

Abstract
Oxidative stress is associated with many physiological and pathological processes, as well as xenobiotic metabolism, leading to the oxidation of biomacromolecules, including DNA. Therefore, efficient detection of DNA oxidation is important for a variety of research disciplines, including medicine and toxicology. A common biomarker of oxidatively damaged DNA is 8-oxo-7,8-dihydro-2'-deoxyguanosine (8-oxo-dGuo; often erroneously referred to as 8-hydroxy-2'-deoxyguanosine (8-OH-dGuo or 8-oxo-dG)). Several protocols for 8-oxo-dGuo measurement by high pressure liquid chromatography with electrochemical detection (HPLC-ED) have been described. However, these were mainly applied to purified DNA treated with pro-oxidants. In addition, due to methodological differences between laboratories, mainly due to differences in analytical equipment, the adoption of published methods for detection of 8-oxo-dGuo by HPLC-ED requires careful optimization by each laboratory. A comprehensive protocol, describing such an optimization process, is lacking. Here, a detailed protocol is described for the detection of 8-oxo-dGuo by HPLC-ED, in DNA from cultured cells or animal tissues. It illustrates how DNA sample preparation can be easily and rapidly optimized to minimize undesirable DNA oxidation that can occur during sample preparation. This protocol shows how to detect 8-oxo-dGuo in cultured human alveolar adenocarcinoma cells (i.e., A549 cells) treated with the oxidizing agent KBrO3, and from the spleen of mice exposed to the polycyclic aromatic hydrocarbon dibenzo(def,p)chrysene (DBC, formerly known as dibenzo(a,l)pyrene, DalP). Overall, this work illustrates how an HPLC-ED methodology can be readily optimized for the detection of 8-oxo-dGuo in biological samples.
Keywords: Chemistry, Issue 102, Oxidative Stress, DNA Damage, 8-oxo-7, 8-dihydro-2'-deoxyguanosine, 8-hydroxy-2'-deoxyguanosine, Xenobiotic Metabolism, Human Health
Introduction
Reactive oxygen species (ROS), whose steady-state levels can increase during many pathological conditions and xenotoxic metabolism, contribute to an increased frequency of oxidative DNA damage. Among several possible nucleobases oxidation products, oxidative DNA damage can readily be measured using the stable marker 8-oxo-7,8-dihydro-2'-deoxyguanosine (8-oxo-dGuo), which is one of the oxidized forms of 2’-deoxyguanosine (dGuo)1. 8-oxo-dGuo is the most abundant DNA lesion2 and, therefore, has been studied to greater detail as a DNA oxidation biomarker despite the existence of multiple DNA oxidation products 3. In humans, this damage can be repaired via base excision repair by 8-oxoguanine glycosylase 1 (hOGG1)4. If left unrepaired, 8-oxo-dGuo can contribute to the formation of base pair-substitution mutations (i.e., G to T transversions)4. Importantly, 8-oxo-dGuo is an established marker for DNA damage in relation to the initiation and promotion of carcinogenesis2. Therefore, accurate quantification of 8-oxo-dGuo is a useful and desirable biomarker of oxidative DNA damage5.
There is widespread confusion in the literature regarding the correct names for oxidatively-damaged forms of 2-deoxyguanosine and, moreover, the correct name of the compound(s) routinely measured as a biomarker of oxidative DNA damage6. The 6,8-diketo and 6-enol,8-keto tautomeric forms of 8-oxo-dGuo (shown in Figure 1) are the two most prominent tautomers discussed in the literature5,7. The 6,8-diketo form is the most prominent form at physiological pH of 7.4, and is the most prominent DNA oxidation product7. Therefore, 8-oxo-dGuo, rather than 8-hydroxy-dGuo is the most appropriate name for this oxidation product6. It is also important to note that 2-deoxyguanosine (dGuo), rather than nucleobase guanine (Gua) or ribonucleoside guanosine (Guo), respectively, is detected by most methods6.
Accurate detection and quantification of 8-oxo-dGuo is challenging due to: i) variability in the digestion of the DNA sample, ii) adventitious oxidation of dGuo to 8-oxo-dGuo that can occur during sample preparation, and iii) the need for effective validation of the analytical HPLC-ED method8. In this protocol, we aimed at achieving i) by providing conditions, favorable for complete DNA digestion and ii) by the inclusion metal chelator and chelator-treated solutions and a special DNA-isolating reagent, while iii) was only partially addressed by inclusion of positive controls and thus providing that the method is capable of detecting 8-oxo-dGuo in biological samples. Further validation is beyond the scope of this paper. However, we are confident that this protocol will help the prospective users determine the extent to which they need to formally validate the protocol, depending on their purposes. A list of steps required for the formal validation of the method is provided further. During the development and deployment of a method for 8-oxo-dGuo detection, published methods were reviewed and consolidated. Thus, this method eliminates the need to gather information from several published sources that often lack important experimental details while also providing rapid and straightforward means of testing if the method for the detection and quantification of 8-oxo-dGuo has been adopted successfully. This adapted method was employed to successfully analyze DNA samples from cultured cells and murine tissue. This video article will assist other groups in establishing an effective method for reliable detection and quantification of 8-oxo-dGuo by HPLC-ED.
Protocol
Ensure that all animal husbandry, housing, handling and experimentation adhere to local rules and regulations and that experimentation protocols are approved prior to commencing any study. For the described experiments, animal care, handling, and treatment were approved by the Health Canada Animal Care Committee. See the “Reagents table” for the suppliers’ information.
1. Collecting Biological Samples
- Cells or Animal Tissues
- Grow human alveolar adenocarcinoma A549 cells in F12-K media containing 10% fetal bovine serum, 100 units/ml of penicillin and 100 µg/ml of streptomycin.
- Seed cells at approximately 1 million cells per a 10-cm plate. For each experiment, use a set of 12 plates in total (three plates per one biological replicate x four doses) to provide enough DNA (80 μg) for enzymatic digestion and HPLC analysis.
- When cell density becomes greater than approximately 70% of the plate surface area, remove media and wash cells twice with 4 ml of phosphate-buffered saline (PBS), pH 7.4.
- For cultured animal cells, use KBrO3 as a positive control. NOTE: A positive linear relationship between KBrO3 concentration and 8-oxo-dGuo frequency has been reported in the literature9.
- Dissolve KBrO3 in PBS. Ensure that the final concentration in the cell culture medium is greater than 1 mM to obtain a statistically significant increase in 8-oxo-dGuo relative to un-exposed cells. Add the same concentration of KBrO3 to each plate in the series (e.g., 12 10-cm plates).
- Incubate the plates for 3 hr at 37 °C. Remove the media and wash once with PBS.
- Add 1 ml of trypsin solution (stock concentration 2.5 g/ml) and incubate for 3 min at 37 °C.
- Wash with 4 ml PBS, collect the cells from each set into a single 50 ml conical polystyrene centrifuge tube, and centrifuge at 1,000 x g for 5 min at 4 °C.
- Remove PBS and store the cell pellet at -80 °C until further analysis. Artificial DNA oxidation can be further minimized by nuclear isolation, as described elsewhere10.
- Animal Tissues
- Dose animals as required. For this protocol, adult (9 weeks of age) treat male Muta Mouse by oral gavage daily for three consecutive days with 20 mg DBC/kg bw per day dissolved in olive oil. NOTE: This transgenic animal harbors engineered λ-bacteriophage and mutation reporter gene lacZ from E. coli11 is used for transgenic rodent mutation assays12.
- Perform animal necropsy e.g., anesthetized the mice can using isoflurane and then euthanized via cervical dislocation followed by chest cavity opening. Immediately flash freeze tissues in liquid nitrogen. Store tissues at -80 °C until analysis. NOTE: The timing of euthanasia after the treatment may be another important variable (for instance, DNA oxidation was maximal at 72 hr after noise exposure in the rat brain and liver13).
2. DNA Extraction, Precipitation and Wash (For Tissues Proceed Directly to 2.2)
Homogenize the collected cells in a 50 ml conical polystyrene centrifuge tube with 1 ml of DNA isolating agent such as DNAzol. Use a 1,000 μl pipette tip to disperse the cell pellet until the solution is homogeneous. Transfer homogenate to a new 1.5 ml microcentrifuge tube. Incubate on ice for 10-20 min. Proceed to 2.3.
Homogenize 15-20 mg of tissue in a 1 ml handheld nonstick glass homogenizer containing 500 μl DNA isolating agent. Gently raise and lower homogenizer for about 1 min; softer tissues will require less time. Gentle treatment ensures less shearing of DNA. Store the homogenate on ice for about 10-20 min.
Pellet the homogenate by centrifugation for 10 min at 10,000 x g and 4 °C in a 1.5 ml conical microcentrifuge tube.
Carefully transfer the resulting supernatant into a new 1.5 ml microcentrifuge tube paying careful attention to avoid contacting the pellet.
Precipitate DNA from the homogenate by adding 0.5 ml 100% EtOH per 1 ml of homogenate. Invert tubes 10 times to ensure isolating reagent and EtOH are sufficiently mixed.
At this point, DNA precipitate is viscous, spool it onto a plastic pipette tip (e.g., with a maximum holding capacity of 200 μl) and then transferred to a new 1.5 ml conical microcentrifuge tube. NOTE: Quick centrifugation can be employed to remove any remaining lysate from the isolated DNA. (DNA Wash and Solubilization)
Add 1 ml of 75% EtOH to the isolated DNA. Suspend the DNA pellet thoroughly by inverting the tubes 10 times.
Carefully decant the ethanol from the tube. Ensure that the DNA pellet sticks to the side of the tube. Store the tubes vertically for 1-2 min and use a pipette to remove any excess EtOH from the bottom of the tube.
Repeat the DNA wash once more, and either store the sample in EtOH at -20 °C (stable for several months) or immediately proceed to digestion.
Dissolve DNA in digestion buffer (described below) and then quantify using standard spectroscopic methods (i.e., absorbance at 260 nm using NanoDrop spectrophotometer).
3. Enzymatic Digestion
Prepare “digestion buffer” by combining appropriate volumes (depending on the number of samples to be digested) of 50 mM monobasic Na phosphate (containing 2.0 mM KCl, 1.0 mM desferal (DFO) and 200 mM MgCl2) with 50 mM dibasic Na phosphate (also containing 2.0 mM KCl, 1.0 mM DFO and 200 mM MgCl2). Each sample requires 4.0 μl monobasic and 17.0 μl dibasic phosphate solution.
Remove any remaining EtOH from the bottom of the tube and air-dry samples for about 5 min. Ensure that the DNA does not dry out completely.
Dissolve 80 μg extracted DNA in 21.0 μl of the digestion buffer, add 1 unit of DNase I dissolved in 2.0 μl of digestion buffer.
Vortex lightly to achieve thorough mixing and incubate for 1.5 hr at 37 °C.
At the end of the incubation period, add 216.4 μl of dibasic 50 mM Na phosphate solution containing 2.0 mM KCl, 1.0 mM DFO and 200 mM MgCl2.
Prepare “digestion buffer-2” by combining one volume of digestion buffer with nine volumes of dibasic, 50 mM Na phosphate (with 2.0 mM KCl, 1.0 mM DFO and 200 mM MgCl2).
Add 0.025 units of phosphodiesterase (PDE) I enzyme in 16.3 μl of digestion buffer-2. Pipette up and down for several seconds, then vortex lightly to achieve thorough mixing and incubate for 1.5 hr at 37 °C.
Add 0.4 units of alkaline phosphatase (AP) in 8.0 μl of digestion buffer-2. Pipette up and down for several seconds, then vortex lightly to achieve thorough mixing and incubate for 1.5 hr at 37 °C. Add 33.0 μl HPLC grade MeOH.
Run digested DNA samples on HPLC, as described below, right away or store at -80 °C until use to minimize oxidation. NOTE: The 1.5-hr digestion steps suggested here are based on such intervals suggested elsewhere9 and on the findings that similar protocols achieve complete DNA digestion9. Therefore, it was assumed that the only limiting factor to achieve complete digestion was time and that enzyme inhibition and sample inhomogeneity were negligible.
4. HPLC Run: Preparation of Mobile Phase, Instrumentation Setup and Maintenance
Use ultrapure water to prepare all buffer solutions and treated with high bond strength resin (e.g., Chelex 100: styrene divinylbenzene copolymer with iminodiacetate ions that chelate transition metals with high affinity) to minimize metal contamination and DNA oxidation during sample preparation.
Prepare 250 mM Na phosphate buffer, pH 6.2. Use a ratio of 1 : 3.6957 of dibasic sodium phosphate (FW = 177.99 g/mol) to monobasic sodium phosphate (FW = 119.98 g/mol). Example: Therefore, for a 3 L stock solution combine 70.81 g of monobasic and 28.43 g of dibasic powder, add water up to e.g., 2.8 L and verify that the pH of the solution is 6.2. Adjust pH with 250 mM mono or dibasic sodium phosphate solutions, prepared separately and not by concentrated acids or bases. Add 2.24 g of KCl to obtain a concentration of 10 mM KCl.
Prepare mobile phase in three bottles, at least 1 L each, containing: solvent B – HPLC-grade methanol, solvent – B ultrapure water, and solvent C – phosphate buffer from step 4.2. Insert HPLC mobile phase supply tubing into the bottles; remaining tubes must be also dipped in one of the bottles (e.g., solution C) and primed even if they are not required for mixing.
During the run, mix the mobile phase solutions as 6% solvent A, 74% solvent B and 20% solvent C to achieve final solution of 50 mM sodium phosphate buffer (pH 6.2) with 2.0 mM KCl, and 6% MeOH.
Detach the electrode from the electrochemical detector, disassemble it and clean reference and working electrodes with the electrode cleaning solutions and polishing discs recommended by the manufacturer. Rinse the cleaned surfaces with ultrapure water, wipe the water off gently and ensure the detector is dry before turning the system on.
Reassemble the detector and connect the mobile phase inlet to the electrode. Turn on the flow and allow the mobile phase to fill up the electrode before attaching the outlet mobile phase tube. Install a new column prior to starting optimization. Ensure that installation is in the proper direction and follows all manufacturer’s recommendations.
Turn on the HLPC instrumentation well ahead (e.g., 1 hr) of sample analyses to allow equilibration and to reduce baseline noise. See Table 1 for suggested settings; the back pressure limit should be set to 3,000 psi to avoid column damage. Turn on the degasser. Proceed with priming the pumps and setting up solvent gradient.
Dry prime the pumps according to manufacturer’s instructions. Do this to wet the lines when a mobile phase has been replaced or the tubing inlet is exposed to air.
Locate the priming disc and insert a 10-15 ml plastic syringe there. Make sure the syringe is fully inserted before opening the line. To open the lines simply turn the disc counter clockwise for one turn.
Start the prime with the appropriate HPLC interface selection. Gently pull the syringe to draw fluid into the lines. Once fluid is flowing, the prime is complete; allow the program to finish the run. Repeat for all lines (usually four, depending on the instrument).
Alternatively, wet prime when the lines are already wetted from an earlier dry prime (see below). NOTE: When a large amount of time has elapsed between uses (e.g., 2 weeks or more), a dry prime is recommended.
Change the composition of the mobile phase solvent components to 25% each using the HPLC user interface. From the interface, select “Direct Function” option, then select “Wet Prime” option. This will prime all the lines at the same time.
Make sure all the air is out of the system by observing the wet prime line as it enters the waste container. After a good prime, ensure that no bubbles are left in the line. If bubbles persist, repeat priming.
Once the pump is primed, begin moving the HPLC mobile phase. Use the HPLC interface to manually change the composition of the mobile phase to 6% methanol (solvent A) and 94% water (solvent B), and select a flow rate of 0.9 ml/min. Allow 5 min to clear any methanol from the column cleaning step.
After 5 min, change the composition to 6% methanol (solvent A), 74% water (solvent B), 20% buffer (solvent C), increase the flow rate to 1.0 ml/min. Set other parameters as summarized in Table 1, and set the back pressure limit to 3,000 psi to avoid column damage. NOTE: Potential problems with HPLC setup include drifting baseline, split peak and diminished signal intensity. These can usually be overcome by cleaning the column after each use, frequent electrode cleaning, and ensuring that the buffer solutions are free of contamination (e.g., particulate matter or microbial growth).
Run the mobile phase for about 1-1.5 hr to achieve a stable baseline signal.
Using the instrument software interface, select parameters as indicated in Table 1 and set run time to 15 min. Inject the sample. Use increased run times for complex samples (determine this empirically by observing the presence or absence of peaks beyond 15-min interval). NOTE: Instrument software allows programming of sample run conditions and setup for autosampling.
After the runs have been completed, turn off the detector cell using the interface of the detector. It is important to follow manufacturer’s recommendations regarding powering down the detector as improper handling and shut-down can damage the instrument. DO NOT turn off the cell by switching the back of the detector as this could damage the instrument.
Manually change the mobile phase composition to 6% methanol and 94% water. Run for 5 min.
Manually reduce the flow rate to 0.7 ml/min, immediately change the composition to 50% MeOH and 50% water. Run for at least 20 min. Failure to run this step for the recommended time could result in damage to the column and the detector. Turn off the flow, and then turn off the degasser.
5. Preparation of Standards
Prepare mobile phase buffer as outlined in steps 4.1–4.3 above. It is important to use the HPLC mobile phase for preparation of standards to avoid peaks resulting from mixing of dissimilar solutions after sample injection.
Dilute this solution one in five with ultrapure water prior to use, and the final solution must contain 6% MeOH.
- dGuo Standard NOTE: High oxidation potential required for dGuo detection may increase the risk of measuring interfering electroactive compounds. This can be verified by, for example, a standard curve of UV detection of dGuo at 260 nm and comparing it to the standard curve created with EC detection. If both align well (e.g., Supplementary Figure 1) and the sample matrix effects have been taken into account, one may proceed with dGuo detection by UV at 260 nm, rather than EC detection.
- Measure 1.0 mg of dGuo in a 1.5 ml microcentrifuge tube. Add 1.0 ml HPLC grade MeOH.
- Vortex at high speed until dGuo dissolves completely. This is the primary stock; store at -20 °C for several weeks.
- To create the secondary dGuo stock (0.5 mM), dilute 143 µl of primary dGuo stock in 857 µl of HPLC mobile phase. Store on ice until use and prepare fresh daily.
- Make further dilutions to create a standard curve (e.g., Table 2). Store solutions on ice and prepare fresh daily. Run standards in series with experimental samples.
- Prior to run, vary the detector voltage using the highest concentration of the stock to find optimal voltage.
- 8-oxo-dGuo Standard
- Measure 1.0 mg of 8-oxo-dGuo in a 1.5 ml microcentrifuge tube. Add 0.1 ml HPLC grade MeOH. Vortex on high speed until 8-oxo-dGuo dissolves completely. This is the primary stock of 8-oxo-dGuo. Store at -20 °C for several weeks.
- In a separate tube, combine 2.9 μl of primary stock and 997.1 μl of HPLC mobile phase buffer, vortex. This is the secondary stock (10,000 nM).
- To create a working solution for the standard curve (250 nM), combine 24.4 μl of secondary stock and 975.6 μl of mobile phase buffer.
- Perform further dilutions to provide the solutions required to create a standard curve (e.g., Table 3). Store solutions on ice and prepare fresh daily. Run standards with the experimental samples.
- Quantification
- Integrate the area under the curve for both 8-oxo-dGuo and dGuo using standard software (as a part of HPLC-computer interface; see Supplementary Figure 2A).
- Construct standard curve (known concentration of each analyte vs. area under the curve (Supplementary Figure 2B). Using the equation of the standard curve, calculate the ratio for 8-oxo-dGuo/dGuo for each sample.
6. Agarose Gel Electrophoresis
NOTE: Agarose gel electrophoresis may be performed to verify the completeness of DNA digestion.
Prepare 50x Tris-acetate-EDTA (TAE) buffer, by dissolving 242 g Tris base (FW = 121.14) in 750 ml ultrapure water. Add 57.1 ml glacial acetic acid and 100 ml of 0.5 M EDTA (pH 8.0; EDTA will completely dissolve when the solution pH is adjusted to 8.0 with e.g., NaOH) and add ultrapure water to a final volume of 1 L. Store at RT, dilute this solution 50x prior to use.
Weigh out 1 g of agarose and dissolve it in 100 ml 1x TAE buffer, heating in the microwave for 1-2 min. Wear goggles and protective equipment to avoid contact with boiling agarose.
Cool the solution down until approximately 50 °C, add 5 μl ethidium bromide (caution: mutagen) and pour it into the agarose gel running apparatus. Insert the comb.
Wait until the solution solidifies, pour TAE buffer over the gel and load the samples (volume depends on the comb size; typically, 10-20 μl of DNA sample is loaded, mixed with 6x running dye to visualize and facilitate loading).
Run the gel until the dye migrates approximately 2/3 the length of the gel (e.g., 45 min at 150 V). Visualize DNA bands under the UV light.
Representative Results
dGuo was observed to have a retention time of 4.7 min whereas 8-oxo-dGuo had a retention time of approximately 6.4 min (Figure 2A and B). There is about 1,000-fold difference in the peak heights between the two analytes, as seen in Figure 2C. Voltammograms for 8-oxo-dGuo and dGuo were obtained by running standards at a working potential in the range of +0.2 to +1.1 V. The optimum working potential for 8-oxo-dGuo was determined to be +0.5 V, and +0.9 V for dGuo (Figure 3). These potentials are in agreement with other glassy carbon electrodes described in the literature14,15. The limit of detection and dynamic range of 8-oxo-dGuo and dGuo electrochemical detection is reported to be in the femtomole and nanomole range, respectively8,9. Standard curves for dGuo and 8-oxo-dGuo should be run daily to ensure linear detection across a suitable concentration range and proper performance of the instrument (Figure 4). Standard curves are constructed by plotting the peak area as a function of known analyte concentration (Supplementary Figure 2) that allows one to calculate the analyte concentration in the samples from the equation generated from the standard curves.
Complete DNA digestion is required for accurate measurements of 8-oxo-dGua frequency and several genomic DNA digestion methods have been suggested8,15,16. Where necessary, the efficacy of DNA extraction and digestion can be verified using agarose gel electrophoresis (Figure 5A) and HPLC-ED (Figure 5B). Lanes with obvious DNA smear in Figure 5A (e.g., lanes 2, 4, 6, 9, 11, and 13) indicate the presence of undigested DNA while samples with digested DNA fragments run off the gel and were thus undetected. The plateau in Figure 5B indicates that further incubation of DNA with digestion enzymes beyond 1.5 hr does not result in greater amount of dGuo detected, suggesting nearly complete digestion after 1.5 hr. In addition, both agarose gel electrophoresis and HPLC-ED were used to test the utility of the phosphate-based DNA digestion buffer employed here. This buffer is well matched to the HPLC mobile phase and does not give rise to undesirable detector noise attributable to mixing of dissimilar solutions (data not shown). Therefore, the switch from the buffer suggested in the literature (i.e., Tris-HCl8) to sodium phosphate buffer is recommended for this protocol (left versus right side of Figure 5A). Please note that the underlying assumptions for the optimization of DNA digestion were that enzyme inhibition and sample inhomogeneity were negligible and digestion time was the only limiting factor, as stated above. A possibility that incomplete DNA digestion resulted in fragments that are too small to be detected on the agarose gel remains. Even though such a systematic error would affect all samples equally, further experiments (e.g., treatment of cultured cells with a radiolabelled pro-oxidant10) could be conducted to completely rule out such a possibility.
Unlike standard solutions, the digested DNA, whether from isolated cells or mouse tissues, produced several additional peaks (Figure 6A and 6B). These were distant from the 8-oxo-dGuo and dGuo peaks, but spiking experiments as a part of validation exercises outlined in the Discussion are needed to examine if additional peaks (i.e., sample impurity due to matrix effects, for example) interfere with quantification. The 8-oxo-dGuo peak was confirmed by correspondence of the retention time with the standard; and moreover, increase in the 8-oxo-dGuo peak for samples from cells or animals that underwent pro-oxidant treatment (Figure 7). Pro-oxidant KBrO3 participates in one-electron abstraction from guanine that leads to 8-oxo-dGuo formation17. It is noteworthy that in the absence of the pro-oxidant stress, 2.4 molecules of 8-oxo-dGuo per 107 dGuo were detected in A549 cells (Figure 7A). This is comparable to 4.5 molecules of 8-oxo-dGuo/107 dGuo detected in A549 cells observed using an alternative, mass spectrometry-based approach designed to address potential shortcomings of the HPLC-ED method9. DBC treatment (i.e., daily 20.0 mg DBC/kg bw per day for three days) increased 8-oxo-dGuo levels in mouse spleen DNA. This is also an expected result, since ROS is known to be produced during the metabolism of polycyclic aromatic hydrocarbons in vivo8. The relative levels of 8-oxo-dGuo in the spleen of unstressed animals were 8.3-9.4 8-oxo-dGuo/106 dGuo (Figure 7B), which is in excellent agreement with published values of four 8-oxo-dG molecules/106 dGuo measured in murine splenic cells under standard conditions by HPLC-ED18. Figures 6 and 7 show that the levels of 8-oxo-dGuo above the baseline are very small. Thus if DNA is oxidized to lower levels than observed here, the levels of 8-oxo-dGuo may be indistinguishable from baseline, which should be kept in mind by prospective protocol users.
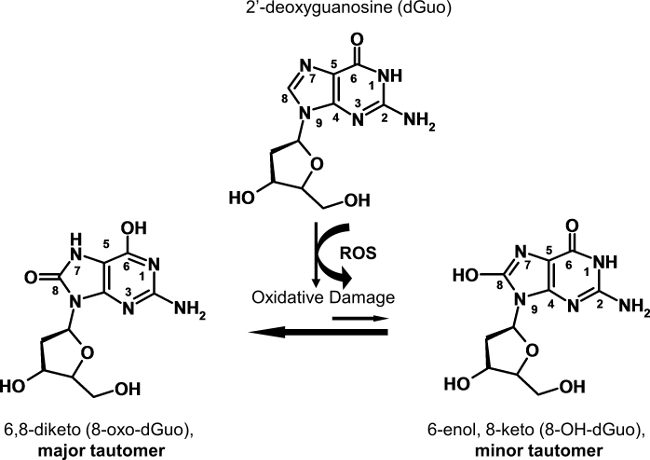 Figure 1. Structure of 8-oxo-dGuo and its tautomer 8-OH-dGuo. Note that 8-oxo-dGuo rather than 8-OH-dGuo is the major tautomer at pH 7.4.
Figure 1. Structure of 8-oxo-dGuo and its tautomer 8-OH-dGuo. Note that 8-oxo-dGuo rather than 8-OH-dGuo is the major tautomer at pH 7.4.
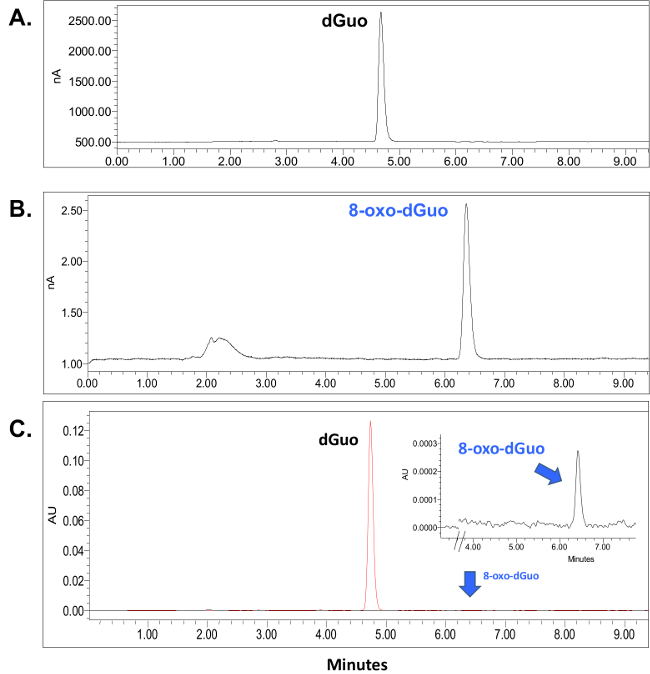 Figure 2. Retention times for dGuo (A) and 8-oxo-dGuo (B). HPLC–ED chromatograms were obtained from a 10.0 μl injection of either dGuo, (1.0 nmol total) or 8-oxo-dGuo (1,000 fmol total) and detected at 0.9 V (A) or 0.5 V (B). The retention peak for dGuo was approximately 4.7 min (A) and that for 8-oxo-dGuo was approximately 6.4 min (B). The broad peak at 2-3 min is likely caused by injection disturbances and is unaffected (i.e., always present at similar intensity) by the concentration of 8-oxo dGuo. (C) Superimposed chromatograms from two separate injections described in (A) and (B) detected by UV (260 nm). The standards were run on the same day using identical run conditions. Please click here to view a larger version of this figure.
Figure 2. Retention times for dGuo (A) and 8-oxo-dGuo (B). HPLC–ED chromatograms were obtained from a 10.0 μl injection of either dGuo, (1.0 nmol total) or 8-oxo-dGuo (1,000 fmol total) and detected at 0.9 V (A) or 0.5 V (B). The retention peak for dGuo was approximately 4.7 min (A) and that for 8-oxo-dGuo was approximately 6.4 min (B). The broad peak at 2-3 min is likely caused by injection disturbances and is unaffected (i.e., always present at similar intensity) by the concentration of 8-oxo dGuo. (C) Superimposed chromatograms from two separate injections described in (A) and (B) detected by UV (260 nm). The standards were run on the same day using identical run conditions. Please click here to view a larger version of this figure.
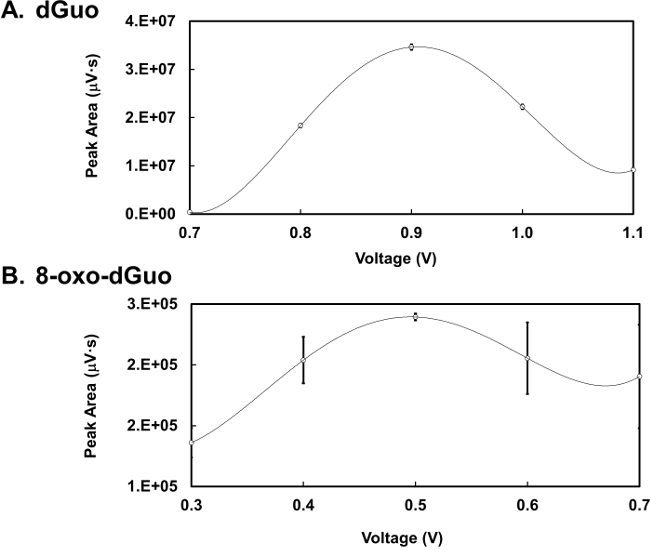 Figure 3. Optimizing detector voltage.(A) Voltammogram of dGuo (final concentration 1 nmol) detected by HPLC-ED analysis at a range of 0.7-1.1 V. (B) Voltammogram of 8-oxo-dGuo (final concentration 500 fmol) detected by HPLC-ED analysis at a range of 0.3-0.7 V. Means of three independent experiments (N=3) ± standard deviation shown. In some cases error bars are smaller than the plotting symbols.
Figure 3. Optimizing detector voltage.(A) Voltammogram of dGuo (final concentration 1 nmol) detected by HPLC-ED analysis at a range of 0.7-1.1 V. (B) Voltammogram of 8-oxo-dGuo (final concentration 500 fmol) detected by HPLC-ED analysis at a range of 0.3-0.7 V. Means of three independent experiments (N=3) ± standard deviation shown. In some cases error bars are smaller than the plotting symbols.
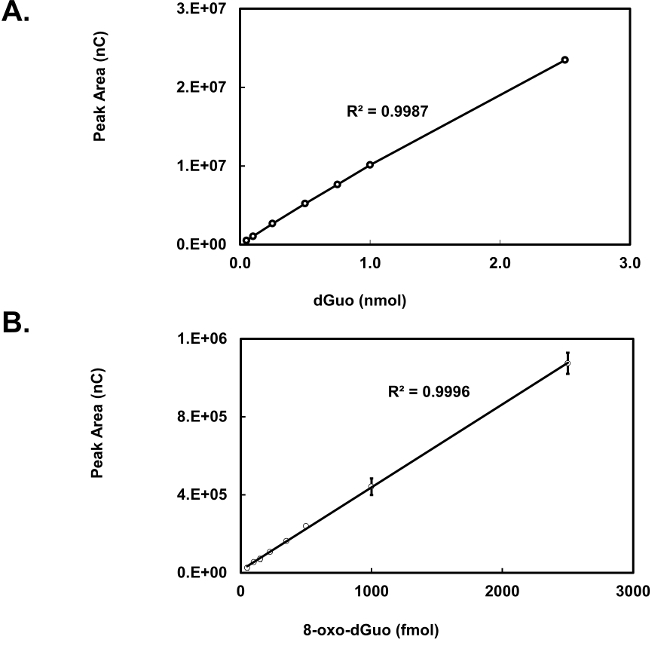 Figure 4. Standard curve for dGuo (A) and 8-oxo-dGuo (B). Ten microliters of either dGuo or 8-oxo-dGuo was injected and measured by HPLC-ED at 0.9 V (A) or 0.6 V (B). Means of three independent experiments (N=3) ± standard deviation shown. In some cases error bars are smaller than the plotting symbols.
Figure 4. Standard curve for dGuo (A) and 8-oxo-dGuo (B). Ten microliters of either dGuo or 8-oxo-dGuo was injected and measured by HPLC-ED at 0.9 V (A) or 0.6 V (B). Means of three independent experiments (N=3) ± standard deviation shown. In some cases error bars are smaller than the plotting symbols.
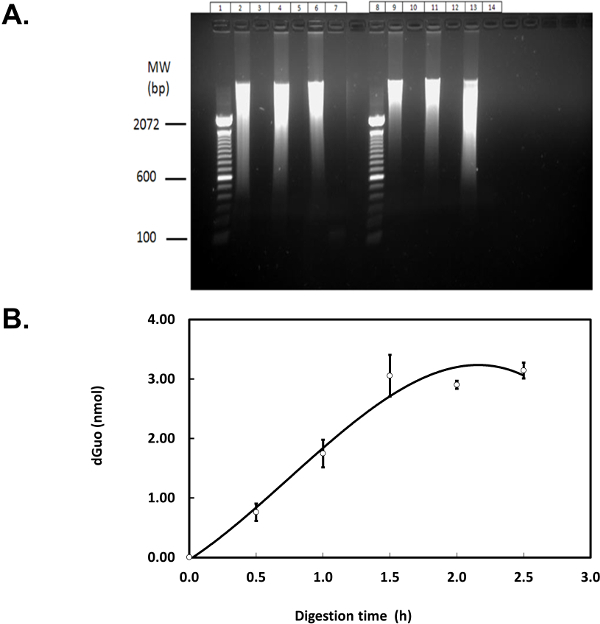 Figure 5. Optimization of DNA digestion. (A) Eighty microgram of salmon testes DNA was digested as described8 in Tris-HCl, glycine-acetate buffer (left) or sodium phosphate buffer (right). Agarose gel electrophoresis (1.0%) was run at 150 V for 45 min. Lane 1 and 8: DNA ladder; lanes 2, 4, and 6: undigested DNA; lanes 3, 5, and 7: digested DNA; lanes 9, 11, and 13: undigested DNA (phosphate buffer); lanes 10, 12, and 14: digested DNA (phosphate buffer). (B) Salmon testes DNA (80 µg) was digested herein by incubating with DNase I, PDE I, and AP for 0.5-2.5 hr per each step, in 50 mM phosphate buffer containing magnesium chloride and DFO. Fifty microliters of each sample, containing 7.3 µg of digested DNA, was analyzed by HPLC (i.e., 0.9 V and 1 ml/min flow) to detect dGuo. Figure shows means of three independent experiments (N=3) ± standard error of the mean. Please click here to view a larger version of this figure.
Figure 5. Optimization of DNA digestion. (A) Eighty microgram of salmon testes DNA was digested as described8 in Tris-HCl, glycine-acetate buffer (left) or sodium phosphate buffer (right). Agarose gel electrophoresis (1.0%) was run at 150 V for 45 min. Lane 1 and 8: DNA ladder; lanes 2, 4, and 6: undigested DNA; lanes 3, 5, and 7: digested DNA; lanes 9, 11, and 13: undigested DNA (phosphate buffer); lanes 10, 12, and 14: digested DNA (phosphate buffer). (B) Salmon testes DNA (80 µg) was digested herein by incubating with DNase I, PDE I, and AP for 0.5-2.5 hr per each step, in 50 mM phosphate buffer containing magnesium chloride and DFO. Fifty microliters of each sample, containing 7.3 µg of digested DNA, was analyzed by HPLC (i.e., 0.9 V and 1 ml/min flow) to detect dGuo. Figure shows means of three independent experiments (N=3) ± standard error of the mean. Please click here to view a larger version of this figure.
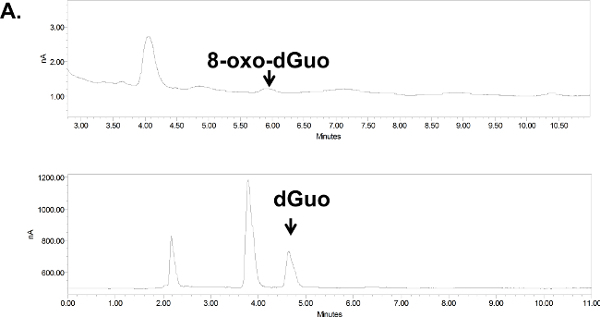
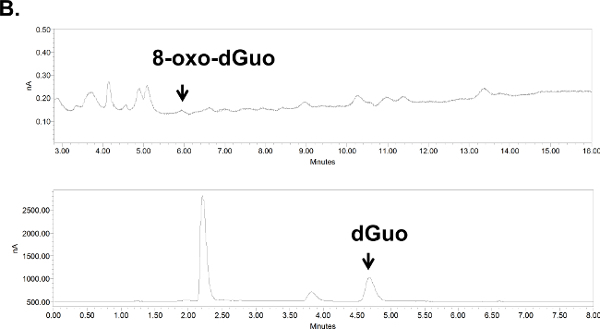 Figure 6. Chromatograms for 8-oxo-dGuo detection in cultured cells (A) or animal tissue (B).(A) Digested DNA from A549 cells, treated or untreated with KBrO3. (B) Digested DNA from the spleen of DBC-treated mice. Peaks shifted to the left due to the removal of guard column (due to pressure build-up in the apparatus; their position was confirmed with standards). Standards and samples were run on the same day using identical run conditions and 10 ml injection volumes, at 1 ml/min flow rate and 0.5 and 0.9 V for dGuo and 8-oxo-dGuo, respectively. Please click here for a larger version of panel A; here for panel B.
Figure 6. Chromatograms for 8-oxo-dGuo detection in cultured cells (A) or animal tissue (B).(A) Digested DNA from A549 cells, treated or untreated with KBrO3. (B) Digested DNA from the spleen of DBC-treated mice. Peaks shifted to the left due to the removal of guard column (due to pressure build-up in the apparatus; their position was confirmed with standards). Standards and samples were run on the same day using identical run conditions and 10 ml injection volumes, at 1 ml/min flow rate and 0.5 and 0.9 V for dGuo and 8-oxo-dGuo, respectively. Please click here for a larger version of panel A; here for panel B.
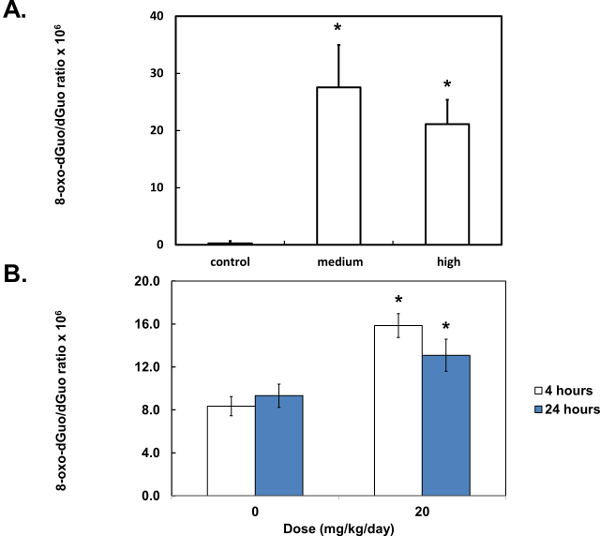 Figure 7. Quantification of 8-oxo-dGuo in biological samples. Peaks were integrated and compared to standard curves (see Supplementary Figure 2 for more details) to yield 8-oxo-dGuo levels in DNA from A549 cells, treated with KBrO3 (A), or 8-oxo-dGuo levels in DNA from the spleen of DBC-treated mice (B). Stars (*) indicate statistical significance (p < 0.05, one-way analysis of variance (ANOVA) (A), Student’s t-test (B)). Means of three (A) or five (B) independent experiments ± standard error of the mean are shown. Samples were collected 4 or 24 hr after the 3-day treatment.
Figure 7. Quantification of 8-oxo-dGuo in biological samples. Peaks were integrated and compared to standard curves (see Supplementary Figure 2 for more details) to yield 8-oxo-dGuo levels in DNA from A549 cells, treated with KBrO3 (A), or 8-oxo-dGuo levels in DNA from the spleen of DBC-treated mice (B). Stars (*) indicate statistical significance (p < 0.05, one-way analysis of variance (ANOVA) (A), Student’s t-test (B)). Means of three (A) or five (B) independent experiments ± standard error of the mean are shown. Samples were collected 4 or 24 hr after the 3-day treatment.
| Mobile phase | 50.0 mM Na phosphate buffer, pH 6.2, containing 6% MeOH and 2.0 mM KCl |
| Mobile phase flow rate | 1.0 ml/min |
| Column type | YMC-BASIC with bonded spherical silica |
| Column length | 15 cm |
| Column inner diameter | 3.5 mm |
| Column temperature | 35 °C |
| Detector temperature | 29 °C |
| Voltage setting for 8-oxo-dGuo | +0.5 V |
| Voltage setting for dGuo | +0.9 V |
| Injection volume | 10.0 ml |
Table 1. Instrument parameters for detection and quantification of 8-oxo-dGuo by HPLC-ED.
| nmoles | 100 nM standard, µl | HPLC Mobile phase, µl | Dilution, fold |
| 5 | 300 | 0 | 1 |
| 2.5 | 150 | 150 | 2 |
| 1 | 60 | 240 | 5 |
| 0.5 | 30 | 270 | 10 |
| 0.25 | 15 | 285 | 20 |
| 0.1 | 6 | 294 | 50 |
Table 2. Preparation of standard curve for dGuo.
| fmoles | 250 nM standard, µl | HPLC Mobile phase, µl | Dilution, fold |
| 2,500 | 300 | 0 | 1 |
| 1,000 | 120 | 180 | 2.5 |
| 500 | 150 | 150 | 5 |
| 250 | 150 | 150 | 10 |
| 100 | 120 | 180 | 25 |
| 50 | 150 | 150 | 50 |
Table 3. Preparation of standard curve for 8-oxo-dGuo.
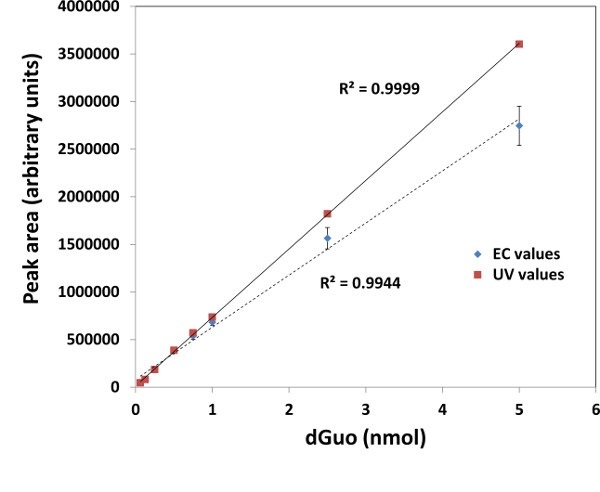 Supplementary Figure 1. Comparison of electrochemical (EC) and UV-based detection of dGuo. Standard curve was created for dGuo detection by electrochemical detection (EC) or UV detection (260 nm). Peak areas were integrated for dGuo peak at approximately 4.7 min by both methods. N=3, means ± standard deviation are shown.
Supplementary Figure 1. Comparison of electrochemical (EC) and UV-based detection of dGuo. Standard curve was created for dGuo detection by electrochemical detection (EC) or UV detection (260 nm). Peak areas were integrated for dGuo peak at approximately 4.7 min by both methods. N=3, means ± standard deviation are shown.
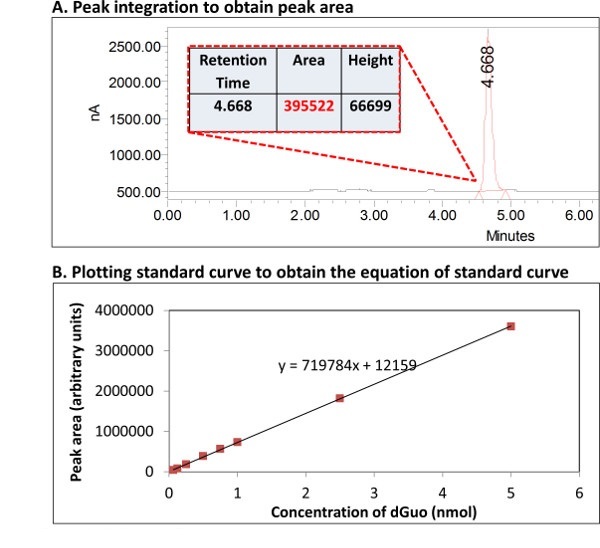 Supplementary Figure 2. Quantification of dGuo from its standard curve. An example of the use of standard curve to determine the concentration of dGuo is shown. (A) Peak area can be determined by integration using standard software. (B) This information is used to construct a standard curve in order to use the equation of the line to calculate the unknown analyte concentration in the sample. Please click here to view a larger version of this figure.
Supplementary Figure 2. Quantification of dGuo from its standard curve. An example of the use of standard curve to determine the concentration of dGuo is shown. (A) Peak area can be determined by integration using standard software. (B) This information is used to construct a standard curve in order to use the equation of the line to calculate the unknown analyte concentration in the sample. Please click here to view a larger version of this figure.
Discussion
Although 8-oxo-dGuo has been reported as a useful biomarker of DNA oxidation, its reliable quantification can pose a challenge. Although several published methods exist, there is a need for a comprehensive, descriptive overview of protocol to permit researchers to deploy the method in their laboratories. Here we present a detailed overview of an HPLC-based protocol that will permit new users to establish an effective method for 8-oxo-dGuo detection and quantification.
Three major methods that have been described for quantification of 8-oxo-dGuo. These include enzyme-linked immunosorbent assay (ELISA)-based commercial kits19, liquid chromatography/mass spectrometry (MS) methods (e.g., as described elsewhere9 and HPLC-ED methods, such as that described herein. ELISA methods can suffer from interferences with certain compounds in biological samples19. In general, results obtained using chromatographic techniques are considered more reliable for 8-oxo-dGuo quantification in comparison with ELISA-based methods20. In comparing the HPLC-ED methods with the MS-based methods, the advantage of the former is relatively inexpensive equipment (i.e., approximately an order of magnitude less costly than equipment for the MS-based methods). In addition, HPLC-MS are superior to HPLC-ED methods as the former are not prone to potential interferences with electroactive compounds and provide unambiguous, quantitative measurements using isotopes21.
Concerns have been raised regarding the use of traditional phenol extraction methods for the extraction of genomic DNA to be used for 8-oxo-dGuo quantification22. Several researchers have noted that the conditions associated with the standard phenol extraction can introduce unwanted dGuo oxidation thus increasing baseline levels1,9. To address this problem, cell and animal genomic DNA can be extracted and purified using a modified DNAzol protocol22. DNAzol contains high concentrations of guanidine thiocyanate, and previous studies9 have noted lower basal level 8-oxo-dGuo formation when DNAzol is used compared to traditional phenol- and NaI-based extraction methods. The iron chelating compound DFO was used to minimize ROS production and DNA oxidation during sample preparation. As noted above, the 8-oxo-dGuo levels in unstressed A549 cells (Figure 7A) were highly similar to what was measured by an MS-based method13. This suggests that incorporation of precautions described for the MS-based methods13 permitted minimization of undesirable DNA oxidation during sample preparation and analysis to achieve accurate 8-oxo-dGuo quantification by HPLC-ED. Timing of necropsy or harvesting culture cells after pro-oxidant treatment may be another important factor, as seen here by declining 8-oxo-dGuo levels in the spleen at 24 hr vs. 4 hr after the last treatment (Figure 7B). This may be due to the activation of the base excision repair to eliminate DNA damage.
The two obvious limitations of the described method are: (1) the requirement for rather large quantities of DNA (about 80 μg per sample to achieve three technical replicates), and (2) potential interference from co-eluting compounds. It was found that only a very small (i.e., about 15 mg) piece of spleen (about 15% of total tissue weight) was sufficient to obtain 80 μg of DNA, leaving a sufficient amount of tissue for other (e.g., gene expression) analyses. The amount of extracted DNA is another critical parameter that affects artifactual DNA oxidation23,24,25. Therefore, working with rather large (~80 mg/sample) that we suggest here is consistent with previous recommendations of using > 30 mg to minimize artifactual DNA oxidation25. For smaller tissues (e.g., the hippocampus), one could scale down the protocol to start with 20-30 μg DNA, since this should be sufficient for 2-3 runs. Larger quantities of DNA would presumably facilitate the resolution of peaks above background noise, but this remains to be experimentally verified The presence of potentially interfering substances can be addressed on a case-by-case basis, and it is essential to always include DNA sample from simultaneous in vitro or in vivo treatments with a positive control that is known to induce oxidative DNA damage (see Figure 7).
Although the phosphate in phosphate buffers has been reported to reduce the activity of alkaline phosphatase, method optimization experiments (Figure 5) confirmed that phosphate buffer could readily be used. Since phosphate buffer is contained in the mobile phase, its use in sample preparation reduced detector noise due to solvent mixing. Thus voltage, buffer composition, DNA extraction and digestion were optimized and positive controls were included. Moreover, a summary outlining how these variables can be further optimized by individual laboratories is provided. Our goal was to produce a method that could be useful for specialist laboratories aimed at developing an assay for 8-oxo-dGuo detection and quantification.
However, we note that formal validation of the method is required, as outlined under the validations steps recommended by the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) for validation of analytical procedures (i.e., ICH Q2(R1)26). Briefly, the elements of quantitative validation should examine the assay’s: 1) accuracy (which can be accomplished by including samples with different amounts of 8-oxo-dGuo spiked-in; 2) range and linearity (linear response across extended analyte concentration; 3) precision (repeatability and reproducibility); 4) limit of detection (generally, the point at which the signal to noise ratio is greater or equal 2-3; this may be matrix-dependent); 5) limit of quantitation (concentration at which the response meets some predetermined levels such as 2 x standard deviation of control; may be matrix-dependent); 6) selectivity/specificity (ability to detect the analyte in complex samples versus neat solutions); 7) reproducibility (the ability to get the same result in different labs with different operators); and 8) robustness and system suitability. As the purpose of the validation recommended by the ICH is to demonstrate that it is “suitable for its intended purposes” and many laboratories will likely have different purposes, the prospective users of this assay would have to validate this to the extent deemed necessary. Accurate quantification of 8-oxo-dGuo is desirable in toxicology and molecular medicine since it can enhance the understanding of how oxidative stress, and more specifically DNA oxidation, is mechanistically and empirically linked to adverse health effects.
Disclosures
No conflict of interest or competing financial interests declared.
Acknowledgments
This research was funded by the Health Canada Genomics Research and Development Initiative (GRDI) and the Canadian Regulatory Strategy for Biotechnology (CRSB). The authors have no conflict of interests.
References
- Helbock HJ, Beckman KB, Shigenaga MK, Walter PB, Woodall AA, Yeo HC, Ames BN. DNA oxidation matters: The HPLC-electrochemical assay of 8-oxo-deoxyguianosine and 8-oxo-guanine. Proc. Natl. Acad. Sci. 1998;95(1):288–293. doi: 10.1073/pnas.95.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2' -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci Health C Environ. Carcinog. Ecotoxicol. Rev. 2009;27(2):120–139. doi: 10.1080/10590500902885684. [DOI] [PubMed] [Google Scholar]
- Cadet J, Bellon S, Douki T, Frelon S, Gasparutto D, Muller E, Pouget JP, Ravanat JL, Romieu A. Radiation-induced DNA damage: formation, measurement, and biochemical features. J Environ Pathol Toxicol Oncol. 2004;23(1):23–23. doi: 10.1615/jenvpathtoxoncol.v23.i1.30. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Goode EL, Ladiges WC, Ulrich CM. Polymorphic variation in hOGG1 and risk of cancer: a review of the functional and epidemiologic literature. Mol. Carcinog. 2005;42(3):127–141. doi: 10.1002/mc.20067. [DOI] [PubMed] [Google Scholar]
- Culp SJ, Cho BP, Kadlubar FF, Evans FE. Structural and Conformational Analyses of 8-hydroxy-2'-deoxyguanosine. Chem. Res. Toxicol. 1989;2(6):416–422. doi: 10.1021/tx00012a010. [DOI] [PubMed] [Google Scholar]
- Cooke MS, Loft S, Olinski R, Evans MD, Bialkowski K, Wagner JR, Dedon PC, Møller P, Greenberg MM, Cadet J. Recommendations for standardized description of and nomenclature concerning oxidatively damaged nucleobases in DNA. Chem. Res. Toxicol. 2010;23(4):705–707. doi: 10.1021/tx1000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YH, Goddard WA3rd, Noyes KT, Sowers LC, Hwang S, Chung DS. First principles calculations of the tautomers and pKa values of 8-oxoguanine: implications for mutagenicity and repair. Chem. Res. Toxicol. 2002;15(8):1023–1035. doi: 10.1021/tx010146r. [DOI] [PubMed] [Google Scholar]
- Park J-H, Gopishetty S, Szewczuk LM, Troxel AB, Harvey RG, Penning TM. Formation of 8-oxo-7,8-dihydro-2'-deoxyguanosine (8-oxo-dGuo) by PAH o-quinones: involvement of reactive oxygen species and copper(ii)/copper(i) redox cycling. Chem. Res. Toxicol. 2005;18(6):1026–1037. doi: 10.1021/tx050001a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangal D, Vudathala D, Park JH, Lee SH, Penning TM, Blair IA. Analysis of 7,8-dihydro-8-oxo-2'-deoxyguanosine in cellular DNA during oxidative stress. Chem. Res. Toxicol. 2009;22(5):788–797. doi: 10.1021/tx800343c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravanat JL, Douki T, Duez P, Gremaud E, Herbert K, Hofer T, Lasserre L, Saint-Pierre C, Favier A. Cellular background level of 8-oxo-7,8-dihydro-2'-deoxyguanosine: an isotope based method to evaluate artefactual oxidation of DNA during its extraction and subsequent work-up. Carcinogenesis. 2002;23(11):1911–1918. doi: 10.1093/carcin/23.11.1911. [DOI] [PubMed] [Google Scholar]
- Gossen JA, De Leeuw WJF, Tan CHT, Zwarthoff EC, Berends F, Lohman PHM, Knook DL, Vijg J. Efficient rescue of integrated shuttle vectors from transgenic mice: A model for studying mutations in vivo. Proc. Natl. Acad. Sci. U.S.A. 1989;86(20):7971–7975. doi: 10.1073/pnas.86.20.7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Test No. 488: Transgenic Rodent Somatic and Germ Cell Gene Mutation Assays. Organisation for Economic Co-operation and Development (OECD); 2015. [2015 Mar 1]. Available from: http://www.oecd-ilibrary.org. [Google Scholar]
- Van Campen LE, Murphy WJ, Franks JR, Mathias PI, Toraason MA. Oxidative DNA damage is associated with intense noise exposure in the rat. Hear Res. 2002;164(1-2):164–161. doi: 10.1016/s0378-5955(01)00391-4. [DOI] [PubMed] [Google Scholar]
- European Standards Committee on Oxidative DNA Damage (ESCODD) Measurement of DNA oxidation in human cells by chromatographic and enzymic methods. Free Radic. Biol. Med. 2003;34(8):1089–1099. doi: 10.1016/s0891-5849(03)00041-8. [DOI] [PubMed] [Google Scholar]
- Rebelo IA, Piedade JA, Oliveira-Brett AM. Development of an HPLC method with electrochemical detection of femtomoles of 8-oxo-7,8-dihydroguanine and 8-oxo-7,8-dihydro-2'-deoxyguanosine in the presence of uric acid. Talanta. 2004;63(2):323–331. doi: 10.1016/j.talanta.2003.10.051. [DOI] [PubMed] [Google Scholar]
- Ravanat J-L, Turesky RJ, Gremaud E, Trudel LJ, Stadler RH. Determination of 8-oxoguanine in DNA by gas chromatography-mass spectrometry and HPLC-electrochemical detection: overestimation of the background level of the oxidized base by the gas chromatography-mass spectrometry assay. Chem. Res. Toxicol. 1995;8(8):1039–1045. doi: 10.1021/tx00050a007. [DOI] [PubMed] [Google Scholar]
- Kawanishi S, Murata M. Mechanism of DNA damage induced by bromate differs from general types of oxidative stress. Toxicology. 2006;221(2-3):172–178. doi: 10.1016/j.tox.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Tahara S, Kaneko T. Susceptibility of mouse splenic cells to oxidative DNA damage by x-ray irradiation. Biol. Pharm. Bull. 2004;27(1):105–108. doi: 10.1248/bpb.27.105. [DOI] [PubMed] [Google Scholar]
- Garratt LW, Mistry V, Singh R, Sandhu JK, Sheil B, Cooke MS, Sly PD. Interpretation of urinary 8-oxo-7,8-dihydro-2'-deoxyguanosine is adversely affected by methodological inaccuracies when using a commercial ELISA. Free Radic. Biol. Med. 2012;48(11):1460–1464. doi: 10.1016/j.freeradbiomed.2010.02.017. [DOI] [PubMed] [Google Scholar]
- Cooke MS, Collins A, Olinski R, Rozalski R, Loft S. Harmonising measurements of 8-oxo-7,8-dihydro-2′-deoxyguanosine in cellular DNA and urine. Free Radic. Res. 2012;46(4):541–553. doi: 10.3109/10715762.2011.644241. [DOI] [PubMed] [Google Scholar]
- Cadet J, Douki T, Ravanat JL. Measurement of oxidatively generated base damage in cellular DNA. Mutat Res. 2011;711(1-2):3–12. doi: 10.1016/j.mrfmmm.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Mackey K, Drews R. DNAzol: a reagent for the rapid isolation of genomic DNA. Biotechniques. 1997;22(3):550–553. doi: 10.2144/97223pf01. [DOI] [PubMed] [Google Scholar]
- Collins AR, Cadet J, Möller L, Poulsen HE, Viña J. Are we sure we know how to measure 8-oxo-7,8-dihydroguanine in DNA from human cells. Arch Biochem Biophys. 2004;423(1):57–65. doi: 10.1016/j.abb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- Badouard C, Ménézo Y, Panteix G, Ravanat JL, Douki T, Cadet J. Determination of new types of DNA lesions in human sperm. Zygote. 2008;16(1):9–13. doi: 10.1017/S0967199407004340. [DOI] [PubMed] [Google Scholar]
- Cadet J, Douki T, Gasparutto D, Ravanat JL. Oxidative damage to DNA: formation, measurement and biochemical features. Mutat Res. 2003;531(1-2):1–2. doi: 10.1016/j.mrfmmm.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Validation of analytical procedures: text and methodology Q2(R1); International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH); 2015 Mar 1; San Diego, CA. 2015. Available at: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf. [Google Scholar]