

Abstract
Background
Large-scale epidemiological evidence on the role of inflammation in early atherosclerosis, assessed by carotid ultrasound, is lacking. We aimed to quantify cross-sectional and longitudinal associations of inflammatory markers with common-carotid-artery intima-media thickness (CCA-IMT) in the general population.
Methods
Information on high-sensitivity C-reactive protein, fibrinogen, leucocyte count and CCA-IMT was available in 20 prospective cohort studies of the PROG-IMT collaboration involving 49,097 participants free of pre-existing cardiovascular disease. Estimates of associations were calculated within each study and then combined using random-effects meta-analyses.
Results
Mean baseline CCA-IMT amounted to 0.74mm (SD = 0.18) and mean CCA-IMT progression over a mean of 3.9 years to 0.011 mm/year (SD = 0.039). Cross-sectional analyses showed positive linear associations between inflammatory markers and baseline CCA-IMT. After adjustment for traditional cardiovascular risk factors, mean differences in baseline CCA-IMT per one-SD higher inflammatory marker were: 0.0082mm for high-sensitivity C-reactive protein (p < 0.001); 0.0072mm for fibrinogen (p < 0.001); and 0.0025mm for leucocyte count (p = 0.033). ‘Inflammatory load’, defined as the number of elevated inflammatory markers (i.e. in upper two quintiles), showed a positive linear association with baseline CCA-IMT (p < 0.001). Longitudinal associations of baseline inflammatory markers and changes therein with CCA-IMT progression were null or at most weak. Participants with the highest ‘inflammatory load’ had a greater CCA-IMT progression (p = 0.015).
Conclusion
Inflammation was independently associated with CCA-IMT cross-sectionally. The lack of clear associations with CCA-IMT progression may be explained by imprecision in its assessment within a limited time period. Our findings for ‘inflammatory load’ suggest important combined effects of the three inflammatory markers on early atherosclerosis.
Keywords: Inflammation, atherosclerosis, meta-analysis
Introduction
High-resolution B-mode ultrasonography has proven to be a valid and reliable method of detecting early atherosclerotic lesions. Ultrasonography allows the assessment of the intima-media thickness (IMT) of the common carotid artery (CCA) as a marker of preclinical atherosclerosis. Increased IMT is correlated with the presence of systemic atherosclerosis and is associated with its clinical sequelae myocardial infarction and stroke.1-3 However, even after accounting for traditional cardiovascular risk factors, most of the variance in IMT remains unexplained.4,5 To improve our understanding of the pathophysiology of early atherosclerosis development, it is important to identify additional determinants related to IMT.
Inflammatory markers have been shown to be predictive of future cardiovascular risk,6,7 and inflammation may have an important role in the development and progression of atherosclerosis.8 Studies of inflammatory biomarkers, such as high-sensitivity C-reactive protein (hsCRP),9,10 fibrinogen11 and leucocyte count12 have lent clinical credence to this concept, but not without controversy.13,14 There is uncertainty concerning the nature of the association of inflammatory biomarkers with the extent and progression of atherosclerosis, 15 and whether this association is independent of other cardiovascular risk factors that are also related to inflammation. Several studies showed an association between hsCRP and fibrinogen with measures of atherosclerosis like IMT or ankle-brachial index16-18 but these associations were weakened if adjusted for conventional cardiovascular risk factors.12,19
To help clarify the conflicting evidence from either single studies or meta-analyses based on published literature, we conducted an individual-participant data meta-analysis based on 49,097 individual records derived from 20 large prospective cohort studies within the PROG-IMT collaboration.3,20 Our aims were four-fold. First, to quantify cross-sectional and longitudinal associations of inflammatory markers hsCRP, leucocyte count and fibrinogen with CCA-IMT, taking into account potential confounding by traditional cardiovascular risk factors. Second, to characterize the shape of any dose–response relationships between inflammatory markers and CCA-IMT. Third, to compare the strength of these associations across clinically relevant subgroups. Fourth, to study the impact of elevations in multiple inflammatory markers (‘inflammatory load’).
Methods
Design of the PROG-IMT collaboration
Details on study identification and eligibility criteria for the PROG-IMT collaboration have been published previously. 3,20 The present analysis used individual records from prospective cohort studies that met the following criteria: (1) participants from the general population; (2) concomitant information on CCA-IMT, plus at least one of the inflammatory markers hsCRP, leucocyte count or fibrinogen; (3) well-defined inclusion criteria and recruitment strategy; and (4) at least two ultrasound visits with assessment of CCA-IMT. Datasets of the contributing studies were carefully checked at the coordinating centre, and implausible values were cleared with the investigators and data managers of the individual studies. The data were harmonized, so that variables were uniformly named, transformed to SI units, and ordinal variables were recoded into binary categories with balanced distributions. The study complies with the Declaration of Helsinki, and the ethics committee of the University Hospital Frankfurt has approved the research protocol. Informed consent has been obtained from all subjects within the individual studies that were included.
Measurement of CCA-IMT and inflammatory markers
For each study, CCA-IMT was calculated as the mean of all mean CCA-IMT measurements available (i.e. from left and right CCA, near and far wall, and/or different insonation angles). For the Bruneck Study and the Chin-Shan Community Cardiovascular Cohort Study, information on mean CCA-IMT was not available and we therefore used the maximum CCA-IMT instead (defined as mean of all maximum CCA-IMT measurements available). From the carotid ultrasound data at two visits, we calculated the yearly CCA-IMT progression rate as the difference in CCA-IMT divided by the time interval in years between the visits. The inflammatory markers (hsCRP, fibrinogen, leucocyte count) were available at both visits in a subset of studies. The methods used in individual studies to assess CCA-IMT and inflammatory markers are provided in online Supplementary Table 1.
Statistical analysis
The statistical analyses followed a pre-specified plan. C-reactive protein was log-transformed to obtain an approximately normal distribution. Age- and sex-adjusted partial correlation coefficients between baseline inflammatory markers were calculated for each study, Fisher’s z-transformation used to obtain a normal distribution, and combined across studies with random-effects meta-analysis. Repeatability correlations of inflammatory markers adjusted for age and sex were calculated within each study by regressing the baseline on the follow-up measurement and were combined similarly.
The principal analysis consisted of three linear regression components: (1) the association of the baseline levels of each inflammatory marker with baseline CCA-IMT; (2) the association of the baseline level of each inflammatory marker with CCA-IMT progression; and (3) the association of the change in each inflammatory marker with CCA-IMT progression. Analyses involved a two-stage approach. For each inflammatory marker, estimates of association were calculated separately within each study before pooling across studies by random-effects meta-analysis. Since the principal analysis consisted of nine regression models, we controlled for the risk of false-positive results by using more stringent criteria for p values (i.e. p < 0.005) in each analysis before claiming convincing evidence of associations. Analyses of components (1) and (2) were adjusted for age, sex and baseline information on selected traditional risk factors (i.e. systolic blood pressure, total cholesterol, history of diabetes, current smoking and use of anti-hypertensive medication). Analyses for component (3) were adjusted for age, sex, mean CCA-IMT, plus the means and changes in the selected traditional risk factors. Participants who had suffered coronary heart disease and/or cerebrovascular disease before the baseline visit were excluded from the principal analyses. Participants who developed first-ever coronary heart disease and/or cerebrovascular disease between the first and the second ultrasound visit were excluded from components (2) and (3) (although sensitivity analyses included these participants). Furthermore, we evaluated association shapes by calculating mean differences across quintiles of inflammatory markers, combining them by multivariate meta-analysis, and plotting them against the respective mean level of inflammatory marker within that category. Ninety-five per cent confidence intervals (95% CIs) were calculated from the variances that correspond to the amount of information underlying each group (including the reference group).21 The I2 statistic was used as a measure of heterogeneity in estimated regression coefficients across studies.22 SDs and quintiles were defined within each study.
Subsidiary analyses compared the associations across clinically relevant pre-defined subgroups, that is, sex, baseline history of diabetes, baseline history of hypertension (defined as systolic blood pressure >140 mmHg, diastolic blood pressure >90 mmHg, or use of antihypertensive medication), statin use at baseline and prevalent cardiovascular disease (CVD) at baseline (these patients being excluded from the principal analyses). For subgroup analyses, data were restricted to studies with some participants in each subgroup. Due to multiple comparisons, we defined a significance level of p-value<0.001 in this analysis.
Finally, analyses were conducted that compared the association with CCA-IMT according to number of elevated inflammatory markers (hsCRP, fibrinogen, leucocyte count) at baseline (‘inflammatory load’). We prespecified that levels of inflammatorymarkerswere regarded elevated if they were in the upper two quintiles of the study-specific distribution. For each study, mean differences in CCA-IMT were estimated across participants with no, one, two or three elevated inflammatory markers and combined by multivariate meta-analysis. Analyses were carried out with Stata software (Stata Corporation, College Station, Texas, USA, Release 12.1).
Results
Overall, the present analysis included individual data from 49,097 participants in 20 studies of the PROG-IMT collaboration (Table 1). Nineteen studies shared data on hsCRP, 13 studies on fibrinogen and 13 studies on leucocyte count. The mean time between first and second CCA-IMT measurement was 3.9 years (SD 1.5 years). The combined CCA-IMT was 0.74mm at baseline and 0.77mm at follow-up. The mean CCA-IMT progression amounted to 0.011 mm/year. Inflammatory marker distributions were similar at baseline and follow-up. Study-specific values of CCA-IMT, inflammatory markers and traditional cardiovascular risk factors are summarized in online Supplementary Tables 2 and 3. The correlation between baseline levels of the inflammatory markers was moderate to low. Age- and sex-adjusted partial correlation coefficients were 0.45 between log hsCRP and fibrinogen (95% CI, 0.39 to 0.50), 0.23 between log hsCRP and leucocyte count (0.19 to 0.27) and 0.25 between fibrinogen and leucocyte count (0.21 to 0.28). Repeatability correlations adjusted for age and sex were 0.62 for CCA-IMT (95% CI, 0.57 to 0.68); 0.58 for log hsCRP (0.52 to 0.64), 0.48 for fibrinogen (0.38 to 0.57) and 0.57 for leucocyte count (0.43 to 0.70).
Table 1.
Baseline and follow-up information available on inflammatory markers and common carotid artery intima-media thickness (CCA-IMT).
| Baseline information
|
Follow-up information (a mean of 3.9 years later)
|
|||
|---|---|---|---|---|
| No. of studies/participants | Mean (SD) | No. of studies/participants | Mean (SD) | |
| CCA-IMT, mm | 20/49,097 | 0.74 (0.18) | 20/36,528 | 0.77 (0.18) |
| CCA-IMT progression, mm/year | – | 20/36,528 | 0.0111 (0.0389) | |
| hsCRP, mg/dl | 19/28,090 | 0.18 (0.22)* | 11/15,934 | 0.19 (0.21)* |
| Fibrinogen, mg/dl | 13/35,096 | 310 (72) | 6/10,941 | 318 (72) |
| Leucocyte count, 103/μl | 13/39,541 | 6.3 (1.9) | 8/25,034 | 6.1 (1.9) |
CCA-IMT: common carotid artery intima-media thickness; hsCRP: high-sensitivity C-reactive protein; SD: standard deviation
Geometric mean (approximate SD).
Cross-sectional associations of inflammatory markers with CCA-IMT
In cross-sectional analyses adjusted for age and sex, we observed positive linear associations between baseline inflammatory markers and baseline CCA-IMT (Figure 1(a)). Associations were somewhat weaker when also adjusting for other traditional risk factors: on average, a one-SD higher baseline level of log hsCRP was associated with 0.0082mm higher baseline CCA-IMT (0.0062 to 0.0103 mm; p < 0.001) (Figure 2(a)). The corresponding mean differences for one-SD higher fibrinogen and leucocyte count were 0.0072mm (0.0047 to 0.0097 mm; p < 0.001) and 0.0025mm (0.0002 to 0.0048 mm; p = 0.033), respectively. Heterogeneity across studies was sometimes high, with I2 statistics ranging from 24% to 74%. Forest plots depicting study-specific effect estimates are provided in online Supplementary Figure 1. Results were similar upon further adjustment for ethnicity, socio-economic status, lipid-lowering treatment, log creatinine or body mass index (online Supplementary Table 4).
Figure 1.
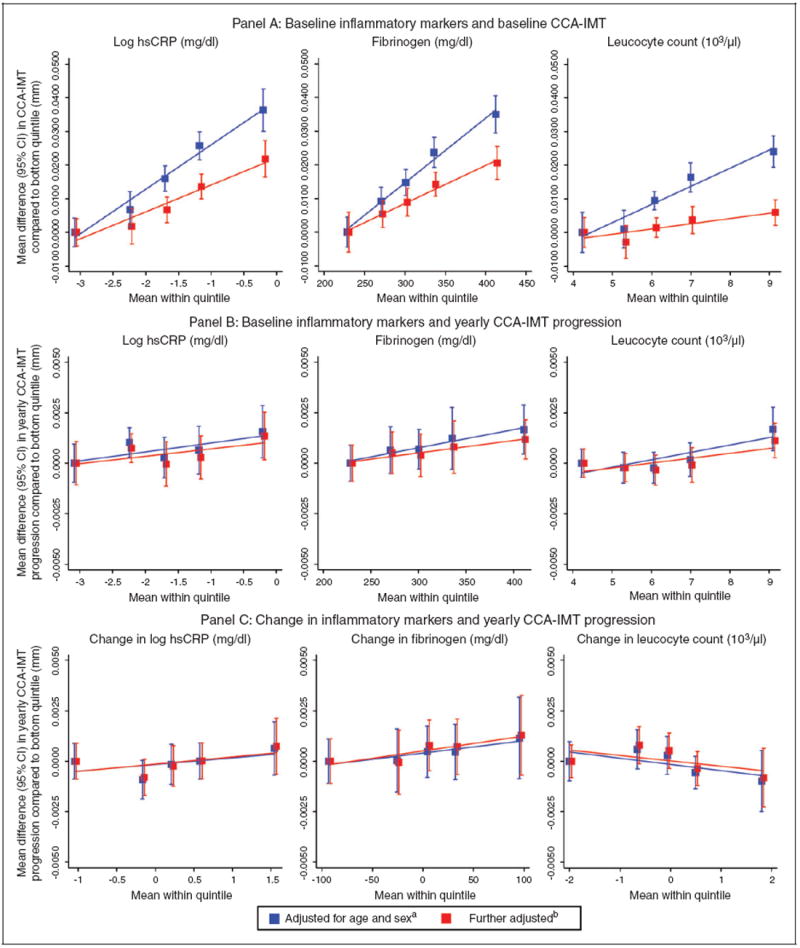
Shape of association of inflammation markers with common carotid artery intima-media thickness (CCA-IMT). Panel (a): baseline inflammatory markers and baseline CCA-IMT; panel (b): baseline inflammatory markers and yearly CCA-IMT progression; panel (c): change in inflammatory markers and yearly CCA-IMT progression.
aModels in panel (c) were additionally adjusted for mean CCA-IMT.
bModels in panels (a) and (b) were further adjusted for baseline traditional risk factors (i.e. systolic blood pressure, total cholesterol, history of diabetes, current smoking, use of anti-hypertensive medication), models in panel (c) for means and changes in traditional risk factors.
hsCRP: high-sensitivity C-reactive protein; CI: confidence interval
Figure 2.
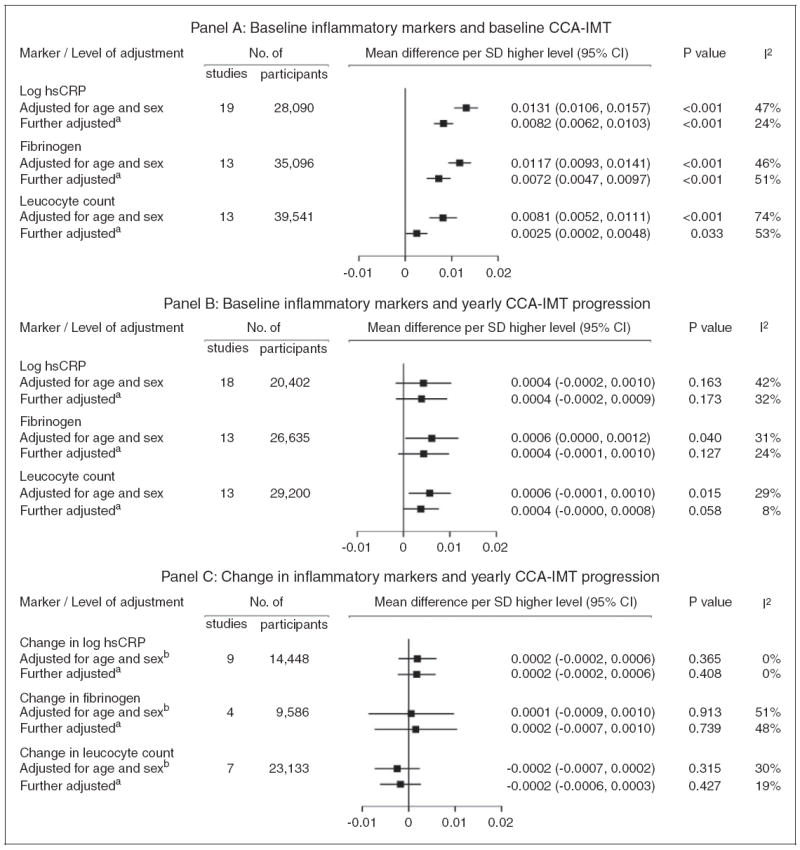
Association of inflammation markers with common carotid artery intima-media thickness (CCA-IMT). Panel (a): baseline inflammatory markers and baseline CCA-IMT; panel (b): baseline inflammatory markers and yearly CCA-IMT progression; panel (c): change in inflammatory markers and yearly CCA-IMT progression.
aModels in panels (a) and (b) were further adjusted for baseline traditional risk factors (i.e. systolic blood pressure, total cholesterol, history of diabetes, current smoking, use of anti-hypertensive medication), models in panel (c) for means and changes in traditional risk factors. The number of participants contributing to the analysis in panel (c) is less than in Table 1 because of missing values in the variables for which the analysis was adjusted.
bModels in panel (c) were additionally adjusted for mean CCA-IMT.
CI: confidence interval
We then investigated whether cross-sectional associations of baseline inflammatory marker concentrations and baseline CCA-IMT differed across pre-specified subgroups (online Supplementary Figure 2). There was some evidence for a stronger association in men compared with women for all three inflammatory markers (mean differences 0.0076mm for log hsCRP (p = 0.001), 0.0095mm for fibrinogen (p = 0.002) and 0.0031mm for leucocyte count (p = 0.031)). Furthermore, the association of leucocyte count appeared to be somewhat stronger in participants with hypertension (0.0037 mm; p = 0.006), and the associations for fibrinogen and leukocyte count appeared stronger in normal compared with obese participants (body mass index ≥ 30 kg/m2). We did not find evidence for heterogeneity in findings across studies grouped according to the methods used to assess inflammatory markers and CCA-IMT (all meta-regressions p > 0.05).
Longitudinal associations of inflammatory markers with CCA-IMT progression
Associations between inflammatory marker concentrations and CCA-IMT progression were at most weak after adjustment for traditional risk factors. Neither baseline inflammatory markers (Figures 1(b) and 2(b)) nor their changes between baseline and follow-up (Figures 1(c) and 2(c)) were significantly associated with CCA-IMT progression. Study-specific estimates for these analyses are shown in online Supplementary Figures 3 and 4. We observed similar findings in sensitivity analyses that additionally included participants with an incident CVD event between the baseline and follow-up surveys. There was no evidence for a difference in associations by inflammation and CCA-IMT assessment methods or by the length of time between baseline and follow-up survey (all meta-regressions p > 0.05).
Inflammatory load and CCA-IMT
Of 14,200 participants with concomitant baseline information on all three inflammatory markers and baseline CCA-IMT, 32% had no, 32% had one, 23% had two and 13% had three elevated inflammation markers (Figure 3(a)). There was a positive linear association between the number of elevated inflammation markers and baseline CCA-IMT (p < 0.001 for trend). For instance, participants with three elevated markers had on average a 0.0194mm higher CCA-IMT compared with participants in the reference group with no elevated markers (0.0110 to 0.0277 mm; p < 0.001). Furthermore, in an analysis involving 13,435 participants, CCA-IMT progression was significantly higher in participants with three elevated inflammatory markers (mean difference 0.0032 mm/year; p = 0.015) as compared with the reference group, whereas participants with one or two elevated inflammatory markers did not differ from the reference group in their CCA-IMT progression (Figure 3(b)).
Figure 3.
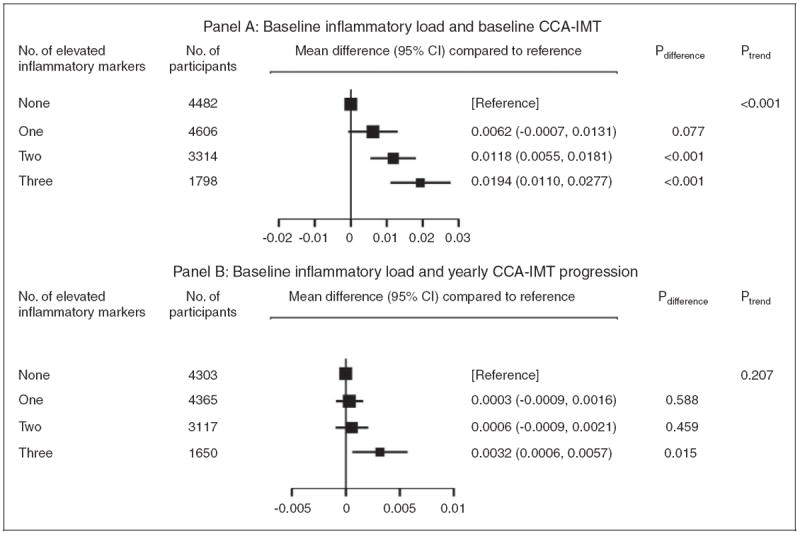
Associations between number of elevated inflammation markers at baseline and common carotid artery intima-media thickness (CCA-IMT). Panel (a): baseline inflammatory load and baseline CCA-IMT; panel (b): baseline inflammatory load and yearly CCA-IMT progression. For each of the three inflammatory markers (high-sensitivity C-reactive protein, fibrinogen, leucocyte count), levels were deemed to be elevated if they were in the top two fifths of the study-specific distribution. People with no elevated inflammatory markers served as the reference group. Models were adjusted for baseline age, sex, systolic blood pressure, total cholesterol, history of diabetes, current smoking, and use of anti-hypertensive medication.
CI: confidence interval
Discussion
Recent evidence suggests that inflammation plays an important role in all stages of the atherosclerotic process, 8 but the association of inflammatory markers with the extent and progression of early carotid atherosclerosis has not been characterized in detail. We have analysed data from 20 prospective cohort studies representative of the general population, including information on a total of 49,097 healthy participants. We have been able to undertake comprehensive and standardized analyses of baseline CCA-IMT as well as CCA-IMT progression with three inflammatory markers on the basis of individual participant records. To our knowledge, this is the largest and most comprehensive analysis available so far on this topic.
Inflammatory markers and baseline CCA-IMT
Our analysis demonstrated significant positive and linear associations between baseline CCA-IMT and all examined markers of inflammation (hsCRP, fibrinogen, leucocyte count) at baseline. Associations persisted even when adjusting for several traditional cardiovascular risk factors. Higher levels of these markers were related to higher CCA-IMT, with perhaps slightly stronger associations for hsCRP and fibrinogen than for leucocyte count.
The association between hsCRP and carotid IMT has previously been investigated in several studies but with conflicting results.18 Some cross-sectional studies demonstrated that hsCRP was associated with IMT,23,24 whereas other broad-based community studies suggested that hsCRP failed to be an independent risk factor for early atherosclerosis after adjustment for various risk factors.12,19 In a literature-based metaanalysis, Baldassare et al. have observed a positive association between carotid IMT and hsCRP, although the heterogeneity of published results was high, potentially due to inconsistent adjustment across studies.25
Experimental evidence has shown that fibrinogen is involved early in the formation and growth of atheroma infiltrating the arterial wall.18 Independent cross-sectional associations between fibrinogen levels and carotid IMT have previously been reported in a population-based study of 135 participants free of clinical atherosclerotic disease11 and study of 597 volunteers with impaired glucose tolerance.26 Again, a literature-based meta-analysis by Baldassare et al.25 endorsed the presence of a significant relationship between these two parameters.
Leucocytes play an important role in early and advanced stages of atherosclerosis formation12,26,27 and are key cells at the various stages of cardiovascular disease progression and its complications.28 Cross-sectional studies have observed a positive association between leucocyte count and IMT in subjects with primary dyslipidaemia,12 in middle-aged men27 and in diabetics.26
Inflammatory markers and progression of IMT
In contrast to the clear association between baseline CCA-IMT and inflammation, we found only weak and non-significant associations of baseline inflammatory markers or changes therein with individual CCA-IMT progression after adjusting for traditional risk factors. This finding corroborates some previous studies,29 but contradicts others. For example, Sabeti et al.30 observed a gradual increase in risk of progression of carotid atherosclerosis with higher baseline fibrinogen levels (adjusted hazard ratio 1.83, 2.09 and 2.45, respectively for the second to fourth quartile as compared with the first quartile). Fibrinogen at follow-up was also associated with progressive atherosclerosis. 30 Another study described a close correlation between inflammation and morphological features of rapidly progressive carotid atherosclerosis in a selected high-risk patient population,31 whereas other studies did not observe an independent association29 or only in specific subgroups.32
What are the possible explanations for the lack of clear association between baseline inflammatory status and IMT progression? Heterogeneity in the ultrasound protocols or the duration of ultrasound follow-up of the studies included in PROG-IMT may potentially affect the progression estimates and their precision. However, the definition of CCA-IMT used was consistent in most studies included in the present analysis,3 and we found no difference in association by inflammation and IMT measurement methods or by follow-up periods. Due to the low CCA-IMT progression observed during a follow-up period of an average 3.9 years, the signal-to-noise ratio of IMT progression may limit its precise assessment. The biology of atherosclerosis may also explain the lack of relation between inflammatory markers and IMT progression. Atherosclerosis is a lifelong process that progresses slowly at a young age and may accelerate with accumulation of risk factors. The slow progression of IMT in healthy populations is therefore difficult to detect.
Additionally, focal plaques at vessel sites with the highest IMT can superimpose the diffuse thickening of the intima-media complex. An analysis from the Rotterdam study has shown that hsCRP predicts progression of more advanced atherosclerosis with the use of a composite plaque score.33 A small investigation also detected a relationship between hsCRP and the progression of the number of plaques and plaque score, respectively.34 Thus, it is conceivable that long lasting low-level inflammation is more closely related to more advanced stages of atherosclerosis such as plaque formation, than to early changes such as the diffuse thickening of the intima-media complex. It has also been shown that IMT is influenced by several genetic polymorphisms.35,36 Therefore, it is possible that progression of IMT also depends on genetic characteristics rather than humoral risk factors alone.
Subgroup analyses
We performed a range of pre-specified subgroup analyses. Of note, we observed a somewhat stronger association between inflammation and baseline CCA-IMT in men compared with women for all three inflammatory markers. The influence of sex on the atherosclerotic response to inflammation has so far not been well characterized. Some studies observed a stronger relationship between low-level inflammation and IMT progression for women.32 However, histologic analyses of plaque specimens from endarterectomy of carotid stenosis showed a higher concentration of inflammatory and, in particular, of macrophage foam cells in men than in women,37 indicating important gender differences in the complex interaction of inflammation and atherosclerosis. Interestingly, a similar effect modification has previously been reported by the Emerging Risk Factors Collaboration, demonstrating a stronger association of CRP with coronary heart disease risk and a greater added value of CRP and fibrinogen measurement for predicting cardiovascular risk in men than in women.7,38
We also observed a significantly stronger association of fibrinogen and leucocyte count, but not of hsCRP, with baseline IMT in participants with body mass index (BMI) < 30 kg/m2. It is well known that obesity is associated with higher levels of inflammatory markers39,40 as well as an increased frequency of conventional risk factors and particularly diabetes. Thus it is conceivable that traditional risk factors play a more important role for extent of atherosclerosis than inflammation status in obese patients.
Inflammatory load
One important finding of our analysis is the highly significant and nearly linear relationship of ‘inflammatory load’ with baseline CCA-IMT and to a lesser degree with CCA-IMT progression. Participants with elevation in all three inflammatory markers had a higher baseline CCA-IMT and a greater CCA-IMT progression as compared with subjects without increased inflammatory markers, even after adjustment for several traditional cardiovascular risk factors. We observed only a moderate correlation between the three different baseline measures of inflammation (correlation coefficients between 0.23 to 0.45). Similar correlations have been observed in other studies comparing different inflammatory parameters.26 This finding may imply that these factors reflect different aspects of low-level inflammation in individual subjects and that the use of a composite measure like ‘inflammatory load’ is a better parameter because of reduced variability.37,38,41,42
Our finding of a nearly linear relationship between ‘inflammatory load’ and CCA-IMT that withstands adjustment for major cardiovascular risk factors points to a synergistic effect of these different markers of chronic low-level inflammation for the development of early atherosclerosis and may indicate a more wide-spread inflammatory state or a genetic preposition due to an ‘inflammatory genetic haplotype’ in these participants. 43,44 Measurement of the ‘inflammatory load’ may therefore be a better marker to identify the impact of inflammation on extent and progression of subclinical atherosclerosis. A similar pathophysiological mechanism has been proposed for the observed association between ‘infectious burden’ and atherosclerosis development.13 Several studies described a linear association between the detection and extent of infectious microorganisms and atherosclerosis (increased IMT and carotid plaque thickness) even after adjusting for traditional risk factors.41,45 Interestingly, it has been shown that improvement of microbial periodontal status is related to a decreased IMT progression, most likely due to a reduced inflammatory response.46
During recent years, several studies have evaluated the usefulness of inflammatory parameters (particular hsCRP) for risk stratification, notably in subjects free of cardiovascular events with intermediate risk. In contrast to several older studies that point to an important role of inflammatory markers for risk stratification, 6,7,47 new data using genome-wide association studies48 or Mendelian randomization analysis49 make it unlikely that concentration of CRP or plasma fibrinogen are causal factors for CVD events. Our findings of associations of ‘inflammatory load’ with baseline IMT and IMT progression led us to hypothesise that the concept of ‘inflammatory burden’ may be able to identify individuals at high risk of enhanced and extended atherosclerosis and probably increased cardiovascular risk where anti-inflammatory treatment may prevent further events. However, further studies have to corroborate this hypothesis, especially controlled trials to evaluate whether inflammation is a cause or consequence of atherosclerotic burden.
Limitations and strengths
One possible limitation of our investigation is the differing durations between repeated IMT measurements in the studies included. However, there was no difference in associations between inflammation and CCA-IMT according to length of time between baseline and follow-up survey. Second, we included data from studies with different IMT assessment methods, but this also did not affect the results of our analysis. On the other hand, our study has several strengths. The large number of included studies allowed detailed exploration of associations overall as well as in several subgroup and sensitivity analyses. Moreover, the individuals included were from population-based samples and representative of subjects with a wide spectrum of different cardiovascular risk factors. Finally, to limit the scope of any effects by treatment initiation and to have a more homogenous study population, analyses in each study for CCA-IMT progression were confined to participants that remained free of an incident CVD event until the follow-up survey (although sensitivity analyses including these individuals showed very similar results).
Conclusions
Inflammation was independently associated with CCA-IMT cross-sectionally. The lack of clear associations with CCA-IMT progression may be explained by imprecision in its assessment over only a few years. Our findings for ‘inflammatory load’ suggest an important combined effect of the three inflammatory markers on early atherosclerosis.
Acknowledgments
The work was performed at: the Department of Public Health and Primary Care, University of Cambridge, Cambridge, UK; Department of Neurology, University Hospital Frankfurt, Frankfurt am Main, Germany; the Department of Neurology, Benedictus Krankenhaus Tutzing and Feldafing, Tutzing, Germany; and the Technische Universität München, Munich, Germany. The ARIC data was provided through the NHLBI data repository (BioLINCC). This article does not necessarily convey the opinions or views of the ARIC Study or the National Heart, Lung and Blood Institute. A full list of principal CHS investigators and institutions can be found at CHS-NHLBI.org. Etude sur le vieillissement artériel was organized with an agreement between INSERM and Merck, Sharp, and Dohme-Chibret.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (grant no. DFG Lo 1569/2-1 to the PROG-IMT project); the National Heart, Lung and Blood Institute, Bethesda, MD, USA in collaboration with the Atherosclerosis Risk In Communities (ARIC) investigators (the ARIC Study, for which a restricted access dataset was used) and contracts HHSN268201200036C, HHSN268200800007C, N01 HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, N01HC85084, N01HC35129, N01HC85085, N01HC45133, and grant no. HL080295, with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS) for the data of Cardiovascular Health Study (CHS); the Pustertaler Verein zur Praevention von Herz- und Hirngefaesserkrankungen, Gesundheitsbezirk Bruneck, and the Assessorat fuer Gesundheit (Province of Bolzano, Italy) (to the Bruneck study); the Stiftung Deutsche Schlaganfall-Hilfe (to the Carotid Atherosclerosis Progression Study); the National Institute on Aging (NIA; grant no. AG023629); the National Institute of Neurological Disorders and Stroke (grant no. R37 NS 029993 to the Northern Manhattan Study/The Oral Infections and Vascular Disease Epidemiology Study); the National Institute of Dental and Craniofacial Research, Bethesda, MD, USA (grant no. R01 DE 13094 to the Oral Infections, Carotid Atherosclerosis, and Stroke study; AOK Bayern (to the Interventionsprojekt zerebrovaskuläre Erkrankungen und Demenz im Landkreis Ebersberg study); the Netherlands Foundation for Scientific Research (ZonMw, Vici 918-76-619 to the Rotterdam Study); the Federal Ministry of Education and Research (grant nos. BMBF 01ZZ9603, 01ZZ0103 and 01ZZ0403), the Ministry of Cultural Affairs, and the Social Ministry of the Federal State of Mecklenburg-West Pomerania (to The Study of Health in Pomerania, part of the Community Medicine Research net of the University of Greifswald, Germany).
Footnotes
Conflict of interest
SA has received speaker’s honoraria from Sanofi, Siemens, Pfizer, Boehringer-Ingelheim, Orion Pharma, and Astra Zeneza and is on the advisory board for Astra Zeneca. OHF works in ErasmusAGE, a centre for aging research across the life course funded by Nestlé Nutrition (Nestec Ltd), Metagenics Inc., and AXA. Nestlé Nutrition (Nestec Ltd.), Metagenics Inc. and AXA had no role in design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review or approval of the manuscript. MHO received a Research Grant from Merck & Co., Inc., West Point, PA. None of the other authors report conflicts of interest.
A complete list of collaborators of the PROG-IMT project is shown in the online Supplementary Material.
References
- 1.Gardin JM, Bartz TM, Polak JF, et al. What do carotid intima-media thickness and plaque add to the prediction of stroke and cardiovascular disease risk in older adults? The cardiovascular health study. J Am Soc Echocardiogr. 2014;27:998–1005.e2. doi: 10.1016/j.echo.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Den Ruijter HM, Peters SAE, Anderson TJ, et al. Common carotid intima-media thickness measurements in cardiovascular risk prediction: A meta-analysis. JAMA. 2012;308:796–803. doi: 10.1001/jama.2012.9630. [DOI] [PubMed] [Google Scholar]
- 3.Lorenz MW, Polak JF, Kavousi M, et al. Carotid intimamedia thickness progression to predict cardiovascular events in the general population (the PROG-IMT collaborative project): A meta-analysis of individual participant data. Lancet. 2012;379:2053–2062. doi: 10.1016/S0140-6736(12)60441-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baldassarre D, Nyyssönen K, Rauramaa R, et al. Cross-sectional analysis of baseline data to identify the major determinants of carotid intima-media thickness in a European population: The IMPROVE study. Eur Heart J. 2010;31:614–622. doi: 10.1093/eurheartj/ehp496. [DOI] [PubMed] [Google Scholar]
- 5.Rundek T, Blanton SH, Bartels S, et al. Traditional risk factors are not major contributors to the variance in carotid intima-media thickness. Stroke. 2013;44:2101–2108. doi: 10.1161/STROKEAHA.111.000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Awan Z, Genest J. Inflammation modulation and cardiovascular disease prevention. Eur J Prev Cardiol. doi: 10.1177/2047487314529350. Epub ahead of print 7 April 2014. [DOI] [PubMed] [Google Scholar]
- 7.The Emerging Risk Factors CollaborationC-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. 2012;367:1310–1320. doi: 10.1056/NEJMoa1107477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 9.Ridker PM, Danielson E, Fonseca FA, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: A prospective study of the JUPITER trial. Lancet. 2009;373:1175–1182. doi: 10.1016/S0140-6736(09)60447-5. [DOI] [PubMed] [Google Scholar]
- 10.Sander D, Winbeck K, Klingelhöfer J, et al. Progression of early carotid atherosclerosis is only temporarily reduced after antibiotic treatment of Chlamydia pneumoniae seropositivity. Circulation. 2004;109:1010–1015. doi: 10.1161/01.CIR.0000117232.30832.EC. [DOI] [PubMed] [Google Scholar]
- 11.Martínez-Vila E, Páramo JA, Beloqui O, et al. Independent association of fibrinogen with carotid intima-media thickness in asymptomatic subjects. Cerebrovasc Dis. 2003;16:356–362. doi: 10.1159/000072557. [DOI] [PubMed] [Google Scholar]
- 12.Ortega E, Gilabert R, Nuñez I, et al. White blood cell count is associated with carotid and femoral atherosclerosis. Atherosclerosis. 2012;221:275–281. doi: 10.1016/j.atherosclerosis.2011.12.038. [DOI] [PubMed] [Google Scholar]
- 13.Libby P, Ridker PM, Hansson GK, et al. Inflammation in atherosclerosis: From pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. 2006;83:456S–460S. doi: 10.1093/ajcn/83.2.456S. [DOI] [PubMed] [Google Scholar]
- 15.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 16.Syvänen K, Korhonen P, Jaatinen P, et al. High-sensitivity C-reactive protein and ankle brachial index in a Finnish cardiovascular risk population. Int J Angiol. 2011;20:43–48. doi: 10.1055/s-0031-1272551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magyar MT, Szikszai Z, Balla J, et al. Early-onset carotid atherosclerosis is associated with increased intima-media thickness and elevated serum levels of inflammatory markers. Stroke. 2003;34:58–63. doi: 10.1161/01.str.0000048845.83285.ac. [DOI] [PubMed] [Google Scholar]
- 18.Corrado E, Rizzo M, Coppola G, et al. An update on the role of markers of inflammation in atherosclerosis. J Atheroscler Thromb. 2010;17:1–11. doi: 10.5551/jat.2600. [DOI] [PubMed] [Google Scholar]
- 19.Chapman CML, Beilby JP, McQuillan BM, et al. Monocyte count, but not C-reactive protein or interleukin- 6, is an independent risk marker for subclinical carotid atherosclerosis. Stroke. 2004;35:1619–1624. doi: 10.1161/01.STR.0000130857.19423.ad. [DOI] [PubMed] [Google Scholar]
- 20.Lorenz MW, Bickel H, Bots ML, et al. Individual progression of carotid intima media thickness as a surrogate for vascular risk (PROG-IMT): Rationale and design of a meta-analysis project. Am Heart J. 2010;159:730–736.e2. doi: 10.1016/j.ahj.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Easton DF, Peto J, Babiker AG. Floating absolute risk: An alternative to relative risk in survival and case-control analysis avoiding an arbitrary reference group. Stat Med. 1991;10:1025–1035. doi: 10.1002/sim.4780100703. [DOI] [PubMed] [Google Scholar]
- 22.Higgins JPT, Thompson SG, Deeks JJ, et al. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–560. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winbeck K, Kukla C, Poppert H, et al. Elevated C-reactive protein is associated with an increased intima to media thickness of the common carotid artery. Cerebrovasc Dis. 2002;13:57–63. doi: 10.1159/000047747. [DOI] [PubMed] [Google Scholar]
- 24.Xu M, Bi Y, Chen Y, et al. Increased C-reactive protein associates with elevated carotid intima-media thickness in Chinese adults with normal low density lipoprotein cholesterol levels. J Atheroscler Thromb. 2013;20:575–584. doi: 10.5551/jat.15172. [DOI] [PubMed] [Google Scholar]
- 25.Baldassarre D, De Jong A, Amato M, et al. Carotid intima-media thickness and markers of inflammation, endothelial damage and hemostasis. Ann Med. 2008;40:21–44. doi: 10.1080/07853890701645399. [DOI] [PubMed] [Google Scholar]
- 26.Temelkova-Kurktschiev T, Koehler C, Henkel E, et al. Leukocyte count and fibrinogen are associated with carotid and femoral intima-media thickness in a risk population for diabetes. Cardiovasc Res. 2002;56:277–283. doi: 10.1016/s0008-6363(02)00547-3. [DOI] [PubMed] [Google Scholar]
- 27.Loimaala A, Rontu R, Vuori I, et al. Blood leukocyte count is a risk factor for intima-media thickening and subclinical carotid atherosclerosis in middle-aged men. Atherosclerosis. 2006;188:363–369. doi: 10.1016/j.atherosclerosis.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 28.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–166. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lorenz MW, Karbstein P, Markus HS, et al. High-sensitivity C-reactive protein is not associated with carotid intima-media progression: The carotid atherosclerosis progression study. Stroke. 2007;38:1774–1779. doi: 10.1161/STROKEAHA.106.476135. [DOI] [PubMed] [Google Scholar]
- 30.Sabeti S, Exner M, Mlekusch W, et al. Prognostic impact of fibrinogen in carotid atherosclerosis: Nonspecific indicator of inflammation or independent predictor of disease progression? Stroke. 2005;36:1400–1404. doi: 10.1161/01.STR.0000169931.96670.fc. [DOI] [PubMed] [Google Scholar]
- 31.Schillinger M, Exner M, Mlekusch W, et al. Inflammation and Carotid Artery – Risk for Atherosclerosis Study (ICARAS) Circulation. 2005;111:2203–2209. doi: 10.1161/01.CIR.0000163569.97918.C0. [DOI] [PubMed] [Google Scholar]
- 32.Sander K, Horn CS, Briesenick C, et al. High-sensitivity C-reactive protein is independently associated with early carotid artery progression in women but not in men: The INVADE Study. Stroke. 2007;38:2881–2886. doi: 10.1161/STROKEAHA.106.481531. [DOI] [PubMed] [Google Scholar]
- 33.Van Der Meer IM, De Maat MPM, Hak AE, et al. C-reactive protein predicts progression of atherosclerosis measured at various sites in the arterial tree: The Rotterdam Study. Stroke. 2002;33:2750–2755. doi: 10.1161/01.str.0000044168.00485.02. [DOI] [PubMed] [Google Scholar]
- 34.Hashimoto H, Kitagawa K, Hougaku H, et al. C-reactive protein is an independent predictor of the rate of increase in early carotid atherosclerosis. Circulation. 2001;104:63–67. doi: 10.1161/hc2601.091705. [DOI] [PubMed] [Google Scholar]
- 35.Armstrong C, Abilleira S, Sitzer M, et al. Polymorphisms in MMP family and TIMP genes and carotid artery intima-media thickness. Stroke. 2007;38:2895–2899. doi: 10.1161/STROKEAHA.107.491696. [DOI] [PubMed] [Google Scholar]
- 36.Paternoster L, Martínez González NA, Lewis S, et al. Association between apolipoprotein E genotype and carotid intima-media thickness may suggest a specific effect on large artery atherothrombotic stroke. Stroke. 2008;39:48–54. doi: 10.1161/STROKEAHA.107.488866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sangiorgi G, Roversi S, Biondi Zoccai G, et al. Sex-related differences in carotid plaque features and inflammation. J Vasc Surg. 2013;57:338–344. doi: 10.1016/j.jvs.2012.07.052. [DOI] [PubMed] [Google Scholar]
- 38.Kaptoge S, Di Angelantonio E, et al. Emerging Risk Factors CollaborationC-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: An individual participant meta-analysis. Lancet. 2010;375:132–140. doi: 10.1016/S0140-6736(09)61717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casula M, Tragni E, Zambon A, et al. C-reactive protein distribution and correlation with traditional cardiovascular risk factors in the Italian population. Eur J Intern Med. 2013;24:161–166. doi: 10.1016/j.ejim.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 40.Rana JS, Arsenault BJ, Després JP, et al. Inflammatory biomarkers, physical activity, waist circumference, and risk of future coronary heart disease in healthy men and women. Eur Heart J. 2011;32:336–344. doi: 10.1093/eurheartj/ehp010. [DOI] [PubMed] [Google Scholar]
- 41.Elkind MSV, Luna JM, Moon YP, et al. Infectious burden and carotid plaque thickness: The northern Manhattan study. Stroke. 2010;41:e117–e122. doi: 10.1161/STROKEAHA.109.571299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ridker PM, Danielson E, Fonseca FAH, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 43.Markus HS, Labrum R, Bevan S, et al. Genetic and acquired inflammatory conditions are synergistically associated with early carotid atherosclerosis. Stroke. 2006;37:2253–2259. doi: 10.1161/01.STR.0000236637.72124.3f. [DOI] [PubMed] [Google Scholar]
- 44.Humphries SE, Morgan L. Genetic risk factors for stroke and carotid atherosclerosis: Insights into pathophysiology from candidate gene approaches. Lancet Neurol. 2004;3:227–235. doi: 10.1016/S1474-4422(04)00708-2. [DOI] [PubMed] [Google Scholar]
- 45.Espinola-Klein C, Rupprecht HJ, Blankenberg S, et al. Impact of infectious burden on progression of carotid atherosclerosis. Stroke. 2002;33:2581–2586. doi: 10.1161/01.str.0000034789.82859.a4. [DOI] [PubMed] [Google Scholar]
- 46.Desvarieux M, Demmer RT, Jacobs DR, et al. Changes in clinical and microbiological periodontal profiles relate to progression of carotid intima-media thickness: The Oral Infections and Vascular Disease Epidemiology study. J Am Heart Assoc. 2013;2:e000254. doi: 10.1161/JAHA.113.000254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Danesh J, Lewington S, et al. Fibrinogen Studies CollaborationPlasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: An individual participant meta-analysis. JAMA. 2005;294:1799–1809. doi: 10.1001/jama.294.14.1799. [DOI] [PubMed] [Google Scholar]
- 48.Sabater-Lleal M, Huang J, Chasman D, et al. Multiethnic meta-analysis of genome-wide association studies in >100 000 subjects identifies 23 fibrinogen-associated Loci but no strong evidence of a causal association between circulating fibrinogen and cardiovascular disease. Circulation. 2013;128:1310–1324. doi: 10.1161/CIRCULATIONAHA.113.002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wensley F, Gao P, et al. C Reactive Protein Coronary Heart Disease Genetics CollaborationAssociation between C reactive protein and coronary heart disease: Mendelian randomization analysis based on individual participant data. BMJ. 2011;342:d548. doi: 10.1136/bmj.d548. [DOI] [PMC free article] [PubMed] [Google Scholar]