eur. J. 2014;20:9130–9136. doi: 10.1002/chem.201302815. [DOI] [PubMed] [Google Scholar]
- 6a.Takeda H, Ohashi M, Tani T, Ishitani O, Inagaki S. Inorg. Chem. 2010;49:4554–4559. doi: 10.1021/ic1000914. [DOI] [PubMed] [Google Scholar]
- 6b.Yui T, Takeda H, Ueda Y, Sekizawa K, Koike K, Inagaki S, Ishitani O. ACS Appl. Mater. Interfaces. 2014;6:1992–1998. doi: 10.1021/am405065a. [DOI] [PubMed] [Google Scholar]
- 7.Yokoi T, Yoshitake H, Tatsumi T. J. Mater. Chem. 2004;14:951–957. [Google Scholar]
- 8.Förster TZ. Discuss. Faraday Soc. 1959;27:7–17. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information