

Abstract
Objective
To evaluate extended dosing intervals (EDIs) with lanreotide Autogel 120 mg in patients with acromegaly previously biochemically controlled with octreotide LAR 10 or 20 mg.
Design and methods
Patients with acromegaly had received octreotide LAR 10 or 20 mg/4 weeks for ≥6 months and had normal IGF1 levels. Lanreotide Autogel 120 mg was administered every 6 weeks for 24 weeks (phase 1); depending on week-24 IGF1 levels, treatment was then administered every 4, 6 or 8 weeks for a further 24 weeks (phase 2). Hormone levels, patient-reported outcomes and adverse events were assessed. Primary endpoint: proportion of patients on 6- or 8-week EDIs with normal IGF1 levels at week 48 (study end).
Results
107/124 patients completed the study (15 withdrew from phase 1 and two from phase 2). Of 124 patients enrolled, 77.4% were allocated to 6- or 8-week EDIs in phase 2 and 75.8% (95% CI: 68.3–83.3) had normal IGF1 levels at week 48 with the EDI (primary analysis). A total of 88.7% (83.1–94.3) had normal IGF1 levels after 24 weeks with 6-weekly dosing. GH levels were ≤2.5 μg/l in >90% of patients after 24 and 48 weeks. Patient preferences for lanreotide Autogel 120 mg every 4, 6 or 8 weeks over octreotide LAR every 4 weeks were high.
Conclusions
Patients with acromegaly achieving biochemical control with octreotide LAR 10 or 20 mg/4 weeks are possible candidates for lanreotide Autogel 120 mg EDIs. EDIs are effective and well received among such patients.
Introduction
Acromegaly is an uncommon disease that is almost always due to a benign pituitary tumour and which is characterized by increased levels of growth hormone (GH) and insulin-like growth factor-1 (IGF1). The hormonal imbalance leads to multiple significant comorbidities – particularly cardiovascular complications, diabetes mellitus, bone and joint disease and sleep apnoea (1) – and an increased mortality rate (2). Medical therapy, one approach for the treatment of acromegaly, is usually recommended for patients for whom pituitary surgery was not curative or for whom a surgical approach was contraindicated or refused (1). For these patients, the first-line medical treatment is generally a long-acting somatostatin analogue (3, 4), such as lanreotide Autogel (known as lanreotide Depot in the USA), which has been shown to reduce GH and IGF1 excesses and pituitary tumour volume, ameliorate symptoms and improve health-related quality of life (HRQoL) (5, 6, 7). It has also been suggested in a meta-analysis that mortality rates are reduced by high biochemical remission rates and the use of somatostatin analogues (8).
As chronic therapy will be required for most patients receiving somatostatin analogues, it is important to consider how to optimize treatment acceptability. Some patients may be able to reduce the burden of frequent clinic visits by taking advantage of the pre-filled ready-to-use syringes available with lanreotide Autogel and administer treatment by self- or partner injection (where home administration is approved, such as in Europe) (9, 10, 11). The ready-to-use mode of administration for lanreotide can, when provided by healthcare practitioners (HCPs; as in the USA), provide time savings for improving other aspects of patient care (12). The burden of frequent clinic visits could also be reduced if, as advocated by the Acromegaly Consensus Group, individuals achieving good biochemical control with long-acting somatostatin analogues received treatment less frequently (3). Pharmacologically effective levels of lanreotide are sustained beyond the standard 4-week dosing interval (13), and initial studies indicate that some patients may thus be able to switch to 6- or 8-weekly dosing intervals without loss of efficacy (14, 15). While benefiting patients, both self-/partner or HCP injections together with extended dosing intervals (EDIs) also have the potential to reduce the economic burden of the disease. Currently, lanreotide Autogel is the only long-acting somatostatin analogue to have been approved for use with EDIs (both in Europe and the USA).
The Lanreotide Extended Autogel Duration (LEAD) study was a large international clinical trial evaluating the efficacy, safety and patient-reported outcomes (PROs) of 6- and 8-week EDIs with lanreotide Autogel 120 mg in patients with acromegaly previously biochemically controlled with 10 or 20 mg of octreotide LAR every 4 weeks.
Patients and methods
Patients
Patients eligible for the study were adults (aged >18 years) who had been diagnosed with acromegaly on the basis of elevated IGF1 and/or GH levels. Patients had additionally received octreotide LAR 10 or 20 mg every 4 weeks for ≥6 months previously and achieved normal (age- and sex-adjusted) IGF1 levels on two consecutive occasions (separated by an interval of ≥2 months). For patients receiving concomitant dopamine agonist therapy, treatment was stable for ≥4 months, with no change in dopamine-agonist medication expected during the study.
Patients were excluded from the study if they had received any previous pituitary radiation therapy, they were likely to require pituitary surgery or radiation therapy during the study, or they had received GH receptor antagonist treatment within the previous 3 months. Eligible patients could not have abnormal laboratory findings or medical conditions that may have jeopardized their safety or interfered with the study, or a history of hypersensitivity to lanreotide or drugs with similar chemical structure, and they could not have used an unlicensed drug in the previous 30 days. Women were excluded if they were not using acceptable methods of contraception or were pregnant or lactating.
Patients were withdrawn from the study at any time if there was evidence of worsening acromegalic symptoms requiring treatment other than lanreotide Autogel, signs of tumour progression requiring surgery or at the discretion of investigators for safety reasons. Patients could also withdraw consent at any time.
Trial design and interventions
The LEAD study was an open-label, non-comparative trial conducted between October 6, 2008 and May 20, 2013 in 14 countries (Brazil, Denmark, Finland, France, Greece, Latvia, The Netherlands, Norway, Poland, Romania, Russia, Serbia, South Korea, and Sweden). There were two 24-week phases in the study (Fig. 1). In phase 1, patients received five injections of lanreotide Autogel 120 mg (Beaufour Ipsen Industrie, Dreux, France) with a 6-week EDI. In phase 2, patients received further injections of lanreotide Autogel 120 mg with a 4-week dosing interval, or 6- or 8-week EDI depending on their IGF1 levels at the end of phase 1. Patients with IGF1 levels >130% of the upper limit of normal (ULN) at the end of phase 1 did not continue into phase 2. Lanreotide Autogel was administered by deep subcutaneous injection. Injections at baseline and week 24 were administered at the study centre; other injections could be administered in the patient's home as part of the patient's regular care.
Figure 1.

Study design and schedule for treatment with lanreotide Autogel 120 mg in the LEAD study. DI, dosing interval; EDI, extended dosing interval; IGF1, insulin-like growth factor-1; ULN, upper limit of normal.
Before the study started, the protocol, its amendments, the consent form, the study questionnaires and the patient information leaflet were approved by independent ethics committees in each country. The trial was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines and all local regulatory requirements and it was registered with EudraCT (2007-005838-37) and ClinicalTrials.gov (NCT00701363). All patients provided written informed consent before participating in the study.
Assessments
Study visits were scheduled at baseline (week 0), at 6-weekly intervals in phase 1 (up to week 24), and according to the dosing interval to which patients were assigned in phase 2 (up to week 48) (Fig. 1). The end-of-study visit was scheduled at week 48; patients discontinuing prematurely from the study attended an early withdrawal visit.
Hormone levels were determined centrally from blood samples taken under fasting conditions at baseline, and at the end of phases 1 and 2 (or at early withdrawal, if applicable). IGF1 levels were assessed from single samples using a solid-phase enzyme-labelled chemiluminescent immunometric assay (Immulite 2000 IGF1/Siemens Healthcare Diagnostics, Los Angeles, CA, USA) with an analytical sensitivity of 20 μg/l, intra-assay precision of 2.3–3.9% and interassay precision of 3.7–8.1%. Multiple batches rather than a single batch of the Immulite assay were used to test the samples. In addition, due to supply difficulties with this IGF1 assay towards the end of the study, IGF1 levels for five samples (three patients) were assessed using the iSYS IGF1 immunoassay (Immunodiagnostic Systems, Boldon, UK). Comparison of the two methods (scatterplot with regression analysis) showed that the IGF1 levels were highly consistent between the two assays up to 200 μg/l. As the range of IGF1 levels across the five samples assessed with the iSYS assay was 85–188 μg/l, neither sensitivity nor po st hoc analyses were considered necessary. GH levels were assessed from three samples taken at 1-h intervals using a simultaneous one-step immunoenzymatic assay (Access Ultrasensitive hGH, Beckman Coulter, Inc., Nyon, Switzerland). The GH assay had a lower limit of detection of 0.004 μg/l, intra-assay precision of 1.9–3.8% and interassay precision of 2.4–3.9%.
The PROs measured were symptom severity, HRQoL and treatment preference. The patient-assessed acromegaly symptom questionnaire (PASQ) was used to report the severity of five symptoms and signs of acromegaly at baseline and at the end of phases 1 and 2 (severity scale ranged from 0 (no symptoms) to 8 (severe, incapacitating symptoms); symptoms and signs assessed were headache, excessive perspiration, fatigue, soft tissue swelling and arthralgia) (16). PASQ symptoms and signs were also recorded as adverse events (AEs) if they were considered serious. HRQoL was assessed with the AcroQoL questionnaire (17) and the SF-36v2® health survey (18); these were completed by patients at baseline and at the end of phases 1 and 2 (patients from Finland, Latvia and Serbia did not complete the AcroQoL questionnaire as validated translations were not available). Scores ranged from 0 (worst possible HRQoL) to 100 (best possible HRQoL). Patients were asked by the investigators to state their treatment preferences for lanreotide Autogel vs octreotide LAR at the end of phases 1 and 2.
Safety assessments comprised: AEs collected throughout the study; physical examination, vital signs, haematology and biochemistry assessed at baseline and at the end of phases 1 and 2 (or early withdrawal, if applicable); and gallbladder ultrasound performed at baseline and at the end of phase 2 (or early withdrawal).
Endpoints
The primary endpoint was the proportion of patients who were on the 6- or 8-week EDI and had a normal IGF1 level at week 48 (study end). Related secondary endpoints were the proportion of patients who had a normal IGF1 level on the 6-week EDI at week 24 (end of phase 1), the proportion who were on the 8-week EDI and had a normal IGF1 level at week 48, and the proportion who were on the 6- or 8-week EDI (regardless of IGF1 level) at week 48. Other secondary endpoints based on IGF1 levels were: IGF1 level changes from baseline to week 48 overall and by dosing-interval group; baseline IGF1 levels for patients with normal IGF1 levels at week 48 by dosing-interval group; and baseline IGF1 levels for patients with vs without normal IGF1 levels at week 24.
Secondary endpoints based on GH control were: proportions of patients with GH levels ≤2.5 μg/l at weeks 24 and 48, and GH levels at baseline and weeks 24 and 48. These a priori endpoints were supplemented post hoc by the proportions of patients with GH levels <1.0 μg/l and the proportions with GH levels ≤2.5 μg/l and a normal IGF1 level at week 24.
Other secondary endpoints included: PASQ scores at baseline and weeks 24 and 48 (the end of phases 1 and 2); changes in HRQoL scores (AcroQoL global score and SF-36v2® physical (PCS) and mental (MCS) component summary scores) at week 24 and 48; the proportion of patients with normal IGF1 levels and no deterioration in AcroQoL global score (deterioration defined as a decrease of at least one point from baseline) at week 48; the correlation between AcroQoL global score changes and IGF1 level (% of ULN) changes at weeks 24 and 48; treatment preferences at weeks 24 and 48; and safety. In accordance with guidelines for the use of the SF-36v2® health survey, the a priori analysis using a total score was replaced post hoc by two separate analyses using PCS and MCS scores.
Statistical analysis
The proportion of patients entering the study who were on a 6- or 8-week EDI with a normal IGF1 level at week 48 (primary endpoint) was initially assumed to be 50%. Following an interim review of the data, the point estimate of this proportion was revised to 70%. Accordingly, a corrected total of 127 patients were needed to estimate this revised proportion with a precision of ±8.0% based on a two-sided 95% CI.
The primary analysis for the primary endpoint was based on the intention-to-treat (ITT) population, which was defined as all patients who received at least one injection (i.e., patients entering into phase 1 regardless of whether or not they entered phase 2). Supportive analyses for the primary endpoint were based on the modified ITT population (patients from the ITT population who were allocated to a phase-2 dosing interval group) and the per-protocol (PP) population (patients from the ITT population without major protocol deviations). Analyses of between-group differences in IGF1 levels in phase 2 were based on the modified ITT population. All other secondary efficacy analyses were based on both ITT and modified ITT populations and results were similar in all cases; thus, only data from analyses on the ITT population are reported. Missing data were not replaced, but last post-baseline value available assessments were calculated for all efficacy endpoints. Safety analyses were based on the safety population (defined as per the ITT population).
Between-group comparisons of mean IGF1 levels were performed using either an analysis of covariance with injection interval group as the main factor and baseline IGF1 level as the covariate (changes in IGF1 levels) or a student's t-test (baseline IGF1 levels). Correlations between HRQoL (AcroQoL score) and IGF1 level changes were examined using a non-parametric Spearman correlation test. Descriptive statistics were used for safety and other secondary efficacy endpoints. Statistical analyses were performed using SAS software version 9.2 or higher (SAS Institute, Inc., Cary, NC, USA).
Results
Patient disposition and baseline characteristics
A total of 124 patients entered phase 1 of the study and received treatment (ITT/safety population). Of these, 15 patients were withdrawn prematurely during phase 1 (including one patient withdrawn because of a lack of efficacy) (Fig. 2). Of 109 patients entering phase 2 (modified ITT population), 107 completed the study. The PP population (n=118) excluded two patients with violation of the inclusion criteria, two with missing IGF1 measurements at week 24, one who had received out-of-date study medication and one who had received a prohibited therapy (dose of dopamine agonist changed during the study). Of 124 patients enrolled, 96 (77.4%) were allocated to 6- or 8-week EDIs in phase 2.
Figure 2.
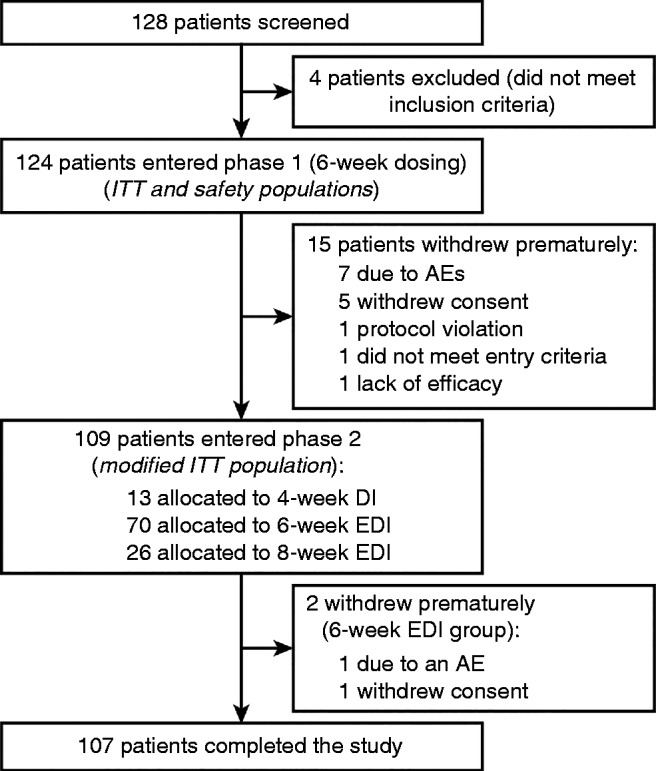
Flow of patients through the study. AE, adverse event; DI, dosing interval; EDI, extended dosing interval; ITT, intention to treat.
Baseline demographic and disease characteristics were generally well balanced among the final dosing-interval groups, except for a slightly higher proportion of women in the 8-week EDI group compared with the 4-week dosing-interval and 6-week EDI groups (Table 1). The mean (s.d.) duration of previous treatment with octreotide LAR was 2.5 (2.2) years and 80.5% of patients had been receiving octreotide 20 mg (among patients in phase 2, 100, 84.3 and 65.4% in the 4-, 6- and 8-week dosing groups, respectively, had been receiving octreotide LAR 20 mg before the study). Nine patients were receiving concomitant dopamine agonists (bromocriptine, n=6; cabergoline, n=3).
Table 1.
Baseline demographic and disease characteristics. Data are from the intention-to-treat population and are expressed as mean (s.d.) unless specified otherwise.
| Phase 1 only a (6-week EDI) (n=15) | Phase 2 | All patients (n=124) | |||
|---|---|---|---|---|---|
| 4-week DI (n=13) | 6-week EDI (n=70) | 8-week EDI (n=26) | |||
| Age (years) | 55.2 (15.3) | 55.0 (10.1) | 53.2 (10.4) | 57.0 (9.8) | 54.4 (10.9) |
| Men:women, n (%) | 6 (40.0):9 (60.0) | 5 (38.5):8 (62.5) | 29 (41.4):41 (58.6) | 6 (23.1):20 (76.9) | 46 (37.1):78 (62.9) |
| BMI (kg/m2) | 28.4 (2.9) | 29.2 (4.1) | 29.5 (6.5) | 27.0 (4.3) | 28.8 (5.6) |
| Time from diagnosis (years) | 6.9 (5.3) | 10.2 (5.9) | 8.2 (5.3) | 10.2 (7.4) | 8.7 (5.9) |
| Time since surgery (years) | (n=10) | (n=10) | (n=54) | (n=24) | (n=98) |
| 6.3 (4.3) | 9.6 (6.0) | 7.6 (4.3) | 9.2 (7.3) | 8.1 (5.4) | |
| Octreotide LAR treatment: duration (years) | (n=14) | (n=13) | (n=70) | (n=26) | (n=123) |
| 2.2 (2.2) | 2.5 (2.4) | 2.6 (2.4) | 2.5 (1.8) | 2.5 (2.2) | |
| Dose, n (%) | |||||
| 10 mg | 4 (28.6) | 0 | 11 (15.7) | 9 (34.6) | 24 (19.5) |
| 20 mg | 10 (71.4) | 13 (100) | 59 (84.3) | 17 (65.4) | 99 (80.5) |
| IGF1 level (% ULN) | (n=15) | (n=12) | (n=70) | (n=26) | (n=123) |
| 93.3 (76.7) | 98.7 (14.6) | 67.7 (29.6) | 52.5 (25.0) | 70.6 (39.2) | |
| GH level (μg/l) b | 1.0 (1.0) | 0.9 (0.7) | 0.9 (1.2) | 1.1 (1.2) | 1.0 (1.1) |
DI, dosing interval; EDI, extended dosing interval; GH, growth hormone; IGF1, insulin-like growth factor-1; ULN, upper limit of normal.
Patients from phase 1 not entering phase 2.
GH levels assessed by local laboratories.
Efficacy
IGF1 control and EDIs
In the primary analysis of the primary endpoint, 94 of the 124 patients (75.8% (95% CI: 68.3–83.3)) in the ITT population were on a 6- or 8-week EDI and had a normal IGF1 level at week 48 (end of study; Fig. 3a). The supportive analysis based on the PP population was similar to the primary analysis (PP population: 92/118, 78.0% (70.5–85.4)), whereas the supportive analysis based on the modified ITT population was higher (modified ITT population: 94/109, 86.2% (79.8–92.7)).
Figure 3.
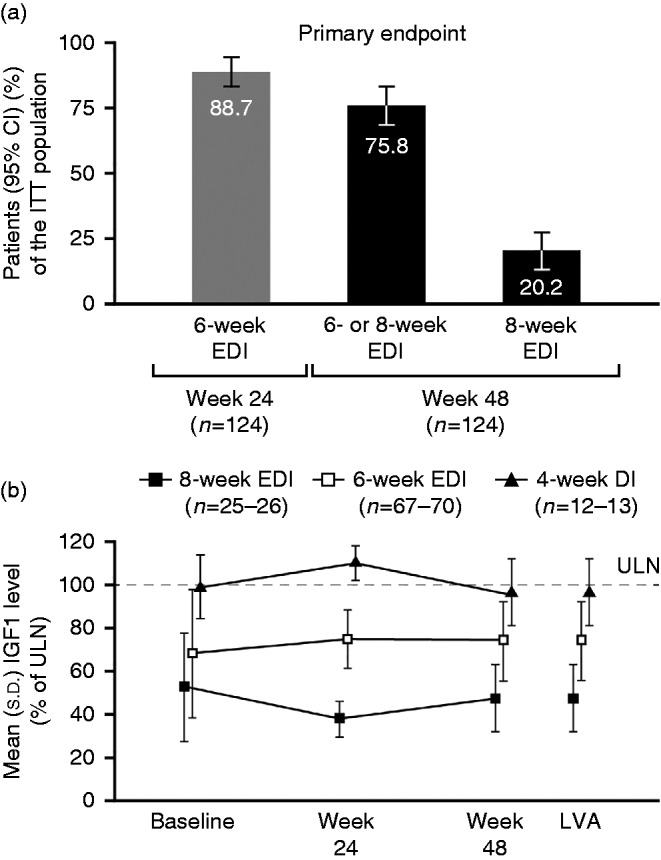
(a) Patients with normal IGF1 levels on EDIs at weeks 24 (end of phase 1) and 48 (end of study). (b) Serum IGF1 levels (% of ULN) according to allocated injection schedule for lanreotide Autogel 120 mg in phase 2. Data are from the ITT population. DI, dosing interval; EDI, extended dosing interval; IGF1, insulin-like growth factor-1; ITT, intention to treat; LVA, last post-baseline value available; ULN, upper limit of normal.
For the related IGF1 and EDI secondary endpoints, 110 of the 124 patients (88.7% (95% CI: 83.1–94.3)) in the ITT population had a normal IGF1 level with the 6-week EDI at week 24 (end of phase 1), 25/124 patients (20.2% (95% CI: 13.1–27.2)) on the most extended dosing schedule (8 weeks) had a normal IGF1 level at week 48 (Fig. 3a) and 97/124 patients (78.2% (95% CI: 71.0–85.5)) were on a 6- or 8-week EDI at week 48 (regardless of IGF1 level). Within the individual dosing-interval groups, normal IGF1 levels were achieved at week 48 by 13/13 patients with a 4-week dosing interval, 67/70 with the 6-week EDI and 25/26 with the 8-week EDI. Of the patients receiving concomitant dopamine agonist treatment, 7/9 had a normal IGF1 level at week 24; all seven patients receiving a concomitant dopamine agonist and continuing into phase 2 were on a 6- or 8-week EDI at week 48 (four patients on the 6-week schedule and three on the 8-week schedule).
Figure 3b shows serum IGF1 levels according to the allocated injection interval schedule in phase 2. Compared with the 4-week dosing-interval group, adjusted mean changes in IGF1 levels (% ULN) from baseline to week 48 were significantly better (smaller increases or larger decreases) for the 6- or 8-week EDI groups among patients with normalized IGF1 levels at week 48 (4- vs 6-week: 18.7 (95% CI: 7.5–29.9), P=0.0013; 4- vs 8-week: 42.7 (29.5–55.8), P<0.0001; Table 2). Compared with the 4-week dosing-interval group, mean baseline % ULN IGF1 levels were also significantly lower in 6- and/or 8-week EDI groups in these patients (4- vs 6-week: 30.9 (13.1–48.8), P=0.0009; 4- vs 8-week: 47.4 (31.7–63.1), P<0.0001; 4- vs 6- and 8-week: 35.1 (18.1–52.7), P<0.0001; Table 2). The mean (s.d.) baseline % ULN IGF1 level at week 24 for patients who went into phase 2 (i.e., with a normal IGF1 levels at week 24) was significantly lower (67.5 (30.0) (n=108)) than that for patients without normalized IGF1 levels (95.9 (78.9) (n=14); mean (95% CI) difference: −28.4 (−50.0 to −6.8), P=0.0103).
Table 2.
Differences between dosing-interval groups for IGF1 levels (% ULN) in patients with normalized levels at week 48 (end of study; secondary endpoints). Data are from the modified intention-to-treat population.
| 4-week DI (n=13) | 6-week EDI (n=70) | 8-week EDI (n=26) | Between-group differences | ||||
|---|---|---|---|---|---|---|---|
| 4-week DI vs 6-week EDI | 6-week EDI vs 8-week EDI | 4-week DI vs 8-week EDI | 4-week DI vs 6-+8-week EDI | ||||
| Baseline a , mean (s.d.) | (n=12) | (n=67) | (n=25) | ||||
| 98.7 (14.6) | 67.8 (30.3) | 51.3 (24.7) | 30.9 (13.1–48.8)‡ | 16.5 (3.0–29.9)* | 47.4 (31.7–63.1)‡ | 35.1 (18.1–52.7)‡ | |
| Change from | (n=12) | (n=67) | (n=25) | ||||
| baseline to week 48 b , adjusted mean (s.e.m.) | 24.8 (5.3) | 6.1 (2.1) | −17.9 (3.5) | 18.7 (7.5–29.9)† | 24.0 (15.8–32.1)‡ | 42.7 (29.5–55.8)‡ | N/A |
*P<0.05, † P<0.01, ‡ P<0.001. DI, dosing interval; EDI, extended dosing interval; IGF1, insulin-like growth-factor 1; NA, not applicable; ULN, upper limit of normal.
Between group differences are presented as mean (95% CI) using student's t-test (two-sided).
Between group differences are presented as adjusted mean (95% CI) of the analysis of covariance with injection interval group as main factor and baseline level as covariate.
GH control and combined IGF1 and GH control
Most patients achieved GH control (≤2.5 μg/l) and combined IGF1 and GH control (normal IGF1 levels and GH levels ≤2.5 μg/l) after 24 weeks of the 6-week EDI (i.e. end of phase 1). Specifically, 105/112 patients with data available (93.8% (95% CI: 89.3–98.2)) had GH levels ≤2.5 μg/l (Fig. 4a) and 103/112 (92.0% (86.9–97.0)) had both normal IGF1 levels and GH levels ≤2.5 μg/l. The majority of patients in phase 1 also achieved the more stringent measure of GH control (<1.0 μg/l) at the same time point (77/112 patients, 68.8% (60.2–77.3). Levels of GH control remained high after a further 24 weeks (i.e., end of phase 2) both within dosing-interval groups and for phase-2 participants overall (Fig. 4a). Specifically, for phase-2 patients overall at week 48, 101/107 patients (94.4% (95% CI: 90.0–98.8)) had GH levels ≤2.5 μg/l.
Figure 4.
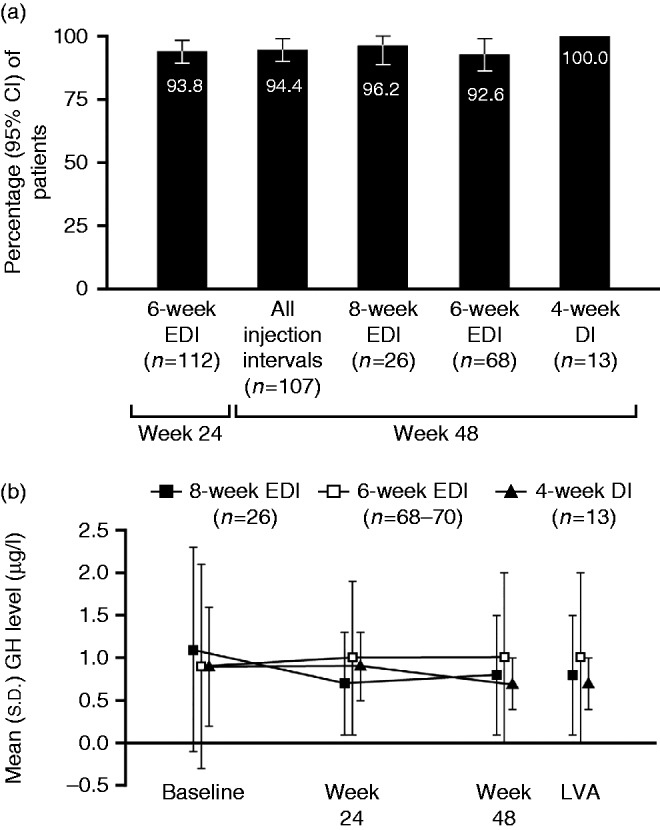
(a) Patients with GH control, showing patients with GH levels ≤2.5 μg/l at weeks 24 (end of phase 1) and 48 (end of study). (b) Serum GH levels according to allocated injection schedule for lanreotide Autogel 120 mg in phase 2. Data are from the intention-to-treat population. DI, dosing interval; EDI, extended dosing interval; GH, growth hormone.
Mean serum GH levels were relatively stable throughout the study, particularly for the 6-week EDI group (Fig. 4b).
Acromegaly symptoms and HRQoL
Mean (s.d.) total PASQ symptom scores changed little between baseline (12.9 (7.8)) and weeks 24 (12.8 (8.6)) and 48 (14.0 (8.5)). Individual PASQ symptom scores were also generally stable throughout the study (Supplementary Figure 1, see section on supplementary data given at the end of this article), and different dosing intervals had no clear effect on PASQ symptom scores (data not shown).
Among patients completing the AcroQoL questionnaire (i.e., in countries in which validated translations were available), adjusted mean changes from baseline to weeks 24 and 48 in the global score were not significantly different in pairwise comparisons of dosing-interval groups (Supplementary Table 1, see section on supplementary data given at the end of this article). Of 83 patients who had data for the global score at baseline and week 48, 37 had a normal IGF1 level at week 48 and a stable or improved global score (44.6% (95% CI: 33.9–55.3)). Overall, there was no correlation between changes from baseline to weeks 24 or 48 in the AcroQoL global score and changes in IGF1 levels (Spearman correlation, −0.01 (95% CI: −0.23 to 0.20) n=85 and (−0.01 (−0.22 to 0.21) n=82, respectively).
Adjusted mean changes from baseline to weeks 24 and 48 in the MCS and PCS scores of the SF-36v2® were not significantly different in pairwise comparisons of dosing-interval groups (Supplementary Table 1).
Treatment preference
At week 24 (end of phase 1), more patients preferred lanreotide Autogel 120 mg with a 6-week EDI than octreotide LAR every 4 weeks (lanreotide: 99/112 patients, 88.4% (95% CI: 82.5–94.3); octreotide: 10/112 patients, 8.9% (3.6–14.2); three patients had no preference).
At week 48 (end of phase 2), almost all patients preferred lanreotide Autogel 120 mg over octreotide LAR in the 8-week EDI group (24/26 patients, 92.3% (82.1–100.0) vs 2/26 patients, 7.7% (0.0–17.9) respectively). In the other two groups, more than three-quarters of patients preferred lanreotide Autogel 120 mg over octreotide LAR (6-week EDI group: 53/68 patients, 77.9% (68.1–87.8) vs 10/68 patients 14.7% (6.3–23.1), respectively (one further patient indicated a preference for lanreotide 4-week dosing interval and four patients had no preference); 4-week dosing interval: 10/13 patients, 76.9% (54.0–99.8) vs 2/13 patients, 15.4% (0.0–35.0) (one patient had no preference)).
Safety
A total of 341 AEs were reported for 91 (73.4%) of the 124 patients in the safety population. The proportions of patients experiencing AEs were similar across dosing interval groups and between phases 1 and 2, and AEs were mild or moderate in most of these patients (Table 3). The most common AEs were cholelithiasis (11.3%) and diarrhoea (10.5%). Fifty-four patients (43.5%) experienced treatment-related AEs. Eight patients experienced AEs leading to withdrawal; of these, four patients withdrew because of AEs considered related to treatment (one patient experienced diarrhoea, vomiting, dizziness, aesthenia, dehydration and hyponatraemia; and one patient in each case experienced stress, flatulence and migraine). Of 18 serious AEs in 11 patients, six were considered related to treatment and were all experienced by the single patient who was withdrawn due to multiple AEs. One of the other serious AEs (lung cancer) led to death but was not considered to be related to treatment.
Table 3.
Adverse events during the study. Data are number (%) of patients from the safety population.
| Phase 1 only a (6-week EDI; n=15) | Phase 2 | All patients (n=124) | |||
|---|---|---|---|---|---|
| 4-week DI (n=13) | 6-week EDI (n=70) | 8-week EDI (n=26) | |||
| Any AE b | 12 (80.0) | 10 (76.9) | 49 (70.0) | 20 (76.9) | 91 (73.4) |
| Related to treatment | 7 (46.7) | 3 (23.1) | 32 (45.7) | 12 (46.2) | 54 (43.5) |
| Severe/moderate/mild | 5 (33.3)/8 (53.3)/9 (60.0) | 3 (23.1)/3 (23.1)/9 (69.2) | 6 (8.6)/23 (32.9)/43 (61.4) | 3 (11.5)/8 (30.8)/17 (65.4) | 17 (13.7)/42 (33.9)/78 (62.9) |
| Leading to withdrawal c | 7 (46.7) | 0 | 1 (1.4) | 0 | 8 (6.5) |
| Any serious AE d | 3 (20.0) | 2 (15.4) | 4 (5.7) | 2 (7.7) | 11 (8.9) |
| Related to treatment | 1 (6.7) | 0 | 0 | 0 | 1 (0.8) |
| AEs in >10% of patients | |||||
| Dizziness | 3 (20.0) | 0 | 1 (1.4) | 0 | 4 (3.2) |
| Flatulence | 2 (13.3) | 0 | 2 (2.9) | 1 (3.8) | 5 (4.0) |
| Constipation | 2 (13.3) | 0 | 0 | 0 | 2 (1.6) |
| Cholelithiasis | 1 (6.7) | 2 (15.4) | 7 (10.0) | 4 (15.4) | 14 (11.3) |
| Diarrhoea | 1 (6.7) | 0 | 11 (15.7) | 1 (3.8) | 13 (10.5) |
| Dyslipidaemia | 1 (6.7) | 0 | 6 (8.6) | 3 (11.5) | 10 (8.1) |
| Headache | 1 (6.7) | 2 (15.4) | 2 (2.9) | 2 (7.7) | 7 (5.6) |
| Nasopharyngitis | 0 | 2 (15.4) | 3 (4.3) | 0 | 5 (4.0) |
| Gallbladder polyp | 0 | 2 (15.4) | 0 | 1 (3.8) | 3 (2.4) |
| Osteoarthritis | 0 | 2 (15.4) | 0 | 0 | 2 (1.6) |
| Abdominal discomfort | 0 | 1 | 1 (1.4) | 3 (11.5) | 4 (3.2) |
AE, adverse event; DI, dosing interval; EDI, extended dosing interval.
Patients from phase 1 not entering phase 2.
Any AE refers to treatment-emergent AEs; that is, all AEs that occurred during the study.
None of the AEs leading to withdrawal occurred in more than one patient; four patients had events leading to withdrawal that were considered to be related to treatment.
No individual serious AEs occurred in more than one patient.
Gallbladder ultrasound showed that five patients developed new gallbladder sludge during the study and eight subjects developed new gallbladder lithiasis during the study. None of the patients had both new sludge and new cholelithiasis. There were no clinically relevant changes for laboratory parameters or vital signs.
Discussion
In the LEAD study, high levels of biochemical control were achieved with lanreotide Autogel 120 mg 6- and 8-week EDIs in patients with acromegaly who had been previously well controlled on octreotide LAR 10 or 20 mg every 4 weeks. Specifically, 75.8% of the patients entering the study and receiving at least one injection of lanreotide Autogel maintained IGF1 control 48 weeks after switching to lanreotide Autogel 120 mg EDIs (24 weeks at a 6-week EDI with a further 24 weeks at a 6- or 8-week EDI). Moreover, after only 24 weeks of the 6-week EDI (phase 1 of the study), 88.7% of patients maintained IGF1 control. A favourable IGF1 response to a lanreotide Autogel EDI was more likely in patients with lower IGF1 levels at baseline. Most patients were considered to have GH control, with levels ≤2.5 μg/l in over 90% of patients at 24 and 48 weeks. Treatment had no adverse effects on symptom control or HRQoL, and patient preference for lanreotide Autogel 120 mg at 4-, 6- or 8-week intervals was higher than that for octreotide LAR every 4 weeks, particularly among patients receiving lanreotide at 8-week intervals. The 120-mg dose of lanreotide was well tolerated during the study regardless of the injection interval. The most frequently reported AEs were those typically associated with somatostatin analogue treatment of acromegaly, and most were mild or moderate in severity.
The extension of dosing intervals (or dose reductions) is recommended by the Acromegaly Consensus Group for patients achieving good biochemical control with long-acting somatostatin analogues (3). Currently, however, lanreotide Autogel is the only such agent that has been approved for use with EDIs, supported by data from initial open-label studies (14, 15, 19, 20). The LEAD study data add to this body of published evidence. While the LEAD data are consistent with findings from earlier trials also involving patients previously receiving octreotide LAR every 4 weeks, greater proportions of patients achieved biochemical control in the LEAD study. In an Italian study of 23 patients with at least a partial response (in terms of IGF1 and/or GH levels) to previous treatment with octreotide LAR (10 mg, n=2; 20 mg, n=10; 30 mg, n=11), patients were assigned to lanreotide Autogel 120 mg every 4, 6 or 8 weeks based on GH levels after four injections of lanreotide Autogel 120 mg every 6 weeks (14). Ten patients continued treatment with a 6- or 8-week EDI and the proportions of patients achieving GH or IGF1 control across dosing-interval groups were similar at the start compared with the end of the study (after 2–3 doses at the assigned injection interval). In a German study of 35 patients with normal IGF1 levels (≤130% ULN) while receiving octreotide LAR, patients were assigned to lanreotide Autogel 120 mg every 8, 6 or 4 weeks initially according to the previous octreotide LAR dose (10 mg, n=7; 20 mg, n=11; 30 mg, n=17) and then based on IGF1 levels after the third injection (15). The proportion of patients maintaining normal IGF1 levels was similar at the start compared with the end of the study (i.e., after the sixth dose of lanreotide Autogel). Almost 50% of patients were successfully treated with lanreotide Autogel EDIs and the EDIs were associated with increased treatment preference vs octreotide LAR. The lower proportions of patients maintaining biochemical control in these two national studies compared with the LEAD study are probably due, in part, to the inclusion in the national studies of patients previously receiving the 30-mg dose of octreotide LAR. These patients may be less responsive to somatostatin analogues than patients well controlled on lower octreotide LAR doses and thus less likely to maintain EDIs. In the LEAD study, lower baseline IGF1 levels were generally predictive of a good biochemical (IGF1) response. This accords well with a previous study of 51 patients in which IGF1 levels at baseline were significantly lower in the EDI groups than the 4-week dosing-interval group (20) and generally with data from the octreotide LAR–lanreotide Autogel switching studies (14, 15). In contrast to the findings in the LEAD study, however, some researchers have noted that baseline GH levels may be predictive of a response to EDIs (19, 20). Again, the apparently discordant results across studies may arise from differences in the proportions of poorer responders in the study cohorts. Poorer responders are likely to have higher baseline GH (and IGF1) levels and thus be assigned to 4-week dosing intervals.
Clinical trials suggest EDIs with octreotide LAR may also be possible with selected patients (21, 22), although such extended intervals have not been approved and data are perhaps more limited. In a study involving 13 patients with normal GH and IGF1 levels while receiving treatment with octreotide LAR 10 or 20 mg at 4-weekly intervals, the injection interval was extended to every 6 weeks, with nine patients maintaining biochemical control (normal IGF1 levels and GH ≤2.5 μg/l) after 36 weeks (21). In a separate study of patients with a mixed treatment history and active acromegaly, 17 of 19 individuals extended octreotide LAR dosing intervals to every 6, 8, 10 or 12 weeks (16 patients with the 20-mg dose and one with the 30-mg dose) (22). As acknowledged by the authors of the second study, pretreatment GH levels were relatively low in their patient cohort; individuals with higher GH levels may have needed more frequent dosing.
The LEAD study was a large international study designed to build on previous smaller national studies and provide robust new data on the profile of patients most likely to successfully maintain lanreotide Autogel EDIs in clinical practice. Nonetheless, there are some limitations that should be noted. First, the large size of the study made it impractical to assess IGF1 levels with just one batch of the relevant immunometric assay (Immulite). Despite this possible source of imprecision for the primary endpoint, differences between dosing-interval groups were readily apparent. Second, in accordance with study aims, patients received the highest approved dose of lanreotide Autogel (120 mg) and were included only if they had achieved biochemical control with 10 or 20 mg of octreotide LAR every 4 weeks. It follows from this that it cannot be assumed similar results would be obtained with other doses of the Autogel formulation or with patients previously achieving biochemical control with the 30-mg dose of octreotide LAR. While it cannot be ruled out that some patients may have different responses to different analogues (13), the requirement for the 30-mg dose of octreotide LAR suggests a greater resistance to somatostatin analogues or a biochemically more active tumour such that there is a lower chance of successfully maintaining a lanreotide Autogel EDI after switching. Finally, few patients in the LEAD study were receiving concomitant dopamine agonists, thus although their results were encouraging, the suitability of individual patients receiving combination therapy for lanreotide Autogel EDIs requires careful clinical assessment. It is also currently unclear whether the duration of prior treatment with somatostatin analogues influenced the likelihood of successfully extending dosing intervals; further research of this aspect may be warranted.
The LEAD study results suggested that patients with acromegaly who previously achieved biochemical control in terms of IGF1 levels while receiving 10 or 20 mg of octreotide LAR every 4 weeks are likely to be good candidates for switching to lanreotide Autogel 120 mg every 6 or 8 weeks. It is important to note baseline IGF1 levels, which may reflect responsiveness to somatostatin analogues, when considering EDI; patients with baseline levels below but close to the ULN may be less suitable candidates who require more careful consideration. After five injections every 6 weeks, it would be possible to determine if an EDI is viable and select the appropriate extension of the dosing interval. The LEAD study results also suggested that such patients are likely to maintain control of IGF1, GH and symptoms for up to at least 1 year without adverse effects on tolerability or HRQoL. Given the marked preference for EDIs among patients, EDIs may improve adherence to treatment. Less frequent dosing should reduce direct drug costs over time, as well as alleviating the burdens on patients and HCPs. In addition, maintenance of similar biochemical control but with potentially improved patient compliance might have positive or at least neutral impact on associated co-morbidities and indirect costs of their management. The current results, therefore, warrant a full health-economic analysis of EDIs in different national contexts, taking into account potential direct and indirect healthcare costs.
Supplementary data
This is linked to the online version of the paper at http://dx.doi.org/10.1530/EJE-15-0215.
Author contribution statement
S JCMM Neggers and A J van der Lely were involved in: concept and design of the study; patient enrolment into the study; analysis, collection and interpretation of the data; and drafting of the manuscript. P Maisonobe and C Sert were involved in: analysis and interpretation of the data; and drafting of the manuscript. V Pronin, I Balcere, M-K Lee, L Rozhinskaya, M D Bronstein and M Gadelha were involved in: patient enrolment into the study; collection and interpretation of the data; and critical review and revision of the paper. All authors approved the final version to be published and agree to be accountable for all aspects of the work.
Acknowledgements
We thank the patients, their families and investigators who participated in this study. We also thank Watermeadow Medical for writing assistance (funded by Ipsen).
LEAD Study Group: Brazil: M Bronstein, M Gadelha; Denmark: M Andersen; J O L Jørgensen; Finland: L Niskanen, M Välimäki; France: F Archambeaud, J Bertherat, F Borson-Chazot, R Cohen, B Delemer, A-M Guedj, A Penfornis, J-L Sadoul, J-L Schlienger, B Verges; Greece: G Krassas, S Tsagarakis, M Tzanela; Latvia: I Balcere; Norway: E Husebye; Poland: A Baldys-Waligorska, M Bolanowski, A Kowalska; Romania: C Ghervan; Russia: L Rozhinskaya, V Pronin; Serbia: S Damjanović, M Medić-Stojanoska, V Popović-Brkić; South Korea: S-Y Kim, E-J Lee, M-K Lee; Sweden: B Ekman, C Höybye, S Karlsson; The Netherlands: S JCMM Neggers, A J van der Lely.
Footnotes
(Details of the LEAD Study group are presented in the Acknowledgements section)
(S JCMM Neggers is now at Section of Endocrinology, Department of Medicine, Erasmus University MC Rotterdam, PO Box 2040, 3000 CA Rotterdam, The Netherlands)
Declaration of interest
S JCMM Neggers has received research grants and consultancy fees from Pfizer and Ipsen. V Pronin declares no conflicts of interest. I Balcere has received research funding from Ipsen. M-K Lee declares no conflicts of interest. L Rozhinskaya has received lecture fees from Novartis, Ipsen, Amgen and Eli Lilly, and has participated as a principal investigator in clinical trials supported by MDS, Novartis, Ipsen and Amgen. M D Bronstein has been an advisory board member for Ipsen, Novartis, Pfizer and Chiasma; has been a speaker for Ipsen and Novartis; and has received research support as principal investigator in clinical trials for Ipsen and Novartis. M Gadelha has been a speaker for Ipsen, Novartis and Pfizer; has been an advisory board member for Novartis; has received research funding from Novartis and Pfizer; and has participated as a principal investigator in clinical trials for Ipsen and Novartis. P Maisonobe and C Sert are employees of Ipsen. A J van der Lely has received research grants and speakers’ and consultancy fees from Novartis, Pfizer and Ipsen.
Funding
This work was supported by Ipsen.
References
- Katznelson L, Atkinson JL, Cook DM, Ezzat SZ, Hamrahian AH, Miller KK. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the diagnosis and treatment of acromegaly – update. Endocrine Practice. 2011;17(Suppl 4):1–44. doi: 10.4158/EP.17.S4.1. [DOI] [PubMed] [Google Scholar]
- Sherlock M, Ayuk J, Tomlinson JW, Toogood AA, Aragon-Alonso A, Sheppard MC, Bates AS, Stewart PM. Mortality in patients with pituitary disease. Endocrine Reviews. 2010;31:301–342. doi: 10.1210/er.2009-0033. [DOI] [PubMed] [Google Scholar]
- Giustina A, Chanson P, Kleinberg D, Bronstein MD, Clemmons DR, Klibanski A, van der Lely AJ, Strasburger CJ, Lamberts SW, Ho KK, et al. Expert consensus document: a consensus on the medical treatment of acromegaly. Nature Reviews. Endocrinology. 2014;10:243–248. doi: 10.1038/nrendo.2014.21. [DOI] [PubMed] [Google Scholar]
- Katznelson L, Laws ER, Jr, Melmed S, Molitch ME, Murad MH, Utz A, Wass JAH. Acromegaly: an endocrine society clinical practice guideline. Journal of Clinical Endocrinology and Metabolism. 2014;99:3933–3951. doi: 10.1210/jc.2014-2700. [DOI] [PubMed] [Google Scholar]
- Caron PJ, Bevan JS, Petersenn S, Flanagan D, Tabarin A, Prevost G, Maisonobe P, Clermont A, PRIMARYS Investigators Tumor shrinkage with lanreotide Autogel 120 mg as primary therapy in acromegaly: results of a prospective multicenter clinical trial. Journal of Clinical Endocrinology and Metabolism. 2014;99:1282–1290. doi: 10.1210/jc.2013-3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melmed S, Cook D, Schopohl J, Goth MI, Lam KS, Marek J. Rapid and sustained reduction of serum growth hormone and insulin-like growth factor-1 in patients with acromegaly receiving lanreotide Autogel therapy: a randomized, placebo-controlled, multicenter study with a 52 week open extension. Pituitary. 2010;13:18–28. doi: 10.1007/s11102-009-0191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron P, Cogne M, Raingeard I, Bex-Bachellerie V, Kuhn JM. Effectiveness and tolerability of 3-year lanreotide Autogel treatment in patients with acromegaly. Clinical Endocrinology. 2006;64:209–214. doi: 10.1111/j.1365-2265.2006.02450.x. [DOI] [PubMed] [Google Scholar]
- Holdaway IM, Bolland MJ, Gamble GD. A meta-analysis of the effect of lowering serum levels of GH and IGF-I on mortality in acromegaly. European Journal of Endocrinology. 2008;159:89–95. doi: 10.1530/EJE-08-0267. [DOI] [PubMed] [Google Scholar]
- Salvatori R, Nachtigall LB, Cook DM, Bonert V, Molitch ME, Blethen S, Chang S, SALSA Study Group Effectiveness of self- or partner-administration of an extended-release aqueous-gel formulation of lanreotide in lanreotide-naive patients with acromegaly. Pituitary. 2010;13:115–122. doi: 10.1007/s11102-009-0207-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatori R, Woodmansee WW, Molitch M, Gordon MB, Lomax KG. Lanreotide extended-release aqueous-gel formulation, injected by patient, partner or healthcare provider in patients with acromegaly in the United States: 1-year data from the SODA registry. Pituitary. 2014;17:13–21. doi: 10.1007/s11102-012-0460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman DT, Liebert KJ, Nachtigall LB, Lamerson M, Bakker B. Acromegaly: the disease, its impact on patients, and managing the burden of long-term treatment. International Journal of General Medicine. 2013;6:31–38. doi: 10.2147/IJGM.S38594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman DT, Burgess A, Davies PR. Evaluation of long-acting somatostatin analog injection devices by nurses: a quantitative study. Medical Devices. 2012;5:103–109. doi: 10.2147/MDER.S37831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troconiz IF, Cendros JM, Peraire C, Ramis J, Garrido MJ, Boscani PF, Obach R. Population pharmacokinetic analysis of lanreotide Autogel in healthy subjects: evidence for injection interval of up to 2 months. Clinical Pharmacokinetics. 2009;48:51–62. doi: 10.2165/0003088-200948010-00004. [DOI] [PubMed] [Google Scholar]
- Ronchi CL, Boschetti M, degli Uberti EC, Mariotti S, Grottoli S, Loli P, Lombardi G, Tamburrano G, Arvigo M, Angeletti G, et al. Efficacy of a slow-release formulation of lanreotide (Autogel) 120 mg) in patients with acromegaly previously treated with octreotide long acting release (LAR): an open, multicentre longitudinal study. Clinical Endocrinology. 2007;67:512–519. doi: 10.1111/j.1365-2265.2007.02917.x. [DOI] [PubMed] [Google Scholar]
- Schopohl J, Strasburger CJ, Caird D, Badenhoop K, Beuschlein F, Droste M, Plöckinger U, Petersenn S, German Lanreotide Study Group Efficacy and acceptability of lanreotide Autogel(R) 120 mg at different dose intervals in patients with acromegaly previously treated with octreotide LAR. Experimental and Clinical Endocrinology & Diabetes. 2011;119:156–162. doi: 10.1055/s-0030-1267244. [DOI] [PubMed] [Google Scholar]
- Neggers SJ, van Aken MO, de Herder WW, Feelders RA, Janssen JA, Badia X, Webb SM, van der Lely AJ. Quality of life in acromegalic patients during long-term somatostatin analog treatment with and without pegvisomant. Journal of Clinical Endocrinology and Metabolism. 2008;93:3853–3859. doi: 10.1210/jc.2008-0669. [DOI] [PubMed] [Google Scholar]
- Webb SM, Badia X, Surinach NL. Validity and clinical applicability of the acromegaly quality of life questionnaire. AcroQoL: a 6-month prospective study. European Journal of Endocrinology. 2006;155:269–277. doi: 10.1530/eje.1.02214. [DOI] [PubMed] [Google Scholar]
- Ware JEJ, Kosinski M, Bjorner JB, Tuner-Bowker DM, Gandek B & Maruish ME. User's Manual for the SF-36v2(R) Health Survey, 2nd edn. Lincoln, RI: QualityMetric, Incorporated, 2007.
- Lucas T, Astorga R. Efficacy of lanreotide Autogel administered every 4–8 weeks in patients with acromegaly previously responsive to lanreotide microparticles 30 mg: a phase III trial. Clinical Endocrinology. 2006;65:320–326. doi: 10.1111/j.1365-2265.2006.02595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi G, Minuto F, Tamburrano G, Ambrosio MR, Arnaldi G, Arosio M, Chiarini V, Cozzi R, Grottoli S, Mantero F, et al. Efficacy of the new long-acting formulation of lanreotide (lanreotide Autogel) in somatostatin analogue-naive patients with acromegaly. Journal of Endocrinological Investigation. 2009;32:202–209. doi: 10.1007/BF03346453. [DOI] [PubMed] [Google Scholar]
- Biermasz NR, van den Oever NC, Frolich M, Arias AM, Smit JW, Romijn JA, Roelfsema F. Sandostatin LAR in acromegaly: a 6-week injection interval suppresses GH secretion as effectively as a 4-week interval. Clinical Endocrinology. 2003;58:288–295. doi: 10.1046/j.1365-2265.2003.01710.x. [DOI] [PubMed] [Google Scholar]
- Turner HE, Thornton-Jones VA, Wass JA. Systematic dose-extension of octreotide LAR: the importance of individual tailoring of treatment in patients with acromegaly. Clinical Endocrinology. 2004;61:224–231. doi: 10.1111/j.1365-2265.2004.02084.x. [DOI] [PubMed] [Google Scholar]