

Abstract
Background
The 14-3-3 family of proteins is implicated in the regulation of several key neuronal processes. Previous human and animal studies have suggested an association between 14-3-3 dysregulation and schizophrenia.
Methods
We characterized the behavioral and functional changes in the transgenic mice that express an isoform-independent 14-3-3 inhibitor peptide in the brain.
Results
We have recently shown that the 14-3-3 functional knockout mice (FKO) exhibit impairments in associative learning and memory. Here, we report that these 14-3-3 FKO mice display other behavioral deficits which correspond to the core symptoms of schizophrenia. These behavioral deficits may be attributed to alterations in multiple neurotransmission systems in the 14-3-3 FKO mice. Particularly, inhibition of 14-3-3 proteins results in a reduction of dendritic complexity and spine density in forebrain excitatory neurons, which may underlie the altered synaptic connectivity in the prefrontal cortical synapse of the 14-3-3 FKO mice. At the molecular level, this dendritic spine defect may stem from dysregulated actin dynamics due to a disruption of the 14-3-3-dependent regulation of phosphorylated cofilin.
Conclusions
Collectively, our data provide a link between 14-3-3 dysfunction, synaptic alterations, and schizophrenia-associated behavioral deficits.
Keywords: 14-3-3 proteins, transgenic mouse model, schizophrenia, prefrontal cortex, neurotransmission, dendritic spines
INTRODUCTION
Schizophrenia is a complex neuropsychiatric disorder affecting approximately one percent of the population worldwide. While the etiology and pathophysiology of schizophrenia remain elusive, genetic risk factors are recognized as an important contributing factor to the pathogenesis of this neuropsychiatric disorder (1). Among them, multiple members of the 14-3-3 family of proteins may be candidate risk genes for schizophrenia (2–4), as supported by the following lines of evidence: 1) linkage analysis studies have identified single nucleotide polymorphisms (SNPs) of individual 14-3-3 isoforms in various schizophrenic populations (4–6); 2) Ywhah, encoding the 14-3-3η isoform, is located within the established 22q12-13 candidate risk chromosomal region (7); 3) postmortem studies have detected decreased neuronal expression of 14-3-3 at both the protein and mRNA levels in the brains of schizophrenic patients (8, 9); and 4) recent exome sequencing has revealed that de novo mutations of Ywhag and Ywhaz are among a group of postsynaptic proteins over represented in schizophrenic populations (10–12).
14-3-3 refers to a family of homologous proteins comprised of seven mammalian isoforms (β, γ, ε, η, ζ, σ, and τ/θ) (13). The 14-3-3 proteins exist as homo- or heterodimers, in which each monomer shares a similar helical structure and contains a concave amphipathic groove for binding to ligands with specific phosphoserine/threonine-containing motifs (14, 15). In the brain, 14-3-3 proteins are abundantly expressed, constituting nearly one percent of the total soluble proteins (16, 17). Certain 14-3-3 isoforms are particularly enriched at the synapse and implicated in the regulation of synaptic transmission and plasticity (18). Additionally, 14-3-3 proteins may play a functional role in other neuronal processes, including neuronal differentiation, migration and survival, neurite outgrowth and ion channel regulation (13, 19–27).
Animal models have previously been established to assess the functions of individual 14-3-3 isoforms in the nervous system. The first in vivo model was the mutant leonardo (leo), a gene encoding one (ζ) of the two 14-3-3 isoforms in Drosophila (22). In mice, deletion of the 14-3-3ε gene (Ywhae) results in severe hippocampal and cortical structural defects (28). More recently, Cheah et al. identified neurodevelopmental abnormalities in the hippocampus of 14-3-3ζ knockout mice (29). Interestingly, both 14-3-3ε deficient and 14-3-3ζ knockout mice exhibit certain behavioral phenotypes associated with schizophrenia (2, 29). However, these mouse models do not account for the functional redundancy of multiple 14-3-3 isoforms expressed in the brain.
Recently, we reported the successful creation of a new mouse model that addresses the collective role of the 14-3-3 family of proteins. These mice transgenically express a 14-3-3 peptide inhibitor that antagonizes the binding of 14-3-3 proteins to their endogenous partners in an isoform-independent manner, thereby disrupting 14-3-3 functions in the brain (30). Here, we show that these 14-3-3 functional knockout (FKO) mice exhibit a variety of behavioral deficits which are reminiscent of the core endophenotypes of established schizophrenia mouse models (31). Our analyses suggest that these behavioral changes in the 14-3-3 FKO mice may arise from schizophrenia-related alterations in neurochemistry and synaptic transmissions. Together, our data provide new evidence that inhibition of 14-3-3 proteins in the brain can lead to pathological changes associated with schizophrenia.
METHODS AND MATERIALS
14-3-3 functional knockout mice
Generation of transgenic 14-3-3 functional knockout (FKO) mice was previously described (10). Briefly, these transgenic mice express the YFP fused difopein (dimeric fourteen-three-three peptide inhibitor) using the Thy-1 promoter. Positive founder line mice were backcrossed to wildtype C57BL/6 mice for at least eight generations before being subjected to behavioral and electrophysiological analyses. All animal procedures were carried out in accordance with the guidelines for the Care and Use of Laboratory Animals of the Florida State University, and approved by the FSU Animal Care and Use Committee (ACUC). Additional details are available in the Supplementary Materials.
Behavioral Assays
Behavioral phenotyping was performed on age-matched adult male mice (8 to 50 weeks; mean ages 39 weeks for both FKO and WT littermates). All procedures were performed in a dimly lit testing room. Prior to testing, mice were habituated to the testing room for 2 h to ensure acclimation. Detailed procedures for locomotor activity, social interaction, working memory, and prepulse inhibition are available in the Supplementary Materials.
Antipsychotic Drug Effectiveness
Effectiveness of atypical (Clozapine, 3 mg/kg, Tocris) and typical (Haloperidol, 0.4 mg/kg, Sigma-Aldrich) antipsychotic drugs was assessed using the open field test. Intraperitoneal (I.P.) injections of clozapine, haloperidol or saline (0.9% NaCl) were delivered 30 min prior to testing. Locomotor activity was recorded over a 30 min period in the open field testing arena.
HPLC Analysis
Punch biopsies of prefrontal cortex (PFC) and striatum were obtained from the brains of 14-3-3 FKO mice and their age- and sex-matched WT littermates. After weighing, the samples were homogenized in ice-cold 0.2 N perchloric acid (HClO4), and centrifuged at 15,000g for 15 min at 4°C. The supernatant was then applied to a C18 reverse phase high-performance liquid chromatography (HPLC) column (Varian) connected to an ESA model 5200A electrochemical detector. Concentrations of dopamine (DA), DA metabolites and other monoamines were determined by comparing the respective peaks to that of the standard reagents (Sigma). HPLC analyses were carried out in the Neurochemistry Core Facility of Vanderbilt University by Dr. Ray Johnson.
Histological Analyses
Immunohistochemistry
Mice were anesthetized and transcardially perfused with 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4 (PBS). After an overnight postfixation in the same fixative at 4°C, the brains were then cut into 40 μm sections on a Vibratome (Leica Microsystems). To evaluate transgene expression in dopaminergic neurons, brain sections were immunostained with a rabbit monocolonal (RabMAb) anti-Tyrosine Hydroxlase (TH) antibody (Epitomics), followed by incubation with Alexa Fluor® 647 donkey anti-rabbit secondary antibodies (Invitrogen). The images were acquired on a Leica TCS SP2 SE laser scanning confocal microscope (Leica Microsystems).
Golgi stain
Golgi staining was performed using the FD Rapid GolgiStain™ Kit (FD NeuroTechnologies Inc.) according to the manufacturer’s instructions. All neurons selected for analysis satisfied the following criteria: (i) the cell body and processes were completely impregnated; (ii) the cell and processes were isolated from surrounding impregnated cells; (iii) the cell body was located in the hippocampus CA1 cell layer, cortex layer III, or cortex layer V (32).
Sholl Analysis
Dendritic morphology was assessed using Sholl Analysis for dendritic complexity. To evaluate the apical dendrite complexity, traced neurons from the cortex and hippocampus were assessed using Sholl analysis (33). Using ImageJ software, concentric circles, with an increasing radius of 5μm, were placed radiating out from the soma over the traced neurons. The number of intersections between the apical dendrite and each overlaid ring was scored.
Spine Density
For dendritic spine counts, 6–7 apical dendrites (at least 50μm in length) were traced from the hippocampus CA1 and cortex layer III and V at 100× magnification for each brain (4–6 animals per group). Traces were then analyzed using ImageJ to measure the length of the dendritic branch and count the total number of spines. The dendritic spine density was calculated as: [(Spines/Length)*10].
Whole-cell Recordings
Whole-cell recordings of spontaneous excitatory and inhibitory postsynaptic currents (sEPSC and sIPSC) and miniature excitatory and inhibitory postsynaptic currents (mEPSC and mIPSC) from medial prefrontal cortex (mPFC) layer V pyramidal neurons were carried out as previously described (34, 35). Additional details are available in the Supplementary Materials.
Western Blot
Mouse hippocampal lysates were prepared as previously described (10, 36). The lysates were sonicated and separated by SDS-PAGE and probed with specific antibodies. The relative amount of GAPDH (probed with anti-GAPDH, EnCor Biotechnology) was used as a loading control for quantification. Protein densities on western blots were then analyzed and quantified with ImageJ software. Additional details are available in the Supplementary Materials.
Statistical Analysis
All data are presented as means ± standard error of the mean (S.E.M.) and were assessed by one-way analysis of variance (ANOVA), two-way ANOVA, or two-tailed t-test for comparisons using Origin version 7 (Originlab, Northampton, MA). A value of *p < 0.05 was considered to be a statistically significant difference.
RESULTS
The 14-3-3 Functional Knockout Mice
To assess the collective functions of the 14-3-3 family of proteins in the brain, we recently generated transgenic mice that are considered to be a functional knockout of 14-3-3 proteins (14-3-3 FKO mice) (10). These 14-3-3 FKO mice transgenically express the YFP-fused difopein (dimeric fourteen-three-three peptide inhibitor) that inhibits the functions of 14-3-3 proteins in an isoform-independent manner, by competitively antagonizing 14-3-3 binding to its endogenous partners (30, 37). To circumvent the embryonic lethality associated with 14-3-3 dysfunction (28), transgene expression was driven by the neuronal specific Thy-1 promoter that initiates gene expression in the perinatal period (38). As expected, Thy-1 driven expression produced several founder lines which have a variety transgene expression patterns that are variable between the different founder lines, but preserved among the descendants of individual founders (Figure S1; Table S1) (39).
In this study, we utilized a battery of behavioral assays to assess the general activity, cognitive abilities, and social behaviors for several different founder lines. Among them, mice of the 132 founder line exhibited the most robust behavioral changes (Figure 1; Table S2). Compared with the other founder lines, the 132 founder line has relatively higher transgene expression in the forebrain, particularly in the cortex and hippocampus (Table S1; Figure S2). Therefore, further characterization was focused on the 132 founder line, which we refer to as the “14-3-3 FKO mice” in this report.
Figure 1. Behavioral and cognitive deficits in the 14-3-3 FKO mice.

(A) 14-3-3 FKO mice (N = 34) have increased distance traveled compared with their WT littermates (N = 22) in 30 min open field testing. (B) Compared with WT (N = 19), the 14-3-3 FKO mice (N = 29) have a reduction in their alternation percentage in Y-maze testing. (C) 14-3-3 FKO mice (N = 30) have decreased PPI percentage compared with WT mice (N = 23). (D) For sociability testing, WT (N = 7) mice spend more time in the chamber containing an unfamiliar mouse, but this preference is not observed in the 14-3-3 FKO mice (N = 11). (E) In subsequent social recognition testing, WT mice spend more time in the chamber with an unfamiliar mouse compared to the familiar mouse. However, the 14-3-3 FKO mice show no preference for any of the chambers. Data are presented as mean ± S.E.M. with statistical significance denoted as: ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001, two tailed t-test (open field), one-way ANOVA (Y-maze, PPI, social interaction).
14-3-3 FKO Mice Display Behavioral Deficits Associated with Schizophrenia
In open field testing, the 14-3-3 FKO mice exhibited a marked increase in locomotor activity compared with their wildtype littermates (WT) (Figure 1A), while no difference was observed in thigmotaxis (data not shown). Additionally, the 14-3-3 FKO mice showed no change in anxiety-like behaviors in light/dark box testing (Table S2). Thus, the hyperactive behaviors of the 14-3-3 FKO mice are not due to a heighted anxiolytic response, but may correspond to the psychotic behaviors present in rodent models of schizophrenia (31, 40).
Schizophrenia is associated with both general cognitive as well as working memory defects (41). Previously, we identified associative learning and memory impairments in the 14-3-3 FKO mice using contextual fear conditioning and passive avoidance tests (Table S2) (10). Here, we assessed the spatial working memory capabilities of the 14-3-3 FKO mice based on their continuous spontaneous alternation activity in the Y-maze (42). Compared with WT, the 14-3-3 FKO mice showed a statistically significant reduction in their alternation percentage during Y-maze testing (Figure 1B), signifying a deficit in working memory.
Sensorimotor gating control is altered in various neuropsychiatric disorders, particularly schizophrenia (43). To determine the sensorimotor gating capabilities of the 14-3-3 FKO mice, we measured their prepulse inhibition (PPI) of the acoustic startle response. Baseline measurements demonstrated that WT and 14-3-3 FKO mice show no difference in their startle amplitude response (Figure S3). However, during PPI testing, 14-3-3 FKO mice exhibited a severe deficit in their PPI percentages compared with WT for all prepulse tone levels tested (Figure 1C).
Next, we utilized a three-chamber social interaction test to assess the social behaviors of the 14-3-3 FKO mice, as social withdrawal is a symptom of schizophrenia (31, 44). During sociability testing, the WT mice preferred to spend more time in the chamber with an unfamiliar mouse, whereas the 14-3-3 FKO mice did not show this preference (Figure 1D). For social recognition testing, while the WT mice preferred the chamber containing a novel mouse, the 14-3-3 FKO mice did not show a clear preference for any chamber (Figure 1E). Based on these results, the 14-3-3 FKO mice exhibit reduced social interaction tendencies, which are characteristic of social withdrawal.
Collectively, this battery of behavioral tests showed that the 14-3-3 FKO mice display a variety of behavioral phenotypes which correspond to the core symptoms of schizophrenia.
14-3-3 FKO Mice Exhibit Hyperactive Dopamine Signaling
Augmented striatal dopamine (DA) transmission may underlie the psychotic symptoms observed in schizophrenic patients and hyperactivity behaviors in relevant mouse models (45). Therefore, we measured the levels of monoamines and their metabolites in both striatal and cortical tissues dissected from the 14-3-3 FKO and WT mice. Compared with WT, the 14-3-3 FKO mice had a higher DA level in the striatum (Figure 2A), but not in the cortex (Figure 2B). While 14-3-3 proteins have been implicated in regulating DA synthesis and transport (46), transgene expression in the 14-3-3 FKO mice was not detected within the dopaminergic neurons of the Ventral Tegmental Area (VTA) and the Substantia Nigra pars compacta (SNc) (Figure 2C), or their processes in the striatum (Figure 2D). Thus, the elevated striatal DA level of the 14-3-3 FKO mice is unlikely a direct consequence of 14-3-3 inhibition in DA producing neurons and their terminals.
Figure 2. Increased striatal dopamine level in the 14-3-3 FKO mice.

(A) HPLC analysis identified a specific increase in striatal DA concentration (ng/mg) in the 14-3-3 FKO (N = 7) mice compared with that of WT (N = 5). (B) In the cortex, no difference in monoamine or metabolite content was detected between WT (N = 4) and 14-3-3 FKO mice (N = 7). (C and D) In 14-3-3 FKO mice, transgene expression (green) does not colocalize with the TH-containing (red) DA neurons of the Substantia Nigra pars compacta (SNc) and Ventral Tegmental Area (VTA) (C), nor their projections to the striatum (D). Scale bar, 100 μm (C) or 50 μm (D). Data are presented as mean ± S.E.M., with statistical significance denoted by *p < 0.05, two-tailed t-test.
Next, we tested the effectiveness of DA receptor targeting drugs to ameliorate the observed psychotic-like behavior of the 14-3-3 FKO mice (Figure 1A). Administration of either atypical (clozapine) or typical (haloperidol) antipsychotics was able to attenuate the hyperactivity of the 14-3-3 FKO mice (Figure 3A). While the drugs had a sedentary effect on WT animals, they did restore activity levels of the 14-3-3 FKO mice to that of saline injected WT animals (Figure 3A). However, these antipsychotics were less effective in attenuating PPI deficits in 14-3-3 FKO mice (Figure 3B).
Figure 3. The effects of antipsychotic drugs on schizophrenia-related behavior deficits of the 14-3-3 FKO mice.

(A) Administration of clozapine (WT, N = 7; 14-3-3 FKO, N = 8) or haloperidol (WT, N = 6; 14-3-3 FKO, N = 8) attenuates the hyperactivity of the 14-3-3 FKO mice in open field testing. (B) Prepulse inhibition (PPI) of the acoustic startle response was measured in WT and 14-3-3 FKO mice after administration of saline (WT, n = 7; 14-3-3 FKO, n = 9), clozapine (WT, n = 7; 14-3-3 FKO, n = 13) or haloperidol (WT, n = 6; 14-3-3 FKO, n = 12). In the 14-3-3 FKO mice, haloperidol increased PPI at 67, 70 and 73 dB prepulse tones to similar PPI percentages of saline injected WT mice. Clozapine did not alter PPI at any prepulse tone levels in the 14-3-3 FKO mice compared with saline injected 14-3-3 FKO mice. Data are presented as mean ± S.E.M., with statistical significance denoted by: *p < 0.05, ***p < 0.001, comparisons between genotypes and drug administration groups were performed using two-way ANOVA.
14-3-3 FKO Mice Show Deficits in Cortical Neurotransmissions
Disturbances in cortical neural circuits may lead to various functional deficits in schizophrenia and other neuropsychiatric disorders (47, 48). In the 14-3-3 FKO mice, the YFP-fused 14-3-3 inhibitor peptide is primarily expressed in the excitatory pyramidal neurons at deep layers of the cortex (Figure S2). Using whole-cell voltage clamp recordings, we first measured spontaneous synaptic activities in layer V pyramidal neurons of the medial prefrontal cortex (mPFC) in WT and 14-3-3 FKO mice (Figures 4A–D). To assess the effects of transgene expression on synaptic activity, we recorded from both transgene expressing (14-3-3FKO-green) and non-transgene expressing (14-3-3FKO-nongreen) neurons in the 14-3-3 FKO mice. In the transgene expressing neurons of the 14-3-3 FKO mice, there was a significant reduction in the frequencies of both spontaneous excitatory and inhibitory postsynaptic currents (sEPSCs and sIPSCs) (Figures 4A–D). Additionally, we observed alterations in miniature excitatory and inhibitory postsynaptic currents (mEPSCs and mIPSCs) in the 14-3-3 FKO mice (Figures 4E–J). For mEPSCs, the average amplitude was smaller in the 14-3-3 FKO transgene expressing neurons compared with that of WT, but no changes in mEPSC frequency were observed (Figures 4E–G). On the other hand, mIPSC recordings revealed a lower frequency but similar amplitudes in the transgene expressing neurons of the 14-3-3 FKO mice relative to WT (Figures 4H–J). As there was no difference in IPSCs and EPSCs between WT and the non-transgene expressing neurons of 14-3-3 FKO mice, the observed changes in synaptic transmissions are likely caused by expression of the 14-3-3 inhibitor in excitatory pyramidal neurons.
Figure 4. Synaptic transmission defects in the mPFC of the 14-3-3 FKO mice.

(A) Representative traces of sEPSCs recorded from mPFC layer V pyramidal neurons. (B) The frequency of sEPSC is lower in transgene expressing neurons (14-3-3 FKO-green, N=7) compared with that of the WT neurons (N=10). No significant difference in sEPSC frequency was observed between the non-transgene expressing (14-3-3 FKO-nongreen, N=8) and WT neurons. (C) Representative traces of sIPSCs in mPFC layer V pyramidal neurons. (D) There is also a decrease in sIPSC frequency in the 14-3-3 FKO-green neurons (N=14), but not 14-3-3 FKO-nongreen (N=12), compared with that of WT neurons (N=6). (E) Representative traces of mEPSCs in mPFC layer V pyramidal neurons. (F–G) Compared with WT (N=7) and 14-3-3-nongreen (N=6), the 14-3-3 FKO-green neurons (N=12) have a reduction in mEPSC amplitude (F) with no change in frequency (G). (H) Representative traces of mIPSCs in the mPFC layer V pyramidal neurons. (I and J) Compared with WT (N=9) and 14-3-3FKO-nongreen neurons (N=7), 14-3-3 FKO-green neurons (N=10) exhibit normal mIPSC amplitude (I), but decreased frequency of mIPSCs (J). ‘N’ represents the number of neurons, and 1–2 neurons were recorded from each animal. Data are presented as mean ± S.E.M. with statistical significance denoted as: *p < 0.05; **p < 0.01, one-way ANOVA.
14-3-3 FKO Mice Exhibit Dendritic Morphological Alterations
To further investigate the impact of transgene expression on the excitatory neurons of the 14-3-3 FKO mice, we characterized the dendritic morphology of pyramidal neurons from the cortex layer V and hippocampus CA1, where the transgenes are extensively expressed (Figure S2). Utilizing Sholl analysis, we identified a specific reduction in distal apical dendritic complexity of both cortical layer V and hippocampal CA1 pyramidal neurons in the 14-3-3 FKO mice (Figures 5A–B, Figure S4). These findings are in line with previous observations of decreased dendritic complexity in both patients and established animal models of schizophrenia (49).
Figure 5. Dendritic morphology and spine density alterations in the 14-3-3 FKO mice.
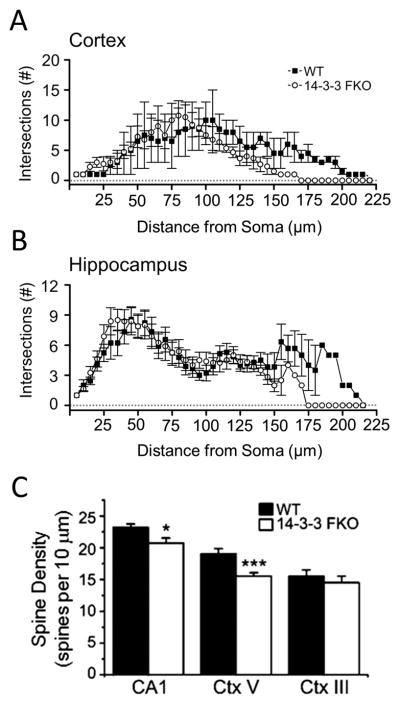
(AB) Sholl analysis revealed a reduction in distal dendritic complexity of apical dendrites from cortical layer V (A; WT, N = 9; 14-3-3 FKO, N = 8) and hippocampal CA1 (B; WT, N = 4; 14-3-3 FKO, N = 4) pyramidal neurons in 14-3-3 FKO mice. (C) In 14-3-3 FKO mice, spine density was reduced in hippocampal CA1 and cortical layer V neurons, but not in cortical layer III neurons, compared with that of WT. For spine counts, 6–7 neurons were analyzed per mouse brain (WT, N = 4; 14-3-3 FKO; N = 4–6) within each brain region. Data are presented as mean ± S.E.M., with statistical significance denoted as: *p < 0.05; ***p < 0.001, one-way ANOVA.
Next, we asked whether transgene expression also leads to alterations in dendritic spine density, another defect associated with schizophrenia pathology (50). Compared with WT, the 14-3-3 FKO mice had lower dendritic spine densities in pyramidal neurons of the cortex layer V and hippocampus CA1 (Figure 5C, Figure S4). In contrast, we found no difference in dendritic spine density between WT and 14-3-3 FKO mice in the cortical layer III pyramidal neurons (Figure 4C, Figure S4), in which the transgenes are minimally expressed (Figure S2). This suggests that the reduction in spine density may be caused by expression of the 14-3-3 inhibitor.
Inhibition of 14-3-3 May Alter Actin Dynamics
To explore the underlying molecular mechanism for the dendritic morphological changes in the 14-3-3 FKO mice, we examined key signaling proteins implicated in the regulation of actin dynamics, which is important for synaptogenesis, spine formation and maintenance. Compared with WT, the 14-3-3 FKO mice had a significantly reduced level of phospho-cofilin in their brain tissues (Figure 6). Cofilin is an actin depolymerizing factor whose phosphorylation may be regulated by 14-3-3 through multiple mechanisms (51, 52). Based on our studies in the heterologous expression system, the effect of 14-3-3 is unlikely mediated by the interaction between 14-3-3 and p-cofilin (Figures S6 & S7). However, we found an increase in the levels of δ-catenin in the 14-3-3 FKO mice (Figure 6). This is consistent with a recent study conducted in the neural progenitor cells of 14-3-3 deficient mice and may provide a potential molecular mechanism for 14-3-3-dependent regulation of actin dynamics and dendritic spines in post-mitotic neurons (53).
Figure 6. Reduction of phosphorylated cofilin in the 14-3-3 FKO mice.

(A) Representative images of western blots from hippocampal lysates of WT and 14-3-3 FKO mice, immunoblotted for cofilin, phosphorylated cofilin (P-cofilin), or δ-catenin. GAPDH was probed as a loading control. (B) There is a specific decrease in the level of phospho-cofilin and an increase in the level of δ-catenin (p = 0.07) in 14-3-3 FKO mice compared with that of WT. For WT (N = 5) and 14-3-3 FKO (N = 6), each sample is a combined pool of tissue from 2–3 animals. Data are presented as mean ± S.E.M., with statistical significance denoted by: *p < 0.05, two-tailed t-test.
DISCUSSION
In this study, we demonstrate that neuronal expression of the 14-3-3 inhibitor peptide leads to a variety of behavioral and cognitive deficits in transgenic mice. The behavioral changes of the 14-3-3 FKO mice are reminiscent of the endophenotypes observed in established schizophrenia mouse models (31). These phenotypes include hyperactivity, social withdrawal, impaired learning and memory, working memory deficit and reduced PPI. Interestingly, among several different founder lines, the behavior alterations are most apparent in the line (#132) with a high level of transgene expression in the cortex and hippocampus, which are the two brain regions most affected in schizophrenia (54). Moreover, administration of antipsychotic drugs attenuates the hyperactive behavior of the 14-3-3 FKO mice. Together, these observations suggest that the 14-3-3 FKO mice may be a new mouse model in which to examine pathophysiologies related to schizophrenia.
Certain schizophrenia-associated behavioral phenotypes have previously been reported in the 14-3-3ε deficient and 14-3-3ζ knockout mice (2, 29). However, the 14-3-3 FKO mice exhibit broader and more severe behavioral deficits compared to these previous models. The differences in range and severity of behavior defects could be attributed to the fact that the 14-3-3 inhibitor peptide antagonizes all 14-3-3 isoforms, thereby eliminating the functional redundancy of multiple 14-3-3 proteins. Moreover, as transgene expression in the 14-3-3 FKO mice is delayed until the perinatal period (38), our study also presents new evidence that disruption of 14-3-3 proteins postnatally can lead to behavior deficits related to schizophrenia. This postnatal transgene expression may also explain why the 14-3-3 FKO mice do not exhibit gross neuroanatomical defects in hippocampal cellular migration and organization (data not shown), which are observed in both 14-3-3ε and 14-3-3ζ deficient mice (28, 29).
Alterations in several different neurotransmitter systems have been linked to the pathogenesis of a variety of neuropsychiatric disorders. Among them, the psychotic symptoms in schizophrenia are thought to arise from hyperactivity of the mesolimbic dopaminergic system (45, 54). In line with the dopamine hypothesis of schizophrenia, we identify a significant increase in the striatal DA content of the 14-3-3 FKO mice. Moreover, the antipsychotic drugs attenuate the heightened locomotor activity of the 14-3-3 FKO mice, likely through antagonizing DA receptors. Recently, similar findings were reported for the 14-3-3ζ knockout mice, including increased striatal DA content and antipsychotic drug effectiveness on locomotor hyperactivity (55). Additionally, the 14-3-3ζ mice were shown to have a reduced level of the DA transporter (DAT), suggesting a direct role for 14-3-3ζ in regulating DA neurotransmission (46). However, in the 14-3-3 FKO mice, transgene expression was not detected in the dopaminergic neurons. Therefore, this increase in striatal DA in the 14-3-3 FKO mice is not likely a direct consequence of 14-3-3 inhibition in dopaminergic neurons. Rather, we propose that expression of 14-3-3 inhibitor peptide in other brain regions, such as the cortex and/or hippocampus, may indirectly alter the regulation of the DA system (45, 56, 57).
In the mPFC, we carried out electrophysiological analyses to examine the potential changes in glutamatergic and GABAergic neurotransmission systems of the 14-3-3 FKO mice. Compared with the WT mice, the 14-3-3 FKO mice show a reduction in the frequency of sEPSCs and sIPSCs recorded from layer V pyramidal neurons. This finding reveals altered neuronal activity in the cortical circuit which could potentially stem from deficits in excitatory or inhibitory neurons of 14-3-3 FKO mice. However, transgene expression is mainly observed in excitatory neurons (Figure. S4A) and there is no reduction in the number of parvalbumin (PV)-containing interneurons in the PFC of the 14-3-3 FKO mice (Figure. S4B). Thus, the changes in cortical network activity are likely due to alterations in excitatory neurotransmission. Consistent with this hypothesis, we identify a transgene-induced difference in synaptic properties of the excitatory neurons in the 14-3-3 FKO mice. Specifically, we found that expression of the 14-3-3 inhibitor in pyramidal neurons leads to reduced amplitude of miniature excitatory postsynaptic currents, which is suggestive of a reduction in the number of synapses and/or functional postsynaptic glutamate receptors (58).
Through morphological analyses, we identified a decrease in dendritic complexity and spine density in cortical and hippocampal pyramidal neurons of the 14-3-3 FKO mice. These observations are in line with previous studies showing a reduction in dendritic complexity and spine density in neurons of both schizophrenic patients and animal models (50, 59). In fact, dendritic deficits have been linked to functional hypoactivity in the brain, which may serve as a potential pathogenic mechanism for schizophrenia (59). Based on our data, we propose that inhibition of 14-3-3 alters dendritic morphology, resulting in a reduction in the number of excitatory synapses in forebrain neurons. This hypothesis presents a framework for understanding how disruption of 14-3-3 in excitatory neurons may perturb synaptic transmission in key forebrain regions, thereby altering behaviors and cognition in the 14-3-3 FKO mice.
While 14-3-3 proteins are known to regulate neuronal migration and axonal outgrowth (13, 26, 29, 60), their involvement in synapse formation and spine maintenance has not been fully investigated. Our study provides the first in vivo evidence of 14-3-3’s role in dendritic spine regulation. Previously, 14-3-3 proteins have been proposed as important regulators of cytoskeleton and actin dynamics, which are critical for synaptogenesis as well as the regulation of the shape, organization and maintenance of dendritic spines (61). Here, we identify a reduction in the level of phosphorylated cofilin in the 14-3-3 FKO mice. Based on our analyses, 14-3-3 does not appear to alter p-cofilin through direct protein-protein interaction, but rather indirectly regulates the level of p-cofilin through the δ-catenin signaling pathway.
In a recent report, we show that the 14-3-3 FKO mice exhibit impairments in associative learning and memory, as well as a deficit in long-term synaptic plasticity of hippocampal synapses (10). These behavioral and synaptic changes are correlated with a reduction in the synaptic N-methyl-D-aspartate (NMDA) receptors in the hippocampus (10). Given that NMDA hypofunction is recognized as one of the pathological mechanisms for schizophrenia (62, 63), this NMDA receptor deficit may also be a contributing factor to the cortical synaptic alterations and other schizophrenia-related behavior changes observed in this study. Moreover, deficits in NMDA receptors are known to result in a decrease of dendritic spines (64), thus, it is possible that the transgene-induced NMDA reduction may also contribute to the dendritic spine defect observed in the 14-3-3 FKO mice (10, 64).
In summary, our study demonstrates that disruption of 14-3-3 functions in key brain regions leads to behavioral abnormalities related to schizophrenia. The 14-3-3 FKO mice support a model in which these behavioral deficits result from alterations in forebrain synaptic connectivity induced by perinatal expression of the 14-3-3 inhibitor peptide. This is an important advance from previous studies that have primarily focused on the role of 14-3-3 proteins in neuronal development during the prenatal period. As 14-3-3 proteins are involved various molecular pathways linked to schizophrenia (65, 66), continued studies of the 14-3-3 FKO mice may facilitate our understanding of the disrupted signaling pathways underlying schizophrenia pathogenesis.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Health Grant NS50355 to Y.Z. We thank Ruth Didier of the Confocal Microscopy Core Laboratory at the FSU Biomedical Sciences Department for her assistance with imaging. We thank Dr. Charles Ouimet at the FSU Biomedical Sciences Department for his expertise and assistance with golgi staining and dendritic morphological experiments. We thank Dr. Pradeep Bhide at the FSU Biomedical Sciences Department for graciously allowing us to use his StereoInvestigator system. We thank Dr. Ray Johnson at the Neurochemistry Core facility of Vanderbilt University for his assistance with HPLC analyses.
Footnotes
FINANCIAL DISCLOSURES
All authors report no biomedical financial interests or potential conflicts of interest.
Author contributions: MF, HQ, KG, YW and YZ designed research; MF, HQ, KG and YW performed research; MF, HQ and KG analyzed data; MF, KG and YZ wrote the paper.
References
- 1.van Os J, Kapur S. Schizophrenia. Lancet. 2009;374:635–645. doi: 10.1016/S0140-6736(09)60995-8. [DOI] [PubMed] [Google Scholar]
- 2.Ikeda M, Hikita T, Taya S, Uraguchi-Asaki J, Toyo-oka K, Wynshaw-Boris A, et al. Identification of YWHAE, a gene encoding 14-3-3epsilon, as a possible susceptibility gene for schizophrenia. Human molecular genetics. 2008;17:3212–3222. doi: 10.1093/hmg/ddn217. [DOI] [PubMed] [Google Scholar]
- 3.Jia Y, Yu X, Zhang B, Yuan Y, Xu Q, Shen Y. An association study between polymorphisms in three genes of 14-3-3 (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein) family and paranoid schizophrenia in northern Chinese population. European psychiatry : the journal of the Association of European Psychiatrists. France. 2004:377–379. doi: 10.1016/j.eurpsy.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Wong AH, Likhodi O, Trakalo J, Yusuf M, Sinha A, Pato CN, et al. Genetic and post-mortem mRNA analysis of the 14-3-3 genes that encode phosphoserine/threonine-binding regulatory proteins in schizophrenia and bipolar disorder. Schizophr Res Netherlands. 2005:137–146. doi: 10.1016/j.schres.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 5.Bell R, Munro J, Russ C, Powell JF, Bruinvels A, Kerwin RW, et al. Systematic screening of the 14-3-3 eta (eta) chain gene for polymorphic variants and case-control analysis in schizophrenia. Am J Med Genet. United States: 2000 Wiley-Liss, Inc. 2000:736–743. [PubMed] [Google Scholar]
- 6.Toyooka K, Muratake T, Tanaka T, Igarashi S, Watanabe H, Takeuchi H, et al. 14-3-3 protein eta chain gene (YWHAH) polymorphism and its genetic association with schizophrenia. American journal of medical genetics. United States. 1999:164–167. [PubMed] [Google Scholar]
- 7.Muratake T, Hayashi S, Ichikawa T, Kumanishi T, Ichimura Y, Kuwano R, et al. Structural organization and chromosomal assignment of the human 14-3-3 eta chain gene (YWHAH) Genomics. United States. 1996:63–69. doi: 10.1006/geno.1996.0426. [DOI] [PubMed] [Google Scholar]
- 8.Middleton FA, Peng L, Lewis DA, Levitt P, Mirnics K. Altered expression of 14-3-3 genes in the prefrontal cortex of subjects with schizophrenia. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2005;30:974–983. doi: 10.1038/sj.npp.1300674. [DOI] [PubMed] [Google Scholar]
- 9.Vawter MP, Barrett T, Cheadle C, Sokolov BP, Wood WH, 3rd, Donovan DM, et al. Application of cDNA microarrays to examine gene expression differences in schizophrenia. Brain research bulletin. United States. 2001:641–650. doi: 10.1016/s0361-9230(01)00522-6. [DOI] [PubMed] [Google Scholar]
- 10.Qiao H, Foote M, Graham K, Wu Y, Zhou Y. 14-3-3 proteins are required for hippocampal long-term potentiation and associative learning and memory. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:4801–4808. doi: 10.1523/JNEUROSCI.4393-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Molecular psychiatry. 2012;17:142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berg D, Holzmann C, Riess O. 14-3-3 proteins in the nervous system. Nature reviews Neuroscience. 2003;4:752–762. doi: 10.1038/nrn1197. [DOI] [PubMed] [Google Scholar]
- 14.Liu D, Bienkowska J, Petosa C, Collier RJ, Fu H, Liddington R. Crystal structure of the zeta isoform of the 14-3-3 protein. Nature. 1995;376:191–194. doi: 10.1038/376191a0. [DOI] [PubMed] [Google Scholar]
- 15.Rittinger K, Budman J, Xu J, Volinia S, Cantley LC, Smerdon SJ, et al. Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Molecular cell. 1999;4:153–166. doi: 10.1016/s1097-2765(00)80363-9. [DOI] [PubMed] [Google Scholar]
- 16.Martin H, Rostas J, Patel Y, Aitken A. Subcellular localisation of 14-3-3 isoforms in rat brain using specific antibodies. J Neurochem. 1994;63:2259–2265. doi: 10.1046/j.1471-4159.1994.63062259.x. [DOI] [PubMed] [Google Scholar]
- 17.Baxter HC, Liu WG, Forster JL, Aitken A, Fraser JR. Immunolocalisation of 14-3-3 isoforms in normal and scrapie-infected murine brain. Neuroscience. 2002;109:5–14. doi: 10.1016/s0306-4522(01)00492-4. [DOI] [PubMed] [Google Scholar]
- 18.Broadie K, Rushton E, Skoulakis EM, Davis RL. Leonardo, a Drosophila 14-3-3 protein involved in learning, regulates presynaptic function. Neuron. 1997;19:391–402. doi: 10.1016/s0896-6273(00)80948-4. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Wu Y, Zhou Y. Modulation of inactivation properties of CaV2.2 channels by 14-3-3 proteins. Neuron. 2006;51:755–771. doi: 10.1016/j.neuron.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Y, Schopperle WM, Murrey H, Jaramillo A, Dagan D, Griffith LC, et al. A dynamically regulated 14-3-3, Slob, and Slowpoke potassium channel complex in Drosophila presynaptic nerve terminals. Neuron. 1999;22:809–818. doi: 10.1016/s0896-6273(00)80739-4. [DOI] [PubMed] [Google Scholar]
- 21.Fu H, Subramanian RR, Masters SC. 14-3-3 proteins: structure, function, and regulation. Annual review of pharmacology and toxicology. 2000;40:617–647. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- 22.Skoulakis EM, Davis RL. Olfactory learning deficits in mutants for leonardo, a Drosophila gene encoding a 14-3-3 protein. Neuron. 1996;17:931–944. doi: 10.1016/s0896-6273(00)80224-x. [DOI] [PubMed] [Google Scholar]
- 23.Roncada P, Bortolato M, Frau R, Saba P, Flore G, Soggiu A, et al. Gating deficits in isolation-reared rats are correlated with alterations in protein expression in nucleus accumbens. J Neurochem. 2009;108:611–620. doi: 10.1111/j.1471-4159.2008.05806.x. [DOI] [PubMed] [Google Scholar]
- 24.Taya S, Shinoda T, Tsuboi D, Asaki J, Nagai K, Hikita T, et al. DISC1 regulates the transport of the NUDEL/LIS1/14-3-3epsilon complex through kinesin-1. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:15–26. doi: 10.1523/JNEUROSCI.3826-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skoulakis EM, Davis RL. 14-3-3 proteins in neuronal development and function. Molecular neurobiology. 1998;16:269–284. doi: 10.1007/BF02741386. [DOI] [PubMed] [Google Scholar]
- 26.Yam PT, Kent CB, Morin S, Farmer WT, Alchini R, Lepelletier L, et al. 14-3-3 proteins regulate a cell-intrinsic switch from sonic hedgehog-mediated commissural axon attraction to repulsion after midline crossing. Neuron. 2012;76:735–749. doi: 10.1016/j.neuron.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 27.Yacoubian TA, Slone SR, Harrington AJ, Hamamichi S, Schieltz JM, Caldwell KA, et al. Differential neuroprotective effects of 14-3-3 proteins in models of Parkinson’s disease. Cell death & disease. 2010;1:e2. doi: 10.1038/cddis.2009.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toyo-oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL, et al. 14-3-3epsilon is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat Genet. 2003;34:274–285. doi: 10.1038/ng1169. [DOI] [PubMed] [Google Scholar]
- 29.Cheah PS, Ramshaw HS, Thomas PQ, Toyo-Oka K, Xu X, Martin S, et al. Neurodevelopmental and neuropsychiatric behaviour defects arise from 14-3-3zeta deficiency. Mol Psychiatry. 2012;17:451–466. doi: 10.1038/mp.2011.158. [DOI] [PubMed] [Google Scholar]
- 30.Masters SC, Fu H. 14-3-3 proteins mediate an essential anti-apoptotic signal. The Journal of biological chemistry. 2001;276:45193–45200. doi: 10.1074/jbc.M105971200. [DOI] [PubMed] [Google Scholar]
- 31.Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nature neuroscience. 2010;13:1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koyama Y, Hattori T, Shimizu S, Taniguchi M, Yamada K, Takamura H, et al. DBZ (DISC1-binding zinc finger protein)-deficient mice display abnormalities in basket cells in the somatosensory cortices. J Chem Neuroanat. 2013;53:1–10. doi: 10.1016/j.jchemneu.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 33.Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- 34.Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li YC, Kellendonk C, Simpson EH, Kandel ER, Gao WJ. D2 receptor overexpression in the striatum leads to a deficit in inhibitory transmission and dopamine sensitivity in mouse prefrontal cortex. Proc Natl Acad Sci U S A. 2011;108:12107–12112. doi: 10.1073/pnas.1109718108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hallett PJ, Collins TL, Standaert DG, Dunah AW. Biochemical fractionation of brain tissue for studies of receptor distribution and trafficking. Curr Protoc Neurosci. 2008;Chapter 1(Unit 1.16) doi: 10.1002/0471142301.ns0116s42. [DOI] [PubMed] [Google Scholar]
- 37.Wang B, Yang H, Liu YC, Jelinek T, Zhang L, Ruoslahti E, et al. Isolation of high-affinity peptide antagonists of 14-3-3 proteins by phage display. Biochemistry. 1999;38:12499–12504. doi: 10.1021/bi991353h. [DOI] [PubMed] [Google Scholar]
- 38.Caroni P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods. Netherlands. 1997:3–9. doi: 10.1016/s0165-0270(96)00121-5. [DOI] [PubMed] [Google Scholar]
- 39.Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. United States. 2000:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 40.Deutsch SI, Hitri A. Measurement of an explosive behavior in the mouse, induced by MK-801, a PCP analogue. Clin Neuropharmacol. 1993;16:251–257. doi: 10.1097/00002826-199306000-00009. [DOI] [PubMed] [Google Scholar]
- 41.Park S, Holzman PS. Schizophrenics show spatial working memory deficits. Arch Gen Psychiatry. 1992;49:975–982. doi: 10.1001/archpsyc.1992.01820120063009. [DOI] [PubMed] [Google Scholar]
- 42.Deacon RM, Rawlins JN. T-maze alternation in the rodent. Nat Protoc. England. 2006:7–12. doi: 10.1038/nprot.2006.2. [DOI] [PubMed] [Google Scholar]
- 43.Powell SB, Weber M, Geyer MA. Genetic models of sensorimotor gating: relevance to neuropsychiatric disorders. Curr Top Behav Neurosci. 2012;12:251–318. doi: 10.1007/7854_2011_195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, et al. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. England. 2004:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- 45.Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophrenia bulletin. 2009;35:549–562. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramshaw H, Xu X, Jaehne EJ, McCarthy P, Greenberg Z, Saleh E, et al. Locomotor hyperactivity in 14-3-3zeta KO mice is associated with dopamine transporter dysfunction. Translational psychiatry. 2013;3:e327. doi: 10.1038/tp.2013.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kehrer C, Maziashvili N, Dugladze T, Gloveli T. Altered Excitatory-Inhibitory Balance in the NMDA-Hypofunction Model of Schizophrenia. Front Mol Neurosci. 2008;1:6. doi: 10.3389/neuro.02.006.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Volk DW, Lewis DA. Prefrontal cortical circuits in schizophrenia. Curr Top Behav Neurosci. 2010;4:485–508. doi: 10.1007/7854_2010_44. [DOI] [PubMed] [Google Scholar]
- 49.Kulkarni VA, Firestein BL. The dendritic tree and brain disorders. Mol Cell Neurosci. 2012;50:10–20. doi: 10.1016/j.mcn.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 50.Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nature neuroscience. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gohla A, Bokoch GM. 14-3-3 regulates actin dynamics by stabilizing phosphorylated cofilin. Current biology : CB. 2002;12:1704–1710. doi: 10.1016/s0960-9822(02)01184-3. [DOI] [PubMed] [Google Scholar]
- 52.Soosairajah J, Maiti S, Wiggan O, Sarmiere P, Moussi N, Sarcevic B, et al. Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. The EMBO journal. 2005;24:473–486. doi: 10.1038/sj.emboj.7600543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toyo-Oka K, Wachi T, Hunt RF, Baraban SC, Taya S, Ramshaw H, et al. 14-3-3epsilon and zeta Regulate Neurogenesis and Differentiation of Neuronal Progenitor Cells in the Developing Brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:12168–12181. doi: 10.1523/JNEUROSCI.2513-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keshavan MS, Tandon R, Boutros NN, Nasrallah HA. Schizophrenia, “just the facts”: what we know in 2008 Part 3: neurobiology. Schizophr Res. 2008;106:89–107. doi: 10.1016/j.schres.2008.07.020. [DOI] [PubMed] [Google Scholar]
- 55.Wang J, Lou H, Pedersen CJ, Smith AD, Perez RG. 14-3-3zeta contributes to tyrosine hydroxylase activity in MN9D cells: localization of dopamine regulatory proteins to mitochondria. J Biol Chem. United States. 2009:14011–14019. doi: 10.1074/jbc.M901310200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lodge DJ, Grace AA. Hippocampal dysfunction and disruption of dopamine system regulation in an animal model of schizophrenia. Neurotoxicity research. 2008;14:97–104. doi: 10.1007/BF03033801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends in pharmacological sciences. 2011;32:507–513. doi: 10.1016/j.tips.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- 59.Glausier JR, Lewis DA. Dendritic spine pathology in schizophrenia. Neuroscience. 2013;251:90–107. doi: 10.1016/j.neuroscience.2012.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kent CB, Shimada T, Ferraro GB, Ritter B, Yam PT, McPherson PS, et al. 14-3-3 proteins regulate protein kinase a activity to modulate growth cone turning responses. J Neurosci. 2010;30:14059–14067. doi: 10.1523/JNEUROSCI.3883-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koleske AJ. Molecular mechanisms of dendrite stability. Nat Rev Neurosci. 2013;14:536–550. doi: 10.1038/nrn3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2012;37:4–15. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- 64.Ramsey AJ, Milenkovic M, Oliveira AF, Escobedo-Lozoya Y, Seshadri S, Salahpour A, et al. Impaired NMDA receptor transmission alters striatal synapses and DISC1 protein in an age-dependent manner. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5795–5800. doi: 10.1073/pnas.1012621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun J, Jia P, Fanous AH, van den Oord E, Chen X, Riley BP, et al. Schizophrenia gene networks and pathways and their applications for novel candidate gene selection. PLoS One. 2010;5:e11351. doi: 10.1371/journal.pone.0011351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Foote M, Zhou Y. 14-3-3 proteins in neurological disorders. International journal of biochemistry and molecular biology. 2012;3:152–164. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.