

Abstract
Purpose.
To investigate the role of C/EBP homologous protein (CHOP), a proapoptotic protein, and the unfolded protein response (UPR) marker that is involved in endoplasmic reticulum (ER) stress-mediated apoptosis in mouse retinal ganglion cell (RGC) death following ischemia/reperfusion (I/R) injury.
Methods.
Retinal I/R injury was induced in adult C57BL/6J wild-type (WT) and CHOP knockout (Chop−/−) mice by raising IOP to 120 mm Hg for 60 minutes. Expression of CHOP and other UPR markers was studied by Western blot and immunohistochemistry. Retinal ganglion cell counts were performed in retinal flat mounts stained with an RGC marker. Retinal ganglion cell function was evaluated by scotopic threshold response (STR) electroretinography.
Results.
In WT mice, retinal CHOP was upregulated by 30% in I/R-injured eyes compared to uninjured eyes 3 days after injury (P < 0.05). Immunohistochemistry confirmed CHOP upregulation specifically in RGCs. CHOP knockout did not affect baseline RGC density or STR amplitude. Ischemia/reperfusion injury decreased RGC densities and STR amplitudes in both WT and Chop−/− mice. However, survival of RGCs in I/R-injured Chop−/− mouse was 48% higher (P < 0.05) than that in I/R-injured WT mouse 3 days after I/R injury. Similarly, RGC density was significantly higher in Chop−/− eyes at 7, 14, and 28 days after I/R injury. Scotopic threshold response amplitudes of Chop−/− mice were significantly higher at 3 and 7 days after I/R than those of WT mice.
Conclusions.
Absence of CHOP partially protects against RGC loss and reduction in retinal function after I/R injury, indicating that CHOP and, thus, ER stress play an important role in RGC apoptosis in retinal I/R injury.
Keywords: apoptosis, CHOP, retinal ganglion cell, retinal ischemia
Ischemia/reperfusion injury induces CHOP expression and leads to RGC death via apoptosis. Knockout of CHOP significantly delays RGC death and partially protects against loss of RGC numbers and function in ischemic retina.
Introduction
Retinal ischemia occurs in many ocular abnormalities, such as anterior ischemic optic neuropathy, retinal and choroidal vessel occlusion diseases, glaucoma, diabetic retinopathy, and retinopathy of prematurity.1 Retinal ischemia causes loss of retinal neurons, including retinal ganglion cells (RGCs), functional degeneration of the retina, and eventual loss of vision.2 Retinal ganglion cells are the final common neurons that collate vision signals in the retina and transmit them to brain visual centers through the optic nerve.3 Retinal ganglion cell death after ischemic injury occurs primarily by apoptosis,4–6 although it may also involve other cell death pathways, such as necrosis, necroptosis, or autophagy.7–9 Loss of RGCs is irreversible; once degenerated, they cannot be replenished.10 Thus, protection against permanent damage to RGCs is of particular importance for vision preservation. Their degenerative mechanisms and protective strategies have been widely studied.11–12
Reduction of oxygen and nutrients from blood supply during ischemia causes tissue hypoxia and hypoglycemia. Hypoxia has been shown to cause aberrant protein folding in neurons, leading to the accumulation of misfolded proteins in the endoplasmic reticulum (ER), which in turn results in ER stress. Endoplasmic reticulum stress triggers a series of adaptive mechanisms, collectively known as the unfolded protein response (UPR), to attempt to restore homeostasis and normal ER function. Unfolded protein response causes dissociation of the molecular chaperone binding immunoglobulin protein (BiP) from the ER stress sensors protein kinase RNA-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1), thereby activating the sensors. Activation of ER stress sensors subsequently leads to induction of the CCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP) gene by activating ATF4, ATF6, and X-box binding protein-1 (XBP1).13,14
CHOP, also known as growth arrest and DNA damage-inducible 153 (GADD153) or DNA damage-inducible transcript 3 (Ddit3) gene, is a proapoptotic molecule and a member of the C/EBP family of transcription factors. CHOP is present in the cytosol under nonstressed conditions, and stress leads to its induction and accumulation in the nucleus. It is ubiquitously expressed at very low levels. However, its expression is several-fold increased under conditions of stress in a wide variety of cells. CHOP plays a pivotal role in ER stress-mediated apoptosis. Induction of CHOP is a signaling event underlying ER stress–induced cell apoptosis. CHOP mediates apoptosis by (1) down-regulation of antiapoptotic proteins such as BCL-2 and BCL-XL; (2) translocation of BAX, a proapoptotic molecule, from the cytosol to the mitochondria15–17; and (3) activation of GADD34 and ER oxidoreductin-1α (ERO-1α). The role of CHOP in the death of ER stressed cells is correlated with its role in oxidative stress inside the ER.18
Because CHOP is a crucial mediator of ER stress–induced apoptosis and is regulated by all three branches of the ER stress pathway, it has been targeted for modulation of ER stress. Previous studies have shown that CHOP knockout (Chop−/−) mice exhibit reduced apoptosis in response to ER stress.19–22 Moreover, deletion of CHOP has been shown to block macrophage apoptosis.23
Involvement of CHOP-mediated apoptosis has been demonstrated in various diseases, including neurodegenerative abnormalities,24–25 cerebral ischemia,26 diabetes,27 and cardiovascular diseases.28 Endoplasmic reticulum stress has been implicated in retinal apoptosis and RGC death.18–19 Robust induction of CHOP has been observed in RGCs after optic nerve axotomy.29 Increased expression of CHOP and other UPR markers has been demonstrated in the nuclei of retinal vascular cells after ischemic injury in mice.30 Most recently, Li et al.31 reported that CHOP expression was upregulated in the rat retina after ischemia/reperfusion (I/R) injury.31
Therefore, the present study was designed to confirm the upregulation of CHOP in RGC apoptosis in ischemic mouse retinas, and in addition, to evaluate whether knockout of CHOP is protective against this insult. We raised the IOP above the systolic blood pressure to induce retinal ischemia. After 60 minutes of ischemia, blood flow was restored. Reperfusion following ischemia induces oxidative stress, leading to inflammation and oxidative damage. This retinal I/R injury model simulates certain mechanisms of RGC degeneration characteristic of retinal ischemic conditions and is widely used to investigate the pathogenesis in RGC death and to examine possible therapeutic interventions for neuroprotection.
In addition to studying the expression of CHOP following retinal I/R injury, we also evaluated the extent of RGC survival in I/R-injured Chop−/− mouse retina. We demonstrated that absence of CHOP partially protects against loss of RGCs and reduction in retinal function after I/R injury. These results indicate that CHOP and, thus, ER stress play an important role in RGC apoptosis in retinal I/R injury.
Materials and Methods
Animals
Adult wild-type (WT) C57BL/6J mice and Chop−/− (B6.129S[Cg]-Ddit3tm2.1Dron/J; stock no. 005530) breeders were obtained from Jackson Laboratory (Bar Harbor, ME, USA). The Chop−/− mice were backcrossed with the C57BL/6J strain for at least 10 generations, making the C57BL/6J strain an appropriate WT control. Chop−/− mice were then bred and subsequently aged at the University of North Texas Health Science Center (UNTHSC, Fort Worth, TX, USA). Animals were housed in conventional cages with 12-hour light/12-hour dark cycle and had ad libitum access to food and water. All mice used for experiments were 9 to 11 weeks old. All experiments were conducted in compliance with the Association for Research in Vision and Ophthalmology statement of the Use of Animals in Ophthalmic and Vision research and were approved by the UNTHSC Institutional Animal Care and Use Committee.
Genotyping
To determine the genotype of mice, standard PCR analysis was performed using primer 1: 5′-ATG CCC TTA CCT ATC GTG-3′ (common); primer 2: 5′-AAC GCC AGG GTT TTC CCA GTC A-3′ (mutant reverse); and primer 3: 5′-GCA GGG TCA AGA GTA GTG-3′ (WT reverse). All three primers were used in the same reaction mixture. The common primer (forward primer upstream of exons 3 and 4) annealed to both the WT allele and the mutant allele. The WT reverse primer was located in the DNA that was replaced in the knockout (i.e., exons 3 and 4) and annealed only to the WT allele. The mutant reverse primer was located in the lacZ construct and annealed to and amplified only the mutant allele. Wild-type genotype was represented by a single band at 544 base pairs (bp), and the mutant (i.e., homozygote Chop−/−) was represented by a single band at 320 bp. Heterozygote was represented by two bands, one at 320 bp and the other at 544 bp.
Ischemic/Reperfusion Injury
Mice were anesthetized with intraperitoneal injections of a solution of ketamine-xylazine-acepromazine (100-10-3 mg/kg, respectively) and placed on a heating pad to prevent hypothermia. Phenylephrine hydrochloride ophthalmic solution (2.5%; Akorn, Inc., Lake Forest, IL, USA) was topically administered to the left eye to dilate the pupil. One drop of 0.5% proparacaine hydrochloride ophthalmic solution (Akorn) was used as a topical local anesthetic. After pupil dilation, the anterior chamber of the left eye was cannulated with a 30-gauge needle (Becton-Dickenson, Franklin Lakes, NJ, USA) connected to a reservoir of phosphate-buffered saline (PBS). By raising the height of the reservoir, IOP was increased to 120 mm Hg for 60 minutes to induce retinal ischemia, which was confirmed by the observed blanching of the retina by using an ophthalmoscope. The cannula was withdrawn after 60 minutes to allow reperfusion. Tobramycin (Tobrex; Alcon, Fort Worth, TX, USA) was then topically administered to prevent ocular infection. Uninjured right eyes served as controls.
Immunoblotting
Immunoblotting was performed to confirm upregulation of CHOP and the UPR markers phosphorylated eukaryotic initiation factor 2α (p-eIF2α) and BiP after I/R injury in WT mice. Retinas were collected from control and I/R-injured eyes at 0, 1, 6, 12, and 24 hours and then at 3, 7, and 14 days after injury (n = 5 retinas per time point). Retinas were then homogenized, and total protein was extracted in mammalian protein extraction reagent buffer containing Halt protease inhibitor cocktail (Thermo Scientific, Rockford, IL, USA). Protein concentrations were determined using the Dc protein assay system (Bio-Rad Laboratories, Richmond, CA, USA) according to the manufacturer's instructions. A total of 20 μg protein per sample was separated using SDS-PAGE and electroblotted onto polyvinylidene difluoride membranes. After blocking, blots were incubated overnight at 4°C with primary antibodies. Primary rabbit antibodies for CHOP (1:1000 dilution, catalog no. NBP2-13172; Novus Biologicals, Littleton, CO, USA); p-eIF2α (1:1000 dilution, catalog no. SAB430022; Sigma-Aldrich Corp., St. Louis, MO, USA); BiP (1:1000 dilution, catalog no. SC-13968; Santa Cruz Biotechnology, Dallas, TX, USA); BCL-2 (1:1000 dilution, catalog no. 2876; Cell Signaling Technology, Beverly, MA, USA); BAX (1:1000 dilution, catalog no. 2772; Cell Signaling Technology); BCL-XL (1:500 dilution, catalog no. 2764; Cell Signaling Technology); cleaved caspase-3 (1:1000 dilution, catalog no. 9664l Cell Signaling Technology); GAPDH (1:1000 dilution, catalog no. 2118S; Cell Signaling Technology), and mouse antibody for β-actin (1:5000 dilution, catalog no. A5316; Sigma-Aldrich Corp.) were used. After primary antibody incubation, blots were washed with PBS and incubated with the respective secondary antibodies for 1 hour at room temperature. Secondary antibodies used were horseradish peroxidase-conjugated goat anti-rabbit (1:10,000 dilution, catalog no. 32460; Thermo Scientific); goat anti-mouse (1:10,000 dilution, catalog no. 32430; Thermo Scientific); and IRDye 800CW-conjugated goat anti-rabbit (1:10,000 dilution, catalog no. 926-32213; LI-COR Biotechnology, Lincoln, NE, USA). Protein bands were detected using Super Signal West Femto maximum sensitivity substrate (Thermo Scientific) ECL detection kit. β-actin or GAPDH was used as the loading control. Visualization and quantification of protein bands was performed using Fluor Chem 8900 imager and software (Alpha Innotech, San Leandro, CA, USA) or Odyssey infrared imaging system and its software (LI-COR Biotechnology).
Immunohistochemistry
To validate upregulation of CHOP in RGCs, colocalization of CHOP with an RGC marker, RNA-binding protein with multiple splicing (RBPMS),32,33 was evaluated. Eyes were enucleated 3 days after I/R injury, fixed in 4% paraformaldehyde for 2 hours at room temperature, submerged in 20% sucrose overnight at 4°C, and then embedded in optimum cutting temperature compound (Tissue-Tek OCT compound; VWR, Radnor, PA, USA) and frozen at −80°C. Transverse 10-μm-thick sections were cut using a cryostat (Leica Biosystems, Buffalo Grove, IL, USA) and placed onto glass slides. Retinal cryosections were hydrated in PBS for 15 minutes and then blocked in PBS containing 10% goat serum and 0.3% Triton X-100 (Fisher Scientific, Richardson, TX, USA) for 1 hour at room temperature. The sections were double-immunolabeled with mouse anti-CHOP antibody (1:50 dilution, catalog no. 2895S; Cell Signaling Technology) and rabbit anti-RBPMS antibody (1:100 dilution, catalog no. GTX118619; GeneTex, Irvine, CA, USA) and incubated overnight at 4°C. Sections were then washed 3 times for 5 minutes each with PBS and incubated for 1 hour with the respective secondary antibodies at room temperature. Alexa Fluor 488 goat-anti-rabbit (1:1000 dilution, catalog no. A11008; Life Technologies, Grand Island, NY, USA) and tetramethylrhodamine goat-anti-mouse immunoglobulin G (IgG; 1:1000 dilution, catalog no. T2762; Invitrogen, Grand Island, NY, USA) secondary antibodies were used. Sections were then washed 3 times for 5 minutes each with PBS and mounted with Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA). Images were acquired using an Eclipse Ti inverted microscope (Nikon Instruments, Inc., Melville, NY, USA) and the CRi Nuance FX multispectral imaging system with Nuance version 3.0 software (Caliper Life Sciences, Hopkinton, MA, USA).
RNA Isolation and qRT-PCR
Retinas were dissected from control and I/R-injured eyes at 3 and 7 days after injury (n = 4–5 per time point). Each retina was collected in 1 mL of Iso-RNA lysis reagent (5Prime, Gaithersburg, MD, USA) and then homogenized using 5-mm steel beads in the TissueLyser LT (Qiagen, Valencia, CA, USA). For RNA isolation, 50 μL of BAN phase separation reagent (Molecular Research Center, Cincinnati, OH, USA) was added to each sample for phase separation and was followed by addition of 0.5 mL isopropanol for RNA precipitation. Reverse transcription was performed using iScript reverse transcription Supermix (Bio-Rad Laboratories) at 25°C for 5 minutes, followed by 42°C for 30 minutes, and then 85°C for 5 minutes. The amount of total RNA used as the template was 500 ng. Complementary DNA products from reverse transcription reactions were quantified using a CFX96 real-time PCR system (Bio-Rad). PCR primers are listed in the Table. The qPCR procedure was initial denaturation at 95°C for 3 minutes, 40 cycles of 95°C for 10 seconds, and 65°C for 30 seconds. Amplicon quantitation was determined by real-time detection of end-point SYBR Green (Bio-Rad Laboratories) fluorescence. Each assay was performed in duplicate. The relevant quantification was calculated by the cycle threshold (ΔΔ Ct) method. Gapdh was used as the endogenous control.
Table.
Primer Sequences for qRT-PCR Analysis
|
Gene Name |
Forward |
Reverse |
| Bcl-2 | 5′-GTCCCGCCTCTTCACCTTTCAG-3′ | 5′-GATTCTGGTGTTTCCCCGTTGG-3′ |
| Bcl-xL | 5′-ATGACTGTGGCTGGTGTGGTTCTG-3′ | 5′-GCTGAAGAGAGAGTTGTGGTGGGG-3′ |
| Bax | 5′-GCGTGGTTGCCCTCTTCTACTTTG-3′ | 5′-AGTCCAGTGTCCAGCCCATGATG-3′ |
| Caspase-3 | 5′-GGGAAACCAACAGTAGTCAGTCCT-3′ | 5′-GCGAGTGAGAATGTGCATAAATTCCPP-3′ |
| Gapdh | 5′-AGAACATCATCCCTGCATCC-3′ | 5′-AGCCGTATTCATTGTCATACC-3′ |
Retinal Flat Mounts
To quantify RGC survival in Chop−/− and WT mice post I/R injury, retina flat mounts were stained with the RGC marker RBPMS, and the number of RBPMS-positive cells were counted. Five mice from each study group were used per time point. Eyes were enucleated and fixed in 4% paraformaldehyde for 2 hours at room temperature. Retinas were then dissected, washed with PBS and then with PBS containing 0.3% Triton X-100 (Fisher Scientific) for 1 hour. Retinas were then incubated with blocking solution (PBS containing 10% goat serum and 0.3% Triton X-100) for 1 hour at room temperature, followed by RBPMS primary rabbit antibody (1:200 dilution, catalog no. GTX 118619; GeneTex) overnight at 4°C. Subsequently, retinas were washed with PBS for 2 hours and then incubated with Alexa Fluor 488 goat-anti-rabbit secondary antibody (1:1000 dilution, catalog no. A11008; Life Technologies) for 1 hour at room temperature. Retinas were washed with PBS for 2 hours and mounted with the RGC layer side up by using Vectashield mounting medium with DAPI. Digital images from the peripheral and midperipheral regions of each of the 4 quadrants (superior, inferior, nasal, and temporal) were obtained at magnifications of 20× and 40×, using an Eclipse Ti inverted microscope and CRi Nuance FX multispectral imaging system. Eight images were taken per retina. The numbers of RBPMS-positive cells were counted in each image. The mean RGC count of the eight images is reported for each retina.
Scotopic Threshold Response (STR) Electroretinography (ERG)
To evaluate RGC function in mice, ERG was performed. Scotopic threshold responses originated from the innermost retina, where RGC bodies are located. Scotopic threshold responses were recorded, and the positive components of STR amplitudes (pSTRs) were analyzed using the handheld multispecies ElectroRetinoGraph instrument (model 2000, HMsERG unit; Ocuscience, Kansas City, MO, USA). Five mice from each time point were used. Mice were dark-adapted overnight before the ERG recordings, and the experiment was performed in dim red light. Mice were anesthetized by continuous flow of 5% isoflurane gas in oxygen into an enclosed induction chamber. Throughout the ERG procedure, mice were placed on a heated plate on a sliding platform, where they were kept sedated by continuous flow of 1.5% isoflurane through a nose cone. Pupils were dilated with 2.5% phenylephrine hydrochloride ophthalmic solution. Silver-embedded thread eye electrodes (Ocuscience) were placed on the cornea, and corneal contact lenses were placed in contact with the electrodes. One drop of 2.5% hypromellose ophthalmic demulcent solution (Akorn) was applied between the eye and the electrode to maximize conductivity of the generated response and maintain corneal hydration. Reference electrodes were inserted subdermally near the base of the eye, and the ground electrode was inserted in the tail. Scotopic threshold responses were recorded by stimulating the retina with a light intensity of 0.03 mcd/s/m2. Responses to ten flashes were averaged. The interval between flashes was 60 seconds. Electroretinography recordings were analyzed using HMsERG software. Positive STR amplitudes were measured from the baseline to the peak of positive deflection.
Statistical Analysis
Statistical analysis for all experiments was performed using Prism version 5.0 software (GraphPad, Inc., La Jolla, CA, USA). Data from two experimental groups were analyzed using unpaired Student t-tests. To analyze three or more experimental groups, one-way analysis of variance (ANOVA) was used, followed by a Tukey post hoc test. Differences were considered statistically significant at a P value of <0.05. Values are expressed as means ± SEM.
Results
Upregulation of CHOP, p-elF2α, and BiP Expression in Mouse Retina After I/R Injury
In order to confirm upregulation of CHOP and selected UPR markers after I/R injury in WT mice, immunoblotting was performed. Retinas were collected from control and I/R-injured eyes at 0, 1, 6, 12, and 24 hours and then at 3, 7, and 14 days after I/R injury, and the mean ± SEM expression level of CHOP was measured at each time point. CHOP was 30% upregulated in I/R-injured eyes (1.30 ± 0.04 arbitrary units [AU]; n = 5) compared to uninjured (control) eyes (1.00 ± 0.07 AU) at 3 days after I/R injury (P < 0.05) (Figs. 1A, 1B). Levels of p-eIF2α and BiP protein also were 22% increased (I/R: 1.35 ± 0.03 AU; uninjured: 1.10 ± 0.04 AU) (Figs. 1C, 1D) and 11% (I/R: 1.31 ± 0.02 AU; uninjured: 1.18 ± 0.03 AU), respectively (both, P < 0.05) (Figs. 1E, 1F). These results indicate that I/R injury triggers UPR, leading to an increase in the expression of UPR-related proteins such as CHOP, p-eIF2α, and BiP in the retina. Retinal ganglion cells accounted for only approximately 2% of total retinal cells, which likely explains the significant although not drastic increases of these proteins in the whole retina.
Figure 1.
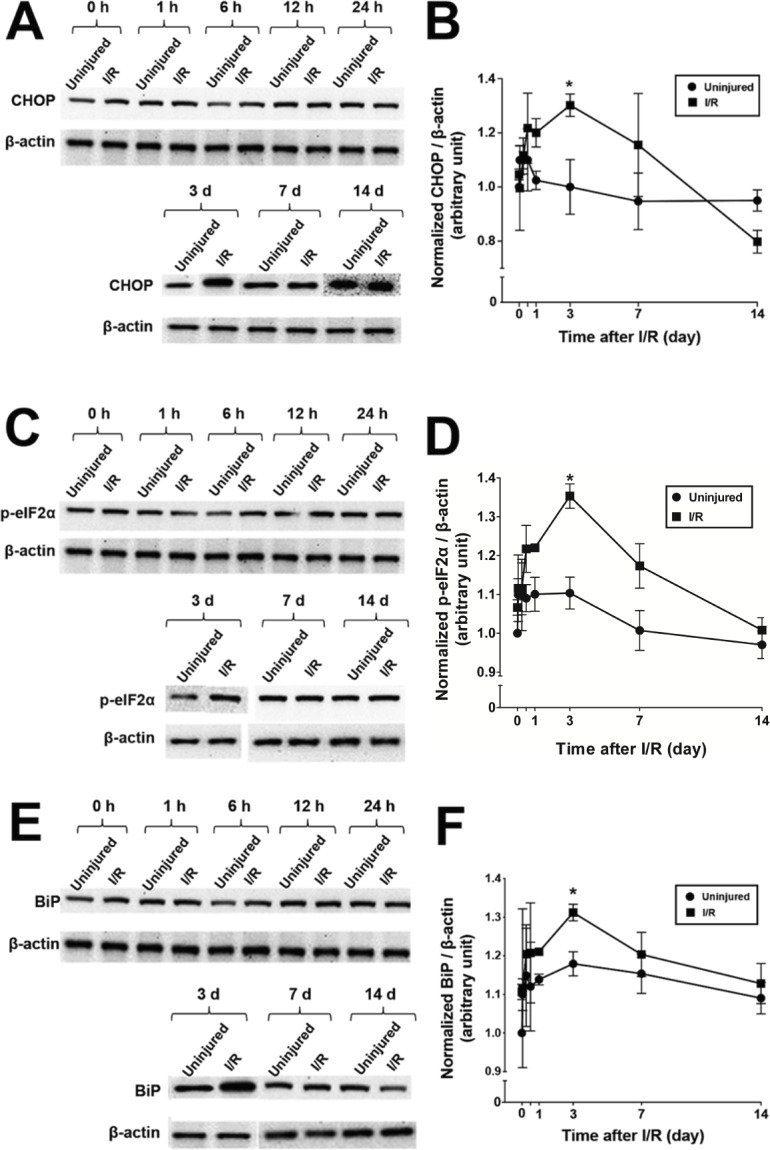
Increase in retinal CHOP, p-eIF2α, and BiP after I/R injury in WT mice. (A, C, E) Representative Western blots of CHOP, p-eIF2α, and BiP at different time points after I/R injury. β-actin was used as a loading control. (B, D, F) Quantitation of CHOP, p-eIF2α, and BiP at different time points after I/R injury. *P < 0.05 versus control according to one-way ANOVA followed by a Tukey test. Data are means ± SEM (n = 5 individual retinas).
Changes in BCL-2, BCL-XL, BAX, and Caspase-3 Levels After I/R Injury
After I/R injury, expression of molecules known to be involved in the CHOP-mediated apoptotic pathway, such as BCL-2, BCL-XL, BAX, and caspase-3, were evaluated by qRT-PCR and immunoblotting. In the retina, mRNA expression of Bcl-2 was reduced by 36 % (uninjured: 1.00 ± 0.05 AU; I/R: 0.64 ± 0.09 AU; n = 5; P < 0.05) at 3 days after I/R injury and by 28% (uninjured: 1.00 ± 0.07 AU; I/R: 0.72 ± 0.13 AU; n = 4) at 7 days after I/R injury (Fig. 2A). The corresponding protein, BCL-2, was downregulated by 51 % (uninjured: 1.00 ± 0.19 AU; I/R: 0.49 ± 0.10 AU; n = 6; P < 0.05) at 3 days after I/R injury and by 26% (uninjured: 1.00 ± 0.08 AU; I/R: 0.74 ± 0.05 AU; n = 7; P < 0.05) at 7 days after I/R injury (Figs. 2B, 2C). BCL-2 is known to be present mainly in Müller cells in adult rodent retina.34,35 The mRNA and protein levels of another anti-apoptotic protein, BCL-XL, which is located in adult RGC,36 was not significantly affected by I/R injury under the study conditions tested (Figs. 2D–F). Moreover, the insult induced a 29% upregulation of Bax mRNA (uninjured: 1.00 ± 0.03 AU; I/R: 1.29 ± 0.07 AU; n = 5; P < 0.01) at 3 days after I/R injury and a 19 % upregulation (uninjured: 1.00 ± 0.08 AU; I/R: 1.19 ± 0.13 AU; n = 4) at 7 days after I/R injury (Fig. 2G). The corresponding BAX protein level was increased by 149% (uninjured: 1.0 ± 0.3 AU; I/R: 2.5 ± 0.6 AU; n = 4; P < 0.05) at 3 days after I/R injury and by 301% (uninjured: 1.0 ± 0.2 AU; I/R: 4.0 ± 1.6 AU; n = 4; P < 0.05) at 7 days after I/R injury (Figs. 2H, 2I). Caspase-3, one of the effector caspases in the apoptotic cell death pathways, was also found to be activated after I/R injury. Cleaved (active form) caspase-3 expression was increased by 194% (uninjured: 1.0 ± 0.3 AU; I/R: 2.9 ± 0.8 AU; n = 6; P < 0.05) at 3 days after I/R injury (Figs. 2K, 2L). Its mRNA expression was also upregulated by 177% (uninjured: 1.0 ± 0.8 AU; I/R: 2.77 ± 0.90 AU; n = 5). These results indicate that I/R-induced CHOP upregulation correlated with the reduced expression of BCL-2, the upregulation of BAX, and the activation of caspase-3 in the retina.
Figure 2.

Changes in retinal expression of BCL-2, BCL-XL, BAX, and caspase-3 after I/R injury in WT mice. (A) Quantitative RT-PCR of Bcl-2 (n = 4–5 individual retinas) at 3 and 7 days after injury. (B) Representative Western blots and (C) quantitative analysis of BCL-2 protein at the same time points (n = 6–7). (D) Quantitative RT-PCR of Bcl-xl (n = 4–5) at 3 and 7 days after injury. (E) Representative Western blots and (F) quantitative analysis of BCL-XL protein at the same time points (n = 4). (G) qRT-PCR of Bax (n = 4–5) at 3 and 7 days after injury. (H) Representative Western blots and (I) quantitative analysis of BAX protein at the same time points (n = 4). (J) Quantitative RT-PCR of Casp3 (n = 5) at 3 days after injury. (K) Representative Western blots and (L) quantitative analysis of cleaved caspase-3 protein at the same time point (n = 6). Gapdh and GAPDH were used to normalize mRNA and protein levels, respectively. Data are means ± SEM. *P < 0.05; **P < 0.01 according to unpaired Student t-test.
Upregulation of CHOP in RGCs After I/R Injury in WT Mice
Immunohistochemical analysis of retinal sections was performed to determine the location of CHOP expression after retinal I/R. In the retina of uninjured eyes, CHOP immunoreactivity was detected in a small number of retinal neurons in the ganglion cell layer (GCL), the inner nuclear layer (INL), and the outer nuclear layer (ONL). The level of CHOP appeared to increase 3 days after I/R injury, primarily in the GCL (Fig. 3). Colocalization of CHOP with the RGC marker RBPMS confirmed that the I/R injury–induced upregulation of CHOP was primarily in RGCs (Fig. 3B).
Figure 3.
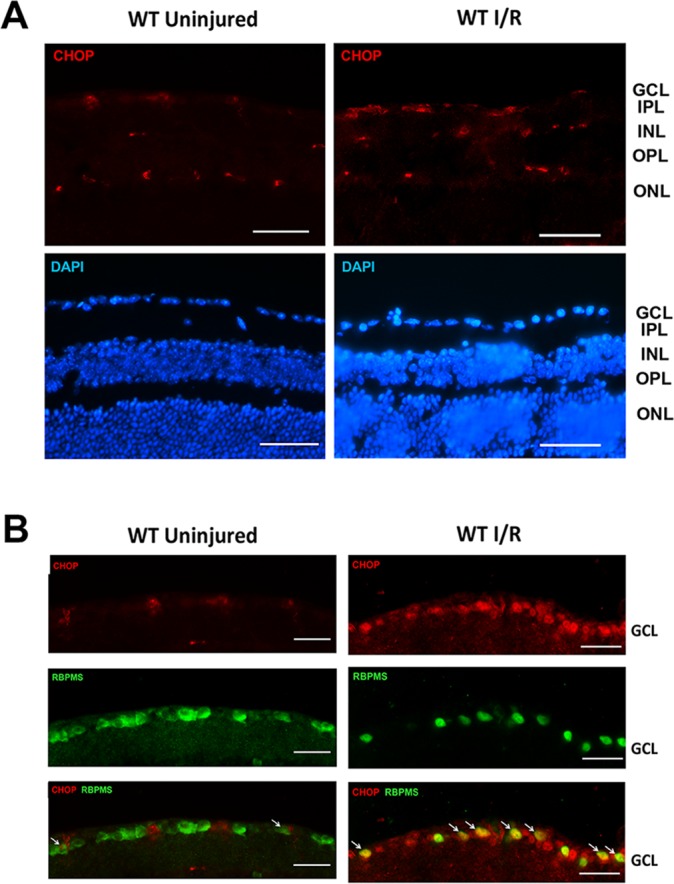
Ischemia/reperfusion injury–induced CHOP upregulation in RGCs. (A) Representative images show CHOP and DAPI staining in WT uninjured and I/R-injured retinas. IPL, inner plexiform layer; OPL, outer plexiform layer. CHOP expression was detected mainly in the GCL, INL, and ONL. In the I/R-injured eye, the CHOP expression appeared to be increased. Scale bar: 100 μm. (B) Representative images of the GCL showing CHOP and RBPMS and DAPI staining in WT uninjured and I/R-injured eyes. Scale bar: 50 μm. Retinal I/R injury increased CHOP expression (red), which was colocalized with RBPMS (a marker of RGC, green) immunoreactivity. Retinal cross-sections were obtained from mouse retinas 3 days after I/R (n = 4).
Partial Protection by CHOP Knockout Against I/R-Injury Induced RGC Loss
Retinal I/R injury has been demonstrated previously to cause RGC apoptosis.37 To evaluate the role that CHOP plays in I/R-induced RGC death, we quantified and compared the survival of RGCs between Chop−/− and WT mice after I/R injury. Retinal whole-mount staining with RBPMS was performed, and the number of RBPMS-positive cells were counted at 3, 7, 14, and 28 days after I/R injury. There were no significant differences observed between RGC counts in the uninjured WT (4394 ± 69 RGC/mm2) and those in Chop−/− (4405 ± 92 RGC/mm2) retinas, indicating that CHOP deficiency did not interfere with the normal development of RGCs (Fig. 4). Similar to our previous findings,37 I/R injury induced a progressive decline in the number of RGCs beginning 3 days after I/R injury through 28 days post I/R injury. Importantly, CHOP knockout partially protected against the I/R injury–induced loss of RGCs; a 48% increase in RGC density was observed in Chop−/− I/R-injured eyes (3337 ± 316 RGC/mm2) compared to WT I/R-injured eyes (2249 ± 226 RGC/mm2; P < 0.05) 3 days after injury. Similarly, the numbers of RGCs were significantly increased by 81%, 99%, and 73% in Chop−/− eyes at 7 days (WT I/R: 1552 ± 233 RGC/mm2; Chop−/− I/R: 2813 ± 393 RGC/mm2; P < 0.05), 14 days (WT I/R: 1507 ± 220 RGC/mm2; Chop−/− I/R: 3000 ± 300 RGC/mm2; P < 0.05), and 28 days (WT I/R: 1848 ± 191 RGC/mm2; Chop−/− I/R: 3206 ± 235 RGC/mm2; P < 0.05), respectively, after I/R injury (Fig. 4). Although CHOP-deficient mice showed increased RGC count, the numbers of RGCs in Chop−/− I/R eyes were significantly less than that in Chop−/− uninjured eyes at all time points. This suggests that knockout of CHOP significantly and only partially protected against I/R injuyr-induced RGC loss.
Figure 4.
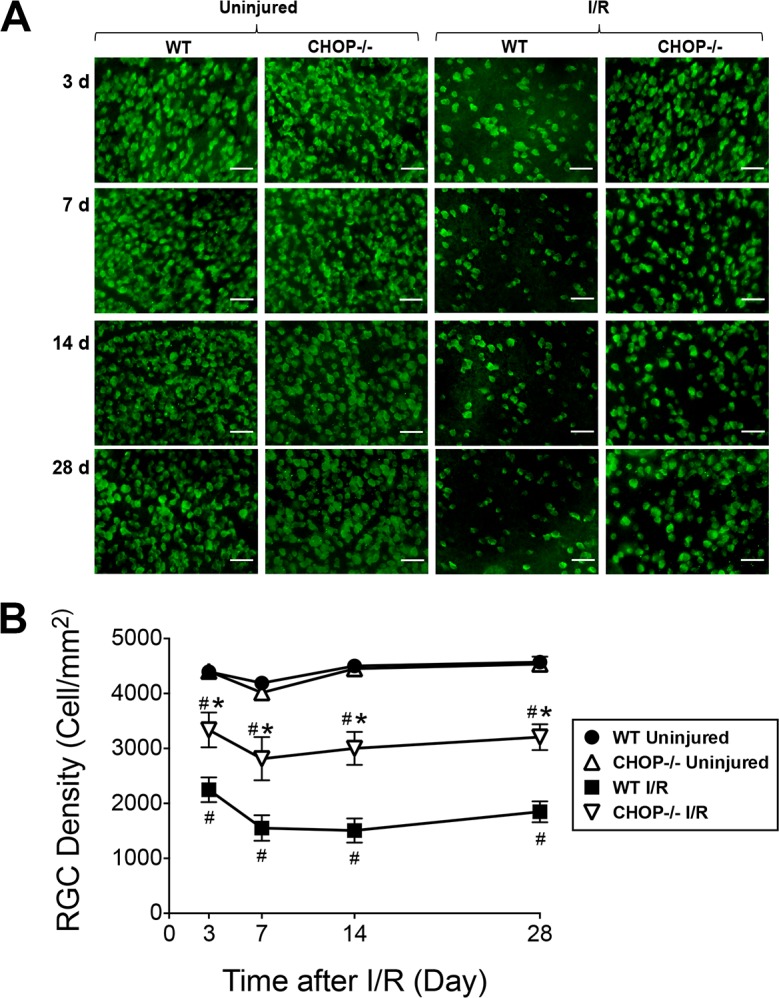
Retinal I/R-induced RGC loss was reduced in CHOP knockout mice. (A) Representative retinal flat mounts show RBPMS-positive cells in uninjured retinas or retinas at 3, 7, 14, and 28 days after I/R injury in WT and Chop−/− mice. (B) Quantitation of RGC counts. There were no significant differences between RGC counts in the WT and those in uninjured Chop−/− retinas. However, the number of surviving RGCs was higher in the Chop−/− than in the WT retinas after I/R injury at 3, 7, 14, and 28 days after I/R injury according to one-way ANOVA followed by a Tukey post hoc test. Data are means ± SEM (n = 5). Scale bar: 100 μm. *P < 0.05 between Chop−/− I/R and WT I/R; #P < 0.05 between I/R and uninjured groups.
Partial Protection by CHOP Knockout Against I/R-Induced Loss of RGC Function
To quantify and compare retinal function in Chop−/− with that in WT eyes, ERG was performed, and STRs were measured at a light intensity of 0.03 mcd/s/m2, and the amplitude of the pSTR was analyzed. Although no differences between pSTRs of WT and those of Chop−/− uninjured retinas were observed, a reduction in pSTRs was noted beginning 3 days after I/R injury in both WT and Chop−/− I/R eyes. Compared to pSTRs in WT I/R retinas, those in Chop−/− I/R retinas were 95% higher at 3 days (WT I/R: 8.8 ± 0.4 μV; Chop−/− I/R: 17.2 ± 0.6 μV; P < 0.05), 56% higher at 7 days (WT I/R: 8.2 ± 0.5 μV; Chop−/− I/R: 12.8 ± 0.6 μV; P < 0.05), 45% higher at 14 days (WT I/R: 10.0 ± 1.1 μV; Chop−/− I/R: 14.6 ± 1.5 μV; P < 0.05), and 47% higher at 28 days (WT I/R: 10.3 ± 1.1 μV; Chop−/− I/R: 15.1 ± 1.7 μV; P > 0.05) after I/R injury (Fig. 5). Therefore, Chop−/− mice had better retinal function, as measured by ERG, after I/R injury than WT mice, indicating that a higher number of functional RGCs were preserved. However, pSTRs from uninjured eyes of both WT and Chop−/− mice were significantly higher than that from Chop−/− I/R eyes, suggesting that deficiency in CHOP significantly and only partially protected against loss of RGC function.
Figure 5.
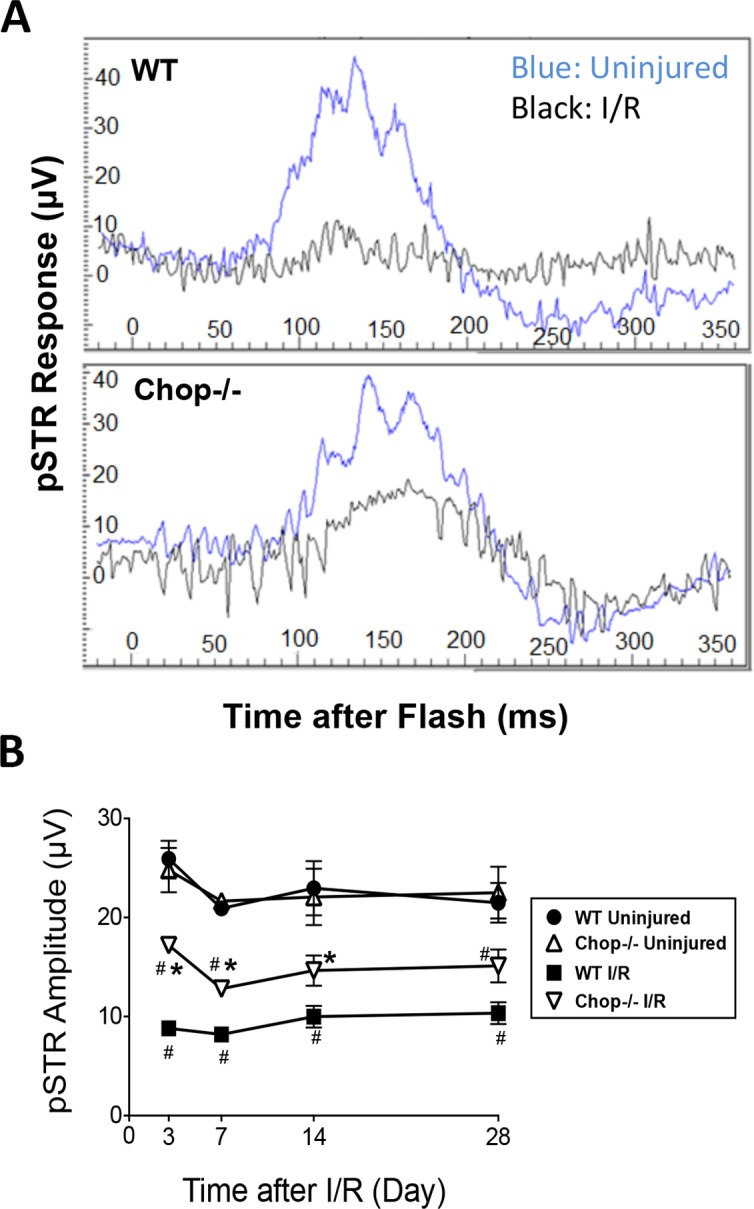
Retinal I/R-induced loss of RGC function was reduced in CHOP knockout mice. (A) Representative images of STR waveforms 3 days after I/R injury. (B) Graph shows pSTR amplitudes (μV) in uninjured and I/R-injured Chop−/− and WT eyes at 3, 7, 14, and 28 days after I/R. Data are means ± SEM (n = 5) analyzed using one-way ANOVA followed by a Tukey post hoc test. *P < 0.05 between Chop−/− I/R and WT I/R; #P < 0.05 between I/R and uninjured groups.
Discussion
Retinal I/R injury results in neuronal degeneration leading to irreversible loss of RGCs and subsequent visual impairment.37–41 Loss of RGCs after retinal ischemia is mainly due to apoptosis,4–6 and CHOP-induced apoptosis has been implicated in RGC death induced by various injuries.19,29,42 Most recently, retinal I/R injury was shown to upregulate CHOP in the rat retina.31 The current study extended these findings and confirmed that CHOP and thus ER stress are involved in RGC apoptosis induced by retinal I/R injury in the mouse. Protein expression of CHOP peaked at 3 days post I/R injury in WT mice. In addition to CHOP, other markers of UPR such as p-eIF2α and BiP were also upregulated at 3 days after retinal I/R injury, suggesting that I/R injury triggers the UPR pathway. Double-immunolabeling of retinal cryosections with CHOP and RBPMS confirmed the upregulation of CHOP in RGCs by I/R injury.
We next examined the molecular mechanisms that are involved in I/R injury and CHOP-mediated cell death. To that end, we tested the expression of antiapoptotic molecules, BCL-2 and BCL-XL, and proapoptotic proteins, BAX and cleaved caspase-3, in the ischemic retina. Previous research findings have indicated that CHOP eliminates the antiapoptotic effect of BCL-2 by blocking its expression.15 Consistent with those findings, our results confirmed that ischemic injury induced significant downregulation of BCL-2 protein in the WT retina. The expression of BCL-2 decreased drastically at 3 days (51%) and 7 days (26%) following I/R injury. Furthermore, it is also known that CHOP mediates the translocation of BAX from the cytosol to the mitochondria to induce apoptosis.16 Along those lines, our current study also demonstrated a significant upregulation of BAX at 3 days (149%) and 7 days (301%) following ischemic injury. These findings suggest that CHOP mediates apoptotic death of retinal cells by blocking BCL-2 expression and increasing BAX expression.
To further confirm that cell death after I/R injury occurs via apoptosis, we examined the protein levels of caspase-3, an effector caspase, in the ischemic retina. We found a 210% increase in cleaved caspase-3 expression 3 days after I/R, suggesting that I/R injury–induced RGC death occurs by apoptosis. These results are consistent with the findings of a previous study in a rodent model of anterior ischemic optic neuropathy model, in which cleaved caspase-3 activation was found, confirming that the apoptotic death of RGCs occurs by caspase activation.43
Because our findings confirmed the involvement of CHOP in RGC apoptosis induced by I/R injury, we further examined the survival of RGCs by using Chop−/− mice. Our retinal flat mount results demonstrated a significant decline in the number of RGCs beginning 3 days and up to 28 days post I/R in WT mice. These results are consistent with our prior study that reported a 30% decrease in RGC numbers at 28 days after I/R injury in C57BL/6J mice.37 Most importantly, compared to the C57BL/6J I/R-injured retina, RGC counts were significantly higher in the ischemic retina of Chop−/− mice at 3 through 28 days post I/R. This suggested that deletion of CHOP prevents loss of RGCs induced by I/R injury. However, the numbers of RGCs were significantly less in the Chop−/− I/R retina than in the contralateral uninjured retina, indicating that deficiency in CHOP renders only partial and not complete protection to RGCs in the ischemic retina. This finding agrees with that in previous studies in which CHOP deficiency increased the survival of axotomized RGCs by 52%.29 Knockout of CHOP was significantly protective against N-methyl-D-aspartate–induced loss of RGCs compared to that in C57BL/6 mice.42
Another important finding in our study was that Chop−/− not only protected against loss of RGC number but also preserved retinal function in ischemic retina at both 3 and 7 days after I/R injury. We found that pSTRs in Chop−/− eyes were significantly higher at 3 (48%) and 7 days (95%) after I/R injury than those in WT I/R-injured eyes. The fact that RGCs contribute to pSTRs has been shown before,44 and many studies have used pSTR to evaluate RGC function in rodent models of ocular diseases.45 Our pSTR findings are in accordance with the retinal flat mount results in which we observed morphological preservation of RGCs in Chop−/− mice beginning 3 days after I/R injury. Functional protection of RGC is clearly represented in the form of a significantly larger pSTR response at 3 and 7 days after I/R in the Chop−/− mice. In addition, although functional preservation of RGCs was not significant at the 14- and 28-day time points, the trend of partial protection was obvious.
To summarize, retinal I/R injury induces CHOP expression and leads to RGC death via apoptosis. Additionally, absence of CHOP significantly delays RGC death and partially protects against loss of RGC numbers and function in the ischemic retina. These findings clearly indicate that CHOP is an important player in the RGC apoptosis induced by retinal I/R.
Acknowledgments
Supported by Department of Defense VISION Grant 10-2-0003 (AFC).
Disclosure: S. Nashine, None; Y. Liu, None; B.-J. Kim, None; A.F. Clark, None; I.-H. Pang, None
References
- 1. Osborne NN, Ugarte M, Chao M, et al. Neuroprotection in relation to retinal ischemia and relevance to glaucoma. Surv Ophthalmol. 1999; 1: S102–S128. [DOI] [PubMed] [Google Scholar]
- 2. Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004; 23: 91–147. [DOI] [PubMed] [Google Scholar]
- 3. Kuehn MH, Fingert JH, Kwon YH. Retinal ganglion cell death in glaucoma: mechanisms and neuroprotective strategies. Ophthalmol Clin North Am. 2005; 18: 383–395. [DOI] [PubMed] [Google Scholar]
- 4. Weise J, Isenmann S, Bahr M. Increased expression and activation of poly (ADP-ribose) polymerase (PARP) contribute to retinal ganglion cell death following rat optic nerve transection. Cell Death Differ. 2001; 8: 801–807. [DOI] [PubMed] [Google Scholar]
- 5. Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly (ADP-ribose) polymerase. J Cereb Blood Flow Metab. 1997; 17: 1143–1151. [DOI] [PubMed] [Google Scholar]
- 6. Tokime T, Nozaki K, Sugino T, Kikuchi H, Hashimoto N, Ueda K. Enhanced poly (ADP-ribosyl)ation after focal ischaemia in rat brain. J Cereb Blood Flow Metab. 1998; 18: 991–997. [DOI] [PubMed] [Google Scholar]
- 7. Rosenbaum DM, Degterev A, David J, et al. Necroptosis, a novel form of caspase independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010; 88: 1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buchi ER. Cell death in the rat retina after a pressure-induced ischaemia reperfusion insult: an electron microscopic study. Ganglion cell layer and inner nuclear layer. Exp Eye Res. 1992; 55: 605–613. [DOI] [PubMed] [Google Scholar]
- 9. Piras A, Gianetto D, Conte D, Bosone A, Vercelli A. Activation of autophagy in a rat model of retinal ischemia following high intraocular pressure. PLoS One. 2011; 6: e22514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Levin LA, Peeples P. History of neuroprotection & rationale as a therapy for glaucoma. Am J Manag Care. 2008; 14: S11–S14. [PubMed] [Google Scholar]
- 11. Pang I-H, Clark AF. Rodent models for glaucoma retinopathy and optic neuropathy. J Glaucoma. 2007; 16: 483–505. [DOI] [PubMed] [Google Scholar]
- 12. Pang I-H, Clark AF. Non-primate models for glaucoma retinopathy and optic neuropathy. In: Pang I-H, Clark AF. eds Animal Models of Retinal Diseases. New York: Humana Press; 2010: 139–164. [Google Scholar]
- 13. Kaur C, Foulds WS, Ling EA. Hypoxia-ischemia and retinal ganglion cell damage. Clin Ophthalmol. 2008; 2: 879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007; 8: 519–529. [DOI] [PubMed] [Google Scholar]
- 15. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001; 21: 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gotoh T, Terada K, Oyadomari S. Mori M. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004; 11: 390–402. [DOI] [PubMed] [Google Scholar]
- 17. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004; 11: 381–389. [DOI] [PubMed] [Google Scholar]
- 18. Shimazawa M, Inokuchi Y, Ito Y, et al. Involvement of ER stress in retinal cell death. Mol Vis. 2007; 13: 578–587. [PMC free article] [PubMed] [Google Scholar]
- 19. Doh SH, Kim JH, Lee KM, Park HY, Park CK. Retinal ganglion cell death induced by endoplasmic reticulum stress in a chronic glaucoma model. Brain Res. 2010; 1308: 158–166. [DOI] [PubMed] [Google Scholar]
- 20. Zode GS, Sharma AB, Lin X, et al. Ocular-specific ER stress reduction rescues glaucoma in murine glucocorticoid-induced glaucoma. J Clin Invest. 2014; 124; 1956–19 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marciniak SJ, Yun CY, Oyadomari S, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stresses endoplasmic reticulum. Genes Dev. 2004; 18: 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zinszner H, Kuroda M, Wang X, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998; 12: 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feng B, Yao PM, Li Y, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003; 5: 781–792. [DOI] [PubMed] [Google Scholar]
- 24. Milhavet O, Martindale JL, Camandola S, et al. Involvement of Gadd153 in the pathogenic action of presenilin-1 mutations. J Neurochem. 2002; 83: 673–681. [DOI] [PubMed] [Google Scholar]
- 25. Prasanthi JR, Larson T, Schommer J, Ghribi O. Silencing GADD153/CHOP gene expression protects against Alzheimer's disease-like pathology induced by 27-hydroxycholesterol in rabbit hippocampus. PLoS One. 2011; 6: e26420. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26. Paschen W, Gissel C, Linden T, Althausen S, Doutheil J. Activation of gadd153 expression through transient cerebral ischemia: evidence that ischemia causes endoplasmic reticulum dysfunction. Mol Brain Res. 1998; 60: 115–122. [DOI] [PubMed] [Google Scholar]
- 27. Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002; 109: 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fu HY, Okada K, Liao Y, et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation. 2010; 122: 361–369. [DOI] [PubMed] [Google Scholar]
- 29. Hu Y, Park KK, Yang L, et al. Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron. 2012; 73: 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li C, Wang L, Huang K, Zheng L. Endoplasmic reticulum stress in retinal vascular degeneration: protective role of resveratrol. Invest Ophthalmol Vis Sci. 2012; 53: 3241–3249. [DOI] [PubMed] [Google Scholar]
- 31. Li H, Zhu X, Fang F, Jiang D, Tang L. Down-regulation of GRP78 enhances apoptosis via CHOP pathway in retinal ischemia-reperfusion injury. Neurosci Lett. 2014; 575: 68–73. [DOI] [PubMed] [Google Scholar]
- 32. Kwong JM, Caprioli J, Piri N. RNA binding protein with multiple splicing: a new marker for retinal ganglion cells. Invest Ophthalmol Vis Sci. 2010; 51: 1052–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rodriguez AR. Sevilla Müller LP, Brecha NC. The RNA binding protein RBPMS is a selective marker of ganglion cells in the mammalian retina. J Comp Neurol. 2014; 522: 1411–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen ST, Garey LJ, Jen LS. Bcl-2 proto-oncogene protein immunoreactivity in normally developing and axotomized rat retinas. Neurosci Lett. 1994; 172: 11–4. [DOI] [PubMed] [Google Scholar]
- 35. Mizutani M, Gerhardinger C., Lorenzi M. Müller cell changes in human diabetic retinopathy. Diabetes. 1998; 47: 445–449. [DOI] [PubMed] [Google Scholar]
- 36. Levin LA, Schlamp CL, Spieldoch RL, Geszvain KM, Nickells RW. Identification of bcl-2 family genes in the rat retina. Invest Ophthalmol Vis Sci. 1997; 38: 2545–2553. [PubMed] [Google Scholar]
- 37. Kim BJ, Braun TA, Wordinger RJ, Clark AF. Progressive morphological changes and impaired retinal function associated with temporal regulation of gene expression after retinal ischemia/reperfusion injury in mice. Mol Neurodegener. 2013; 8: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhu Y, Ohlemiller KK, McMahan BK, Gidday JM. Mouse models of retinal ischemic tolerance. Invest Ophthalmol Vis Sci. 2002; 43: 1903–1911. [PubMed] [Google Scholar]
- 39. Zhu Y, Zhang L, Sasaki Y, Milbrandt J, Gidday JM. Protection of mouse retinal ganglion cell axons and soma from glaucomatous and ischemic injury by cytoplasmic overexpression of nmnat1. Invest Ophthalmol Vis Sci. 2013; 54: 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lafuente MP, Villegas-Pérez MP, Sellés-Navarro I, Mayor-Torroglosa S, Miralles de Imperial J, Vidal-Sanz M. Retinal ganglion cell death after acute retinal ischemia is an ongoing process whose severity and duration depends on the duration of the insult. Neuroscience. 2002; 109: 157–168. [DOI] [PubMed] [Google Scholar]
- 41. Vidal-Sanz M, Lafuente MP, Mayor S, de Imperial JM, Villegas-Pérez MP. Retinal ganglion cell death induced by retinal ischemia. Neuroprotective effects of two alpha-2 agonists. Surv Ophthalmol. 2001; 45: S261–S267. [DOI] [PubMed] [Google Scholar]
- 42. Awai M, Koga T, Inomata Y, Oyadomari S, Gotoh T, Mori M, Tanihara H. NMDA-induced retinal injury is mediated by an endoplasmic reticulum stress-related protein, CHOP/GADD153. J Neurochem. 2006; 96: 43–52. [DOI] [PubMed] [Google Scholar]
- 43. Slater BJ, Mehrabian Z, Guo Y, Hunter A, Bernstein SL. Rodent anterior ischemic optic neuropathy (rAION) induces regional retinal ganglion cell apoptosis with a unique temporal pattern. Invest Ophthalmol Vis Sci. 2008; 49: 3671–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smith BJ, Wang X, Chauhan BC, Cote PD, Tremblay F. Contribution of retinal ganglion cells to the mouse electroretinogram. Doc Ophthalmol. 2014; 128: 155–168. [DOI] [PubMed] [Google Scholar]
- 45. Chuman H. Nonarteritic ischemic optic neuropathy animal model and its treatment applications. Nihon Ganka Gakkai Zasshi. 2014; 118: 331–361. [PubMed] [Google Scholar]