

Abstract
Transposable elements (TEs) comprise a group of repetitive sequences that bring positive, negative, as well as neutral effects to the host organism. Earlier considered as “junk DNA,” TEs are now well-accepted driving forces of evolution and critical regulators the of expression of genetic information. Their activity is regulated by epigenetic mechanisms, including methylation of DNA and histone modifications. The loss of epigenetic control over TEs, exhibited as loss of DNA methylation and decondensation of the chromatin structure, may result in TEs reactivation, initiation of their insertional mutagenesis (retrotransposition) and has been reported in numerous human diseases, including cancer. Accumulating evidence suggests that these alterations are not the simple consequences of the disease, but often may drive the pathogenesis, as they can be detected early during disease development. Knowledge derived from the in vitro, in vivo, and epidemiological studies, clearly demonstrates that exposure to ubiquitous environmental stressors, many of which are carcinogens or suspected carcinogens, are capable of causing alterations in methylation and expression of TEs and initiate retrotransposition events. Evidence summarized in this review suggests that TEs are the sensitive endpoints for detection of effects caused by such environmental stressors, as ionizing radiation (terrestrial, space, and UV-radiation), air pollution (including particulate matter [PM]-derived and gaseous), persistent organic pollutants, and metals. Furthermore, the significance of these effects is characterized by their early appearance, persistence and presence in both, target organs and peripheral blood. Altogether, these findings suggest that TEs may potentially be introduced into safety and risk assessment and serve as biomarkers of exposure to environmental stressors. Furthermore, TEs also show significant potential to become invaluable surrogate biomarkers in clinic and possible targets for therapeutic modalities for disease treatment and prevention.
Keywords: air pollution, DNA methylation, environmental stressors, ionizing radiation, metals, retrotransposition
1. Introduction
1.1. Classification
Repetitive sequences account for about 40-45% of mammalian genomes, with some estimates suggesting 66-69% of the human genomes to be repetitive or repetitive elements-derived [1]. The majority of these repetitive elements are transposable elements (TEs) – repeated and mobile DNA sequences, capable of moving and invading genomes [2]. TEs are represented as retrotransposons and transposons, while the rest of repetitive elements are represented with satellite DNA and tandem repeats, which are immobile (Table 1).
Table 1.
Transposable elements in mammalian genomes.
| Class | Origin of name | Main representatives | Main characteristics | Genome coverage |
|---|---|---|---|---|
|
| ||||
| LINE | Long Interspersed Nucleotide Element | LINE-1 (currently active in mammals), LINE-2, LINE-3 (both are archaic and inactive). | Autonomous retrotransposon; 6Kb length (humans); ~500,000 copies; 100-1,000 are active in humans, ~3,000 are active in mice | ~ 20% (human) |
| ~ 21% (mouse) | ||||
| ~ 23% (rat) | ||||
|
| ||||
| SINE | Short Interspersed Nucleotide Elements | SINE B1, SINE B2 (mouse, rat), Alu (humans) | Non-autonomous retrotransposon (dependent on LINE-1 machinery); 280bp in lengths; ~1,000,000 copies | ~ 5-6% (mouse) |
| ~ 13.7% (human) | ||||
|
| ||||
| SVA | SINE-VNTR-Alu | SVA | Non-autonomous retrotransposon (dependent on LINE-1 machinery); ~3,000 copies | < 1% |
|
| ||||
| ERV (LTR) | Endogenous Retroviruses (Long Terminal Repeats) | ERV1, ERV2, ERV3 (currently active in mice; activity in humans is controversial). | Autonomous retrotransposon; 9-10Kb length; ~450,000 copies | ~ 8% (human) |
| ~ 10% (mouse) | ||||
|
| ||||
| DNA Transposons | Charlie, Mariner, Tigger | DNA Transposons (cut- and-paste mechanism); 80- 3,000bp; ~400,000 copies | ~ 3% | |
TEs comprise a group of repetitive sequences that bring positive, negative, as well as neutral effects to the host organism. Previously considered as “junk DNA,” it has become increasingly evident that TEs carry a set of important gene regulatory functions, including serving as recombinogenic structures for rapid genome remodeling, maintaining centromere and telomere integrity, providing alternative promoters, silencing by transcriptional or RNA interference (RNAi), and creating cryptic splice sites and polyadenylation signals [3-7]. TEs are also considered evolutionary precursors of many genes in mammalian genomes [8]. Of particular concern are the deleterious effects of TEs exerted by their transposition that may result in potential insertions and deletions within the coding sequences that may disrupt gene expression, as well as damaging recombination events [9,10].
All transposable elements can be classified into retrotransposons (or those that relocate via an RNA intermediate in a “copy-and-paste” mechanism, Class I) and transposons (or those that use “cut-andpaste” mechanism, Class II) (Fig. 1). Retrotransposons are clustered into Long Terminal Repeat (LTR) and non-LTR elements, depending on their structure, and can be further subdivided into autonomous (capable of propagating themselves throughout the genome) and non-autonomous (those that use machinery of other TEs). In this review, we will concentrate solely on retrotransposons, the most abundant and only active in mammalian genomes group of repetitive elements.
Figure 1. The structure of the main transposable elements in mammalian genomes.
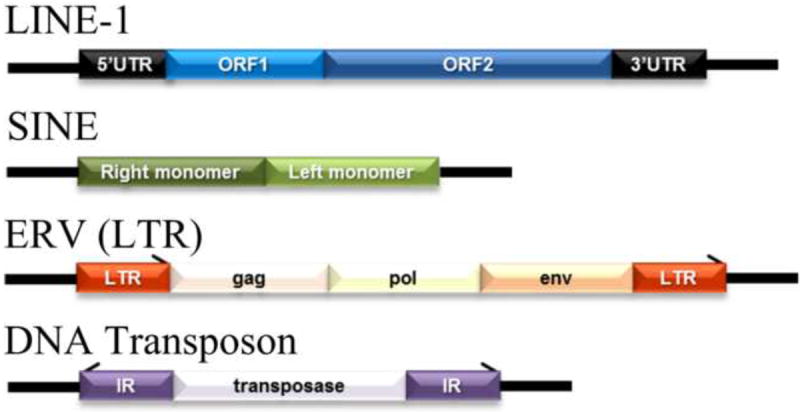
L1 – LINE-1, Long Interspersed Nucleotide Element 1; SINE – Short Interspersed Nucleotide Element; ERV (LTR) – Endogenous Retrovirus (Long Terminal Repeat); UTR – Untranslated Region; ORF – Open Reading Frame; gag, pol, env – encoded genes; IR – Inverted Repeats.
1.1.1. Long Interspersed Nuclear Elements (LINE)
Autonomous Long Interspersed Nuclear Elements (LINE) represent the most abundant group, not only among non-LTR retrotransposons, but of all mammalian TEs, reaching 17 to 23% of their genomes [6,7]. They include low-copy archaic inactive elements such as LINE-2 and LINE-3 and currently active and abundant LINE-1 (L1) elements.
Around 516,000 copies of L1 elements are present in the human genome; however, only about 100 of them are functional full-length (6 Kb long) sequences. The majority of L1s are 5’-truncated (0.9 Kb in length on average), incapable of retrotransposition, elements. Full length L1 contains four functional units: a ~900 bp 5’-untranslated region (UTR), a bicistronic open reading frame that encodes two proteins - ORF1p (a 40 kDa trimeric protein with RNA binding and nucleic acid chaperone activity) and ORF2p (a 150 kDa responsible for retrotransposition protein that encodes an endonuclease, reverse transcriptase, and zinc finger-like protein) and a 3’-UTR with a poly(A) tail [11,12] (Fig. 1). The 5’-UTR of L1 is an important regulatory unit that contains sense and antisense promoters and transcription factors binding sites, including those for p53, YY1, RUNX, SRY, and SOCS1 [13-15]. While the sense promoter regulates the expression of L1, the function of the antisense promoter is still largely unknown. However, some studies report the role of L1 antisense promoter in gene transcription regulation [16,17]. Accumulating evidence also indicates that L1 antisense promoter is involved in suppression of transcription from its own sense promoter, as well as in control over L1 retrotransposition [18,19]. A 3’-UTR is represented as a conserved poly-G tract with unknown functions.
The important feature of mammalian retrotransposons and L1, particularly, is their functional retrotransposition activity by which they are initially transcribed from the genome and then are reverse transcribed into a new location using a transposon-encoded reverse transcriptase [20]. L1 retrotransposition is initiated by transcription of its full-length mRNA from the internal promoter located in the 5’UTR between nucleotide positions +390 and +526 [21], and this process is mediated by the RNA polymerase II (Fig. 2-1). The newly transcribed mRNA is transported to the cytoplasm, where it is translated to L1-encoded proteins ORF1p and ORF2p by the ribosomal machinery (Fig. 2-2). ORF1p and ORF2p then anneal with an mRNA molecule, creating a ribonucleoprotein particle (Fig. 2-3). The ribonucleoprotein particle is formed in a cis preference, meaning that although ORF1p and ORF2p have the ability to bind any cellular mRNA molecule, there is, in fact, a strong bias observed towards annealing with L1 mRNAs specifically [22]. The ORF1p/ORF2p/mRNA ribonucleoprotein particle then enters the nucleus, where reintegration into genomic DNA occurs [12]. At this stage, due to the ORF2p endonuclease activity, a single-stranded nick is produced in genomic DNA. The exposed free 3’-hydroxyl residue serves as a primer, and the associated L1 mRNA is reverse-transcribed into cDNA. This process is referred to as “target-primed reverse transcription” (Fig. 2-4). The end product is a new L1 insertion into genomic DNA (Fig. 2-5). The site of insertion is a function of the endonuclease moiety of ORF2p, with minor grove width and TnAn content of the genomic DNA sequence being major factors [12]. For more detailed mechanisms of retrotransposition, we refer the readers to excellent reviews [20,23,24].
Figure 2. The mechanisms of LINE-1 (L1) and Alu retrotransposition.
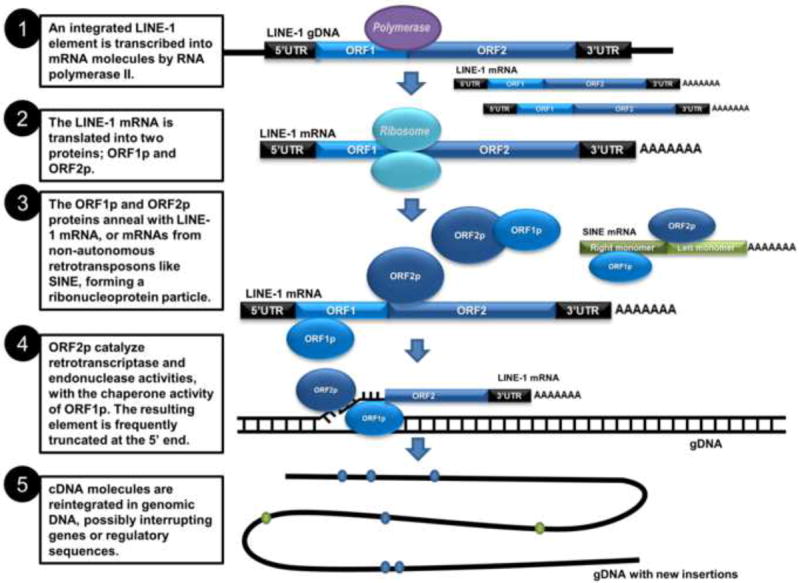
L1 – LINE-1, Long Interspersed Nucleotide Element 1, autonomous retrotransposon; Alu – human SINE element, nonautonomous retrotransposon that utilizes L1 machinery for its own retrotransposition; UTR – Untranslated Region; ORF – Open Reading Frame; AAAAA – polyA tail.
L1 RNA and associated proteins have been detected primarily in germ cells and embryos, while their presence in differentiated cells under normal conditions is rare [25,26]. L1 retrotransposition occurs mainly in embryogenesis and has been associated with somatic mosaicism, but can be also detected in the germline [26]. The estimated rate of retrotransposition in humans is between 1 in 95 and 270 live births [26]. Importantly, L1 retrotransposition usually results in 5’-truncated elements incapable of future retrotransposition [3,27].
1.1.2. Long Terminal Repeats (LTR)
LTRs are named for their long terminal repeats flanking the internal proviral sequence on both sides of the element. They comprise 8% of the human genome and about 10% of the mouse genome [28]. Structurally, LTRs are related to exogenous retroviruses; however, they lack the ability to move between cells and, therefore, are also called endogenous retroviruses (ERVs). LTRs encode gag, pol, pro, and env genes or may use the retroviral genes encoded by other ERVs (Fig. 1). While the activity of LTRs in humans remains controversial, at least two families are reported to be currently active in mice [29,30].
1.1.3. Short Interspersed Nucleotide Elements (SINE)
The cluster of non-autonomous retrotransposons includes Short Interspersed Nuclear Elements (SINE), which in humans are represented as Alu elements, the only active family of primate-specific SINE, and SINE-R, VNTR, and Alu (SVA) [31,32]. These elements lack their own retrotransposition machinery and, thus, utilize L1-encoded proteins for their own mobilization due to homologies between the 5’ ends of the two elements (Fig. 1).
Alu elements comprise up to 13.7% of human genomes (~1.1 million copies) with their de novo insertion rates exceeding those of L1 elements, reaching 1 in 20 live births [33-35]. They are 300 bp in length and contain two almost identical monomer sequences separated by a centrally-located A-rich region (A5TACA6). Alu elements derive from the 7SL RNA gene, are transcribed by RNA Polymerase III, and require L1 ORF1p for their mobilization [35,36]. In rodents they correspond to SINE elements – SINE B1 and SINE B2.
Accumulating evidence clearly demonstrates that SINE/Alu are also important regulators of genetic information, as they may possess an insulator/boundary activity, repress transcription by disrupting contacts between RNA polymerase II and promoter DNA, and cause epigenetic reprogramming of adjacent gene promoters [37-40].
1.2. Regulation of expression of transposable elements
The expression of TEs is regulated by both TE- and host-mediated mechanisms. TE-mediated control is usually linked to the ability to produce truncated TE suppressor copies for transposase-mediated autoregulation or utilization of host regulatory factors for activation [5,41]. Host-mediated mechanisms of control over the TEs are primarily associated with epigenetic mechanisms and include methylation of DNA, histone modifications, and regulation by small RNAs.
DNA methylation is important for normal development and maintenance of cellular homeostasis. It is involved in the regulation of the proper expression of genetic information in a sex-, tissue-, and cell type-dependent manner and serves as a key mechanism in silencing of TEs. DNA methylation is primarily associated with cytosine residues of cytosine and guanine base sequences (so called CpG sites). Approximately 70–90% of CpG sites in the mammalian genome are methylated, where TEs represent the most highly methylated sequences [42,43]. In turn, L1s are among the most highly methylated TEs in mammalian genomes; however, the methylation status of L1 may differ between the families and evolutionary age of the elements [44,45]. On the other hand, tandem repeats are relatively CpG-poor elements, but the extent of their methylation is relatively high given their length.
Earlier studies considered hypermethylation of TEs as a primary mechanism for their silencing. For instance, methylation of the CpG island located within the 5’-UTR of mammalian L1 has been shown to repress the expression of L1 [46,47]. Some studies, however, report reactivation of retrotransposons without alterations in DNA methylation, suggesting that other epigenetic mechanisms are also involved. These findings have led to the development of the hypothesis that methylation may serve rather as a locking mechanism over the other established epigenetic silencing marks [43]. Further studies have also indicated that while DNA methylation may be crucial for maintaining inactive transcription status of some TEs (i.e., LTR/ERVs), other TEs such as L1 may be less dependent on it [5,48]. It has also been demonstrated that different epigenetic controls are necessary for the regulation of TEs in somatic and embryonic stem cells [48,49].
Histone modifications are among the most important epigenetic mechanisms and have recently been recognized to play a key role in regulation of TEs. Covalent histone modifications shape the heterochromatin ubiquitously associated with TEs. Transcriptionally silent TEs are often associated with repressive histone lysine methylation marks (lysines 9 and 27 on histone 3 and lysine 20 on histone 4) and histone H2A.Z; however, different marks are specifically enriched in different TEs and cell types [50-52].
Non-coding RNAs provide another conserved mechanism of suppression of TEs and include control by PIWI-interacting RNA (piRNA), specific for germline and spermatogenesis as well as siRNA-, and miRNA-regulated silencing [42,53,54]. Additionally, some elements, such as L1, can use their antisense promoter as a mechanism of self-regulation [19].
1.3. Transposable elements in human disease
Failure of epigenetic control over TEs is associated with numerous pathological states. Loss of methylation of L1 and Alu/SINE has been reported in virtually all human and rodent experimental cancers (reviewed in [35,42,45]). Global genomic hypomethylation, to which TEs are the main contributors, is now a well-accepted hallmark of cancer. In turn, hypomethylation of 5’-UTR of L1 is frequently associated with its reactivation and increased expression of L1 has been detected in human cancer tissues [55].
Although only small subset of L1 are the full length and capable of retrotransposition elements, their insertions accumulated during the millions years of evolution may have dramatic effects on genomic loci in which retrotransposition occur. In general, L1 are associated with AT-rich and gene-poor regions of the genome [56]; however, many genes acquired L1 insertions either within them or in their proximal vicinity, and these genes are usually lowly expressed [57,58]. Alterations in the methylation status of such truncated and retrotransposition-inactive L1 elements may result in reactivation of these genes, if the insertion occured within the transcription start sites [59]. For instance, hypomethylation of the L1 element located within the MET oncogene results in activation of an alternate transcript of the latter in blade tumors and across the entire uroepithelium in tumor-bearing bladders [60]. Similar findings were reported in patients with colorectal cancer, where hypomethylation of the L1 sequence within the MET gene inversely correlated with induction of MET expression in metastatic tissue [61].
Retrotransposition of a given TE can also result in disruption of the ORF with subsequent inactivation of a functional gene, development of genomic instability and genome amplification, and thus serve as a causative factor in life-threatening human disorders, including cancer. For instance, Miki and colleagues reported insertion of the 3’ portion of L1 into the last exon of the APC gene, leading to the disruption of its function in colorectal cancer [62]. L1 retrotransposition has been reported in a number of other human cancers, including lung, prostate, and ovarian cancers [63,64]. However, it still remains unknown whether retrotransposition serves as a driving force of tumorigenesis or merely a consequence, occurring after tumor initiation. Taking into account the existing evidence, it is plausible to hypothesize that both scenarios may take place. For instance, a retrotransposition event that occurs within a critical gene with its further inactivation, like in the case of APC in colorectal cancer [62], can be considered as a driving mechanism. On the other hand, Solyom and colleagues reported that in about 60% of cases, L1 insertion identified in the first section of the colorectal tumor was not identified in the second section of the same tumor [65], suggesting that this particular case of retrotransposition may play role in tumor heterogeneity, but, most likely, was not a tumor-initiating mechanisms.
L1 retrotransposition is not associated with cancers only. Independent mutagenic L1 insertion into exon 14 of the Factor VIII gene disrupts synthesis of functional coagulation factor and results in hemophilia A [66]. In addition, human diseases, such as diabetes, chronic granulomatous disease, and β-thalassemia are associated with L1 retrotransposition [67,68].
1.4. Transposable elements in response to environmental stressors: detection methodologies
1.4.1. Analysis of TEs methylation
Because of their potential for transposition in human genomes, L1 and Alu/SINE received the most attention in the studies dedicated to the effects of environmental stressors on TEs. The majority of studies have concentrated on the studies of TEs’ methylation, as this epigenetic mechanism is critical for transcriptional silencing of TEs, and because the loss of global DNA methylation is a generally-accepted hallmark of cancer, can be detected in carcinogenesis and even shortly after exposure to carcinogens.
Combined bisulfite restriction analysis (COBRA) is the methodology based on a standard bisulfite modification of genomic DNA, followed by polymerase chain reaction (PCR) amplification and digestion of PCR product with restriction endonculeases. This methodology was extensively utilized in the L1 methylation status analysis. Sodium bisulfite treatment with subsequent PCR amplification results in methylation-dependent creation of new restriction endonucleases sites (Rsa I) or methylation-dependent retention of pre-existing sites (BstUI) within the rat L1 elements. The digested products are then separated on agarose gel and the band intensity serves as a qualitative measurement of L1 methylation. The limitations of COBRA include low sensitivity and limited number of CpG sites that can be evaluated. Methylation-sensitive quantitative PCR (MS qPCR) is also based on the sensitivity of restriction endonucleases to differentially methylated DNA. The digested DNA is amplified with a set of primers specific for the sequence of interest. Utilization of real-time PCR allows for more precise and quantitative assessment of DNA methylation compared to COBRA; however, evaluation will be still limited to CpG sites to which restriction endonucleases are selected. Pyrosequencing is a recently developed sequencebased technology that allows for accurate quantification of locus-specific DNA methylation. It relies on the detection of pyrophosphate release on nucleotide incorporation. Advantages of pyrosequencing is its very high sensitivity and analysis of the methylation status of all CpG sites within the investigated locus, but the methodology is limited to assessment of only short DNA sequences and relatively high costs.
1.4.2. Analysis of retrotransposition
Increases in TEs expression that can be assessed by designing specific assays for the TE of interest and real-time PCR may lead to retrotransposition events. Investigating the mobilization of endogenous TEs is associated with certain difficulties; therefore, an exogenous cell culture retrotransposition assay was developed and adapted for studies on L1 mobilization, first - in cells lines and later – in animal models. The history of development and success of this assay has been described in detail in an outstanding review by Rangwala and Kazazian [69]. Recent technological advances, including next-generation sequencing, now allow for comprehensive analysis of somatic retrotranspositions [70,71]. A number of tools have been developed in the last several years to analyze L1 insertions in both normal tissues and tumors, including ME-Scan [72], droplet digital PCR [73], and Transposeq [74]. Similarly, tools such as RepEnrich have been developed to study genome-wide transcriptional regulation of TEs [75]. Finally, the current knowledge on L1 insertions in humans has been summarized in euL1db – the European database of L1 retrotransposon insertions that contains over 140,000 sample-wise insertions and almost 9,000 distinct merged insertions [76].
Research in the last twenty years has convincingly demonstrated that alterations in TEs may not be only the consequences of the disease, but are among those that are involved in the pathogenesis. Further knowledge that environmental stressors can affect TEs has allowed a connection to be drawn between the environmental exposures, TEs, and disease development. Below, we review the current knowledge on the effects of the most ubiquitous environmental stressors exerted on mammalian TEs. The summary of the reviewed data can be found in Table 2
Table 1.
Effects of exposure to environmental stressors on transposable elements.
| Compound | Source | RE | Results | Methodology | Reference |
|---|---|---|---|---|---|
|
| |||||
| Terrestrial radiation | Rat spleen tissue 24 h and 7 months after localized cranial exposure to 20 Gy of X-rays | L1 | Hypomethylation, increase in expression | Combined bisulfite restriction assays (COBRA) | [74] |
|
| |||||
| RKO human colorectal carcinoma cells AG01522D primary human diploid skin fibroblasts irradiated with 1 Gy X-rays | L1 | AG01522 cells a general trend for hypermethylation was observed, but the RKO cells were more likely to exhibit a trend for hypomethylation | COBRA | [79] | |
|
| |||||
| C57BL/6 mice irradiated with 10 mGy of x-ray peripheral blood, spleen, and liver | L1, SINE B1 IAP | No persistent alterations in methylation | Bisulfite conversion, high resolution melt (HRM) | [81] | |
| Pyrosequencing | |||||
|
| |||||
| C57BL/6 mice irradiated with 1 Gy of x-ray peripheral blood, spleen, and liver | L1, SINE B1 IAP | Strain-, tissue-, sex-, and time-dependent alterations in methylation. hypomethylation of L1 in C57BL/6 mice, while CBA was characterized by hypermethylation of L1 in the spleen tissue shortly after exposure. Methylation of SINE B1 and IAP was affected to a lesser extent | Bisulfite conversion, high resolution melt (HRM) | [82] | |
| Pyrosequencing | |||||
|
| |||||
| Spleen tissue 7 months post localized hippocampal irradiation with two doses of 10cGy X-rays | L1 | Persistent global and REs-associated hypomethylation after exposure to high doses of IR has been observed even in bystander tissue, as evident in the study describing demethylation and reactivation of | COBRA | [74] | |
|
| |||||
| 100 cGy X-rays, AG01522D and RKO cells | L1 and Alu | Hypomethylation in RKO and hypermethylation in AG01522D | COBRA | [79] | |
|
| |||||
| Space radiation | C57BL/6 mice, 56Fe 0.1-0.4Gy | L1 SINE B1 | Dose-dependent hypermethylation in hematopoietic stem and progenitor cells | Methylation-sensitive enzymatic digestion followed qPCR | [76] |
| Bone marrow | |||||
|
| |||||
| C57BL/6 mice 56Fe 0.1-0.4Gy, | L1, SINE B1, Charlie, Mariner, and major and minor satellites | Dose-dependent hypermethylation was observed in the lungs 5 months after exposure | Methylation-sensitive enzymatic digestion followed qPCR | [77] | |
| Lung | Transcriptional silencing of these REs. | ||||
|
| |||||
| C3H/HeN mice irradiated with 10 and 100 cGy 56Fe, Liver and Lung | L1 | Hypomethylation in TF promoter types 7 days after exposure, and hypermethylation 30 days after exposure. | Bisulphite conversion, pyrosequencing | [75] | |
|
| |||||
| AG01522D and RKO cells irradiated with 10 and 100 cGy proton or 56Fe ions | L1 and Alu elements | Hypomethylation at 16-20 population doublings after irradiation | COBRA | [79] | |
|
| |||||
| Human-hamster hybrid cell line GM10115, were exposure to 1 Gy of 56Fe | L1 and Alu | Hypomethylation | COBRA | [78] | |
|
| |||||
| NP-2 human glioma cell line | L1 | Increase L1-RNA transcription | EGFP retrotransposition cassette-tagged L1 element and semi-quantitative PCR | [90] | |
| 0, 1, 2 or 4 Gy of carbon ions | De novo L1 insertions by carbon-ion irradiation were clearly longer than those by X-rays | ||||
|
| |||||
| UV radiation | HaCaT cells, treated with multiple doses (UV)A+B (UVA, 150-200 mJ/cm(2) and UVB, 15-20 mJ/cm(2) × 6) | L1 | Malignant transformation was associated with increase in L1 transcription | Rapid amplification of polymorphic DNA and sequencing | [95] |
|
| |||||
| HeLa cells irradiated with UV (10-20 J/m2) | L1 | Induction and transactivation | Luciferase reporter assay, semiquantitative and quantitative real time PCR | [96] | |
|
| |||||
| Peripheral blood cells from participants and estimates of sun exposure over the previous 6 weeks | L1 | Methylation in L1 decreased with increasing solar UV exposure, does not appear to be influenced or mediated by vitamin D status. | Bisulphite conversion, pyrosequencing | [97] | |
|
| |||||
| Human epidermal melanocytes (NHEM) and in melanoma cell lines irradiated with 10 and 30 mJ/cm2 UVC | HERV-K transcripts rec and np9 | In NHEM, HERV-K rec expression was induced 24 h after UV exposure with 10 mJ/cm2 UVC, and np9 expression increased 6 and 12 h after irradiation with both UV doses. | Quantitative real-time PCR | [102] | |
| In melanoma MEWO cells, G-361 and GR-M rec, and np9 expression was downregulated | |||||
|
| |||||
| Melanoma cell lines, irradiated with UVB (200 mJ/cm2) | HERV-K | Transcriptional activation of the retroviral pol gene as well as in an enhanced expression of the retroviral envelope protein (env) | Quantitative real time PCR and immunofluorescence | [103] | |
| Induced the production of retroviral particles | |||||
|
| |||||
| Melanoma and melanocyte cell lines irradiated with 200 mJ/cm2 of UVB | HERV-K (HML-2) | Decrease in relative expression compared with nonirradiated cells, with loci to loci variations | Quantitative real-time PCR | [101] | |
|
| |||||
| Air pollution | Peripheral blood cells from Ma Ta Phut industrial estate workers in Rayong, Thailand | L1 | Workers showed lower L1 methylation than rural residents | Bisulphite conversion, pyrosequencing | [121] |
|
| |||||
| Peripheral blood cells from workers in an electric furnace steel plant exposed to PM10 | L1 and Alu | PM10 exposure levels were negatively associated with methylation in both Alu and L1 | Bisulphite conversion, pyrosequencing | [122] | |
|
| |||||
| Peripheral blood cells from participants exposed to fine and coarse concentrated ambient particles (CAPs), or HEPA-filtered air | L1 and Alu | Fine CAPs exposure lowered Alu methylation | Bisulphite conversion, pyrosequencing | [123] | |
|
| |||||
| Murine RAW264.7 macrophages exposed to PM10 | L1, SINE B1 SINE B2, major and minor satellites | Increase in the methylation of L1 and SINE B1, increase in expression of SINE B2, major and minor satellites | Methylation-sensitive digestion, qPCR | [125] | |
|
| |||||
| C57BL/CBA mice exposed to ambient air near two steel mills and a major highway | Tandem repeat | 1.6-fold increase in sperm mutation frequency in mice exposed for 10 weeks with a 6 week recovery period; no increases in mutation frequency in DNA collected after 3-10 wks of exposure | PCR | [124] | |
|
| |||||
| Traffic-related particles and diesel exhaust | Peripheral blood cells from elderly participants in the Boston area Normative Aging Study | L1 and Alu | L1 methylation decreased after recent exposure to higher black carbon and PM2.5 | Bisulphite conversion, pyrosequencing | [128] |
| No association was found with Alu methylation | |||||
|
| |||||
| Peripheral blood cells from elderly participants in the Boston area Normative Aging Study | L1 and Alu | An increase in black carbon over a 90-day period was associated with Alu hypomethylation. An increase in SO4 over a 90-day period was associated with L1hypomethylation. The GSTM1-null genotype strengthened the association between black carbon and Alu hypomethylation. | Bisulphite conversion, pyrosequencing | [129] | |
|
| |||||
| Peripheral blood cells from steel workers exposed to metal-rich PM10, gas-station attendants exposed to air benzene, and truck drivers exposed to traffic-derived elemental carbon | L1 and Alu | L1PA2 showed lower DNA methylation in steel workers and gas station attendants; L1Ta showed lower DNA methylation in steel workers; AluYb8 showed higher DNA methylation in truck drivers Stronger effects on older LINEs from PM10 and benzene, and on younger Alu from PM10 | Bisulphite conversion, pyrosequencing | [42] | |
|
| |||||
| Peripheral blood cells from non-smoking asthmatic subjects exposed to filtered air and diesel exhaust | L1 and Alu | Diesel exhaust-associated change was found for CpG sites overlapping with Alu and L1 elements | Human Methylation Array | [133] | |
|
| |||||
| Benzene | Peripheral blood cells from gas station attendants and traffic policemen vs controls in Milan | L1 | Decreased methylation, -2.33% for 10 fold increase in airborne benzene | Bisulphite conversion, pyrosequencing | [138] |
|
| |||||
| Peripheral blood cells from gas station attendants and traffic policemen vs controls in Milan | Alu | Decreased methylation, -1.00% for 10 fold increase in airborne benzene) | Bisulphite conversion, pyrosequencing Bisulphite conversion, pyrosequencing | [138] | |
|
| |||||
| Peripheral blood cells from non-smoking asthmatic subjects exposed to filtered air and diesel exhaust | L1 | Decreased methylation, -2.41% for interquartile range increase in personal airborne benzene | Human Methylation Array | [133] | |
|
| |||||
| Peripheral blood cells from gas station attendants | L1 and Alu | Methylation negatively associated with airborne benzene, and t,t-muconic acid (t,t-MA) in urine. | Bisulphite conversion, pyrosequencing | [139] | |
|
| |||||
| Peripheral blood cells from Bulgarian petrochemical workers vs controls | L1 | Decreased methylation (-0.15% mean methylation level for each interquartile range increase in S-phenylmercapturic acid in urine) | Bisulphite conversion, pyrosequencing | [140] | |
|
| |||||
| 1,3-Butadiene | C57BL/6J male mice inhaling 6.25 and 625 ppm 1,3-butadiene for 2 weeks | L1 | Hypomethylation | Methylation-sensitive digestion, qPCR | [144] |
|
| |||||
| NOD/LtJ, CAST/EiJ, A/J, WSB/EiJ, PWK/PhJ, C57BL/6J, and 129S1/SvImJ male mice inhaling 0 or 625 ppm 1,3-butadiene for 2 weeks | L1, major and minor satellites | L1 DNA methylation was significantly decreased after BD exposure in 129S1/SvImJ and C57BL/6J mice. Decrease in the level of L1 expression in BD-exposed CAST/EiJ mice; Increase in expression of L1 and major and minor satellites in BD-exposed C57BL/6J mice | Methylation-sensitive digestion, qPCR | [145] | |
|
| |||||
| Male C57BL/6 mice inhaling filtered air or 425 ppm of BD by inhalation 2 weeks | SINE B1 and B2, and minor and major satellites | Hypomethylation in the livers and lungs. Non-target organ (kidneys) was not affected | Methylation-sensitive digestion, qPCR | [146] | |
|
| |||||
| Arsenic | Cord blood, Lymphocytes in vitro | L1 | No change in L1 methylation in vivo | Combined bisulfite restriction analysis | [194] |
| Decrease in L1 methylation in lymphocytes exposed to arsenite for 4 hours (not 2 or 8h) | |||||
|
| |||||
| HepG2 cells | L1 | Increased rates of retrotransposition after 12 days | Construct, FACS | [197] | |
|
| |||||
| Peripheral blood cells from elderly men | L1 and Alu | Decrease L1 methylation. Increase Alu methylation in individuals with low plasma folate. | Bisulphite conversion, pyrosequencing, | [195] | |
| Decrease Alu methylation in individuals with high plasma folate | |||||
|
| |||||
| Peripheral blood cells from a case-control study of bladder cancer in New Hampshire | L1 | Levels of arsenic in the 90th percentile were associated with reduced L1 methylation (P = 0.04) | Bisulphite conversion, pyrosequencing, | [193] | |
|
| |||||
| mouse NIH3T3 cell | VL30 | 2.5-20μM sodium arsenite revealed induction of retrotransposition events in a dose- and time-dependent manner, effect is amplified by Hsp70 | Flow cytometry | [198] | |
|
| |||||
| Peripheral blood cells from Spanish participants | L1 | Decrease L1 methylation with increasing toenail arsenic | Bisulphite conversion, pyrosequencing, | [196] | |
|
| |||||
| Cadmium | Peripheral blood cells from nonsmoking women from Argentina | L1 | Urinary cadmium was inversely associated with L1 methylation | Bisulphite conversion, pyrosequencing, | [182] |
|
| |||||
| Lead | Peripheral blood cells from elderly participants in the Boston area Normative Aging Study | L1 and Alu | Patella lead levels were inversely associated with L1 but not Alu | Bisulphite conversion, pyrosequencing, | [216] |
| No association with tibia or blood lead | |||||
|
| |||||
| Umbilical cord blood samples from the biorepository of the Early Life Exposures in Mexico | L1 and Alu | Maternal patellar lead levels were inversely correlated with umbilical cord L1 methylation Maternal tibia lead levels were inversely correlated with Alu methylation. | Bisulphite conversion, pyrosequencing | [217] | |
|
| |||||
| Peripheral blood cells from workers at a battery plant | L1 | Pb exposure significantly decreased the level of L1 methylation | Methylation-specific real-time PCR | [218] | |
|
| |||||
| Mercury | Buccal mucosa cells from dentists | L1 | No significant relationships between urine or hair Hg and L1 DNA methylation | Bisulphite conversion, pyrosequencing | [222] |
|
| |||||
| Human neuroblastoma cell line and three non-neuronal cell lines | L1 | Mercury increased expression of L1 RNA, activity of the L1 5’UTR, and L1 retrotransposition in neuroblastoma cells | Quantitative real-time PCR | [223] | |
|
| |||||
| Cigarette smoke condensate | Normal human small airway epithelial cells and cdk-4/hTERT-immortalized human bronchial epithelial cells (HBEC) | L1 | L1 hypomethylation, Progressive genomic hypomethylation and locoregional DNA hypermethylation induced by CSC coincided with an increase in soft-agar clonogenicity | Bisulphite conversion, pyrosequencing | [154] |
|
| |||||
| Cigarette smoking | DNA from placenta | L1 and Alu | Mean AluYb8 methylation was significantly higher with maternal tobacco use during pregnancy (p < 0.01). | Bisulphite conversion, pyrosequencing | [156] |
|
| |||||
| Tumor and corresponding normal esophageal mucosa in esophageal squamous cell carcinoma | L1 | L1 methylation levels in the normal tissue were inversely correlated with the Brinkman index (daily number of cigarettes × years, p = 0.037) and the alcohol consumption index (p = 0.0076) | Bisulphite conversion, pyrosequencing | [155] | |
|
| |||||
| Persistent Organic Pollutants | Peripheral blood cells from Korean participants | L1 and Alu | Most OC pesticides were inversely and significantly associated with %5-mC in Alu. Most PCBs and PBDEs showed nonsignificant inverse trends with %5-mC in Alu. POPs were not associated with %5-mC in L1 | Bisulphite conversion, pyrosequencing | [170] |
|
| |||||
| Peripheral blood cells from newborns and nine-year old Mexican-American children | L1 and Alu (compared with POP concentrations in blood) | Inverse relationships between prenatal DDT/E exposure and Alu and L1 methylation in cord blood DNA; not significant. Similar trends in nine-year-old children | Bisulphite conversion, pyrosequencing | [171] | |
|
| |||||
| Peripheral blood cells from Greenlandic Inuit | L1 and Alu | Alu inverse linear relationships between POP concentrations in blood and: p,p’-DDT, p,p’-DDE, β-hexachlorocyclohexane, oxychlordane, α-chlordane, mirex, sum of polychlorinated biphenyls, and sum of all POP. L1: no significant changes | Bisulphite conversion, pyrosequencing | [169] | |
|
| |||||
| Benzo(a)pyrene | HeLa cells | L1 | Increased rate of retrotransposition, increased L1 mRNA levels | Tranfection with a reporter, semi-quantitative PCR | [175] |
|
| |||||
| HeLa cells | L1 | Decreased L1 methylation after 96h but not at earlier time-points | Bisulphite conversion, pyrosequencing | [176] | |
|
| |||||
| Vascular smooth muscle cells | L1 | BaP and its 7,8-diol metabolite (0. 03-3 M), activate L1Md in a concentration-dependent manner. Two other metabolites, 3-OH BaP and 3,6-BaP quinone (0.03-3 M) also activated L1Md | Northern analysis | [174] | |
|
| |||||
| MCF-7 and T47-D cells | SINE and LTR | BaP-induced hypomethylation events identified in SINE and LTR elements | Methyl acceptor assay, sequencing | [177] | |
2. Transposable elements and ionizing radiation
Exposure to ionizing radiation (IR), both man-made and natural, constitutes a serious hazard for human health. Of major concern is medical radiation, utilized as a diagnostic and treatment modality because of the ever-growing number of patients routinely being exposed. Recent reports suggest that a significant risk of radiation-induced health effects, including cancer, exists even after medical procedures associated with exposure to low doses of IR [77,78]. While it is generally accepted that the benefits of medical radiation outweigh the risks, there are, however, considerable concerns, since in the US alone tens of millions of unnecessary computed tomography scans are performed annually, and approximately half of cancer patients receive some form of radiotherapy [79-81]. Additionally, large cohorts of people can be exposed to radiation because of accidents, such as Chornobyl and Fukushima-Daiichi.
2.1. Effects of terrestrial radiation on methylation and expression of TEs
Terrestrial radiation includes primarily low- linear energy transfer (LET) photon radiation, such as X- and γ-rays and is characterized by relatively diffuse energy deposition patterns. It is a potent genotoxic agent capable of inducing DNA single- and double-strand breaks, oxidative damage, and mutation induction.
IR is also capable of targeting the cellular epigenome and DNA methylation, in particular. Earlier studies described hypermethylation of tumor-suppressor genes in lung cancers of occupationally exposed workers in Russia [82]. Aside from the gene-specific hypermethylation associated with exposure to IR, a wealth of data from in vivo studies also indicate loss of global DNA methylation [83-90]. Further investigations have revealed that alterations in global DNA methylation are primarily associated with the alterations in the methylation status of TEs in a tissue-, dose-, and radiation quality-dependent manner [91-94].
Less consistency has been observed in DNA methylation response to IR in vitro [95-97]. For instance, Goetz et al reported loss of L1 methylation in RKO human colorectal carcinoma cells after exposure to 1 Gy of X-rays (250 kV peak, 13 mA; half-value layer, 1.65 mm copper; 2 keV/μm; dose rate of 2.4 Gy min−1); however, the same dose of radiation led to hypermethylation of L1 in AG01522D primary human diploid skin fibroblasts [96]. Importantly, these changes were detected at ~20 population doublings after irradiation, suggesting a persistent nature of cell-dependent radiation-induced alterations in L1 methylation. At the same time, no significant changes in methylation of Alu elements were detected in either cell line.
A lack of low dose-associated alterations in methylation of TEs was observed in in vivo studies. Exposure to 10 mGy of X-ray (using an attenuated 140 kVp X-ray beam (8 mm Al half value layer) from a Gulmay D3150 superficial X-ray unit; dose-rate of 13.9 mGy/min) did not result in any persistent alterations in the methylation of L1, SINE B1, and IAP TEs in peripheral blood, spleen, or liver of C57BL/6 mice [98]. Exposure to a higher dose of 1 Gy of X-irradiation led to strain-, tissue-, sex-, and time-dependent alterations in methylation of the TEs [99]. Interestingly, hypermethylation of L1 was identified in relatively radio-resistant C57BL/6 mice at day 1 after irradiation, followed by hypomethylation at day 14. At the same time, radiosensitive BALB/c mice were characterized by hypermethylation of L1 in spleen tissue at 1 and 14 days after exposure. Methylation of SINE B1 and IAP was affected to a lesser extent [99]. A limitation of this study was that the mice of different strains analyzed at different time-points were irradiated at somewhat different conditions. Specifically, C57BL/6 mice, analyzed at day 1 after irradiation, were exposed with 100 kVp Philips RT100 SXR X-ray unit, while the mice analyzed at day 14 after irradiation were irradiated with a 140 kVp Gulmay D3150 X-ray unit. BALB/c mice were exposed using a 6 MeV X-ray beam from a Varian 600 CD Linear Accelerator. Exposures to doses higher than 1 Gy are usually associated with the loss of global and repetitive elements-associated DNA methylation [84,85,87-89]. Persistent global and TEs-associated hypomethylation after exposure to high doses of IR has been observed even in bystander tissues, as evident in a study reporting a 5 to 10% decrease in methylation and 60% increase in expression of L1 element in the spleen tissue 7 months after localized hippocampal irradiation (20Gy (5cGy/s) of X-rays, 90 kV, 5 mA) [91].
2.2. Effects of space radiation on methylation and expression of TEs
The recent introduction of proton radiation into clinical practice and interest in space exploration has inspired the investigation of biological effects and molecular mechanisms of response to space radiation exposure. It is now clear that exposure to proton and heavy ion irradiation, the sources of space radiation, is associated with more complex clustered and often irreparable DNA damage, leading to greater relative biological effectiveness in cell death (reviewed in [100]).
Studies in cell culture have confirmed that space radiation epigenetic effects are also distinct from the effects elicited by terrestrial radiation. Exposure to low mean absorbed doses of protons (150 MeV/n, LET 0.55 keV/μm; dose rate 2 cGy min−1) or 56Fe ions (56Fe+26, 1 GeV/n, LET 150 keV/μm; 10 cGy min−1) lead to hypomethylation of L1 and Alu elements in AG01522D and RKO cells at 16-20 population doublings after irradiation [96]. Similar effects were observed in the human-hamster hybrid cell line GM10115, where exposure to 1 Gy of 56Fe (56Fe+26, 1GeV/amu, 150keV/μm, dose rate 2 cGy min−1) led to hypomethylation in both L1 and Alu [95]. At the same time, exposure to 0.1 Gy of 56Fe did not affect the methylation status of these retrotransposons.
Exposure to mean absorbed doses of 56Fe ions within10-100 cGy; 56Fe+26, LET 175 keV/mm did not affect the methylation status of LINE-1 in the livers of C3H/HeN mice for up to 120 days after exposure [92]. This is a particularly interesting finding because exposure to 56Fe within this range of doses is associated with the development of liver tumors in this mouse strain [101,102], and the global and L1-associated DNA hypomethylation is a well-recognized hallmark of cancer and hepatocellular carcinoma in particular [103]. It is possible that L1 hypomethylation is a signature of later time-points, or high-LET hepatocarcinogenesis may not be necessarily associated with L1 hypomethylation at all. Another explanation is that assays specific for two L1 families – TF and A – were used in this study, while it has been suggested that L1s from different families may differ in their response to external stressors [44].
The same study has reported weak hypomethylation of both TF and A L1s shortly after exposure to 56Fe in the C3H/HeN mice lungs (day 1- day 7) and subtle hypermethylation of TF at day 120 [92]. L1 hypermethylation (26% increase) was also observed in the lungs of C57BL/6 mice 5 months after exposure to 0.4 Gy of 56Fe (56Fe+26, 600 MeV/nucleon), but not after lower doses [94]. Similar effects were observed in other TEs as well, including SINE B1, Charlie, and Mariner (15-25% increase), where the latter two present TEs that are currently transpositionally-inactive in mammals. This hypermethylation was clearly associated with further transcriptional silencing of these TEs. Interestingly, a recent study reported induction of lung tumors in mice exposed only to 56Fe doses below 0.4 Gy [104], suggesting that hypermethylation and silencing of TEs may have a protective effect against heavy ion-induced carcinogenesis.
Of particular interest are the effects of high-LET radiation on TEs in the hematopoietic system because exposure to 56Fe and 28Si has been associated with increased rates of acute myeloid leukemia (AML) in mice [101,102], and studies indicate alterations in L1 and Alu methylation in patients with AML [105,106]. Exposure to 56Fe ion radiation selectively targeted transposable elements in the less differentiated hematopoietic stem and progenitor cells, while the effects in mononuclear cells that are primarily comprised of terminally differentiated cells were miniscule [93]. Interestingly, while global, L1-, and SINE B1-associated dose-dependent hypermethylation was observed in hematopoietic stem and progenitor cells 30 days after exposure (similar to abovementioned findings in the lung by Lima et al. [92] at the same time-point), global hypomethylation (37% decrease) was observed 22 weeks after exposure to 0.4 Gy of 56Fe (56Fe+26, 600 MeV/nucleon) – a dose associated with increased rates of AML [101]. Importantly, this hypomethylation was associated with a dramatic reactivation of both L1 and SINE B1, suggesting a possible link between the epigenetic alterations and development of AML.
2.3. Exposure to ionizing radiation and L1 retrotransposition
Previous studies have indicated that hypomethylation of L1 elements after exposure to radiation is associated with its transcriptional reactivation and may potentially result in its de novo retrotransposition [91]. Indeed, accumulating evidence clearly demonstrates that both low- and high-LET irradiation may induce L1 retrotransposition in vitro; however, radiation quality-dependent differences were noted. Both X-rays (0, 2.5, 5 or 10 Gy; dose rate of 0.86 Gy/min; 200 kV, 20 mA) and carbon-ion beams (0, 1, 2 or 4 Gy; 18.3 MeV/nucleon, 108 keV/μm) similarly increased frequencies of transcription in L1/reporter knock-in human glioma cell line NP2; at the same time, X-ray induced the de novo insertions of 5’-truncated L1s, while carbon-ion beams promoted insertions of full-length or long-sized insertions [107]. Interestingly, radiation-induced retrotransposition has been shown to regulate gene expression in the human hybrid endothelial cell line EA.hy926 with the human L1 [108]. Although it is expected that radiation-induced L1 hypomethylation may serve as one of the key mechanisms for L1 retrotransposition, no studies, to the best of our knowledge, have been performed to analyze simultaneously the methylation, transcription, and retrotransposition of L1 in response to irradiation.
2.4. Effects of UV-radiation on TEs
UV-radiation, derived primarily from sun, is known to target cellular processes in human skin, including induction of inflammation, immune-suppression, cell death, and premature aging and can serve as both tumor initiator and promoter in skin carcinogenesis [109-111]. Chronic exposure to sub-apoptotic doses of UV results in transformation of the normal human keratinocyte cell line, HaCaT, and upregulation of L1 ORF2 [112]. Importantly, among all the exposed cells, increased mRNA transcripts of L1 ORF2 were detected only in transformed cells, suggesting L1’s role in UV-induced tumorigenesis. Another study reported L1 activation in human cervical carcinoma cells (HeLa) upon exposure to UV [113]. Although DNA methylation was not analyzed in any of these studies, the results from the recent report suggest the possibility of hypomethylation-induced reactivation of L1 in response to UV exposure since a decrease in peripheral blood lymphocytes L1 methylation was associated with increased solar UV exposure [114].
Of particular interest are several investigations that report involvement of human endogenous retroviruses (HERV) in melanoma, the most malignant type of skin cancer. Increased activity of the HERV-K(HML-2), an HERV family member, has been suggested to play a critical role in the etiology of melanoma, most probably due to increased promoter activity [115,116]. Additionally, several melanoma cell lines have been characterized by the production of retrovirus-like particles with reverse transcriptase activity [117]. A recent study by Schmitt et al. identified an HML-2 locus transcribed in melanomaderived samples only [118]. At the same time, it has been demonstrated that exposure to UV can activate HERVs, and HML-2 transcripts, in particular [118-120].
2.5. Conclusions
Overall, the exposure to terrestrial radiation at doses above 1 Gy is characterized by the loss of global DNA methylation, which stems from the TEs, and L1, particularly. At the same time, global DNA hypermethylation is characteristic for exposure to space radiation – proton and heavy ions. Limited numbers of studies suggest that this hypermethylation originates primarily from TEs, rather than from specific genes, but more studies are needed to confirm this notion. The principal differences in methylation of TEs in response to either terrestrial or space radiation may underlie the differences in biological responses to these types of radiation. Both terrestrial and space radiation cause long-term alterations in TEs-associated DNA methylation and expression, as has been shown both in vitro and in vivo, and lead to increased rates of L1 retrotransposition, suggesting their involvement in the development of delayed radiation-induced health outcomes. At the same time, it has to be taken into consideration that the data on methylation and expression of TEs is derived from in vitro and in vivo experiments, and the successful studies on retrotransposition were performed in vitro only. Thus, the translational relevance of these studies should be interpreted with caution, since no data, to our knowledge, exist on the effects of exposure to radiation on TEs methylation, expression, or retrotransposition in normal tissues in humans. Another considerable limitation of the studies that used rodent models is utilization of mainly male animals, while sex differences in response radiation exposure, including effects of DNA methylation are well-documented [89]. It also has to be acknowledged that the effects of low doses of radiation that are characterized by non-linear responses remain to be a challenge in assessment of the effects exerted on TEs in experimental systems.
3. Transposable elements and environmental contaminants
3.1. Particulate matter (PM)
The inhalation of atmospheric pollutants has been associated with eye, nose, and throat irritation, wheezing and breathing difficulties, allergies, worsening of respiratory diseases, such as asthma, and chronic obstructive pulmonary disease, lung cancer, increased cardiovascular morbidity, and hospitalization and emergency room visits for heart attack and stroke [121]. In addition, long-term exposure can cause impaired function of the respiratory tract, and significant damage to immune, neurological, and reproductive systems [122,123]. A priori, poor air quality affects everybody; however, specific groups of people (newborn and children, older adults, and those with compromised health) may be more susceptible to air pollution [124].
Ambient PM is a spatiotemporally variant and heterogeneous suspension of tiny solid particles and liquid droplets. The size, shape, and chemical content of PM depend on emissions from various sources and physicochemical transformations during atmospheric transport [125]. Exposures to different particle types and sizes [10-2.5 μm: PM10-2.5 (thoracic/coarse), 2.5-0.1 μm: PM2.5 (fine), and; >0.1 μm: PM0.1 (ultrafine)] are associated with premature mortality, increased hospitalizations, and the development/progression of sub-clinical and clinical disease in the respiratory and circulatory tracts as well as in unexpected organs, such as liver and brain [126-129]. The International Agency for Research on Cancer (IARC) has recently classified outdoor air pollution as carcinogenic to humans (Group 1 carcinogen) [130]. PM originating from traffic depletes antioxidants, with transition and heavy metals and oxidized organic compounds being the primary determinants of the observed responses [131-133]. PM from biomass burning appears to proceed through the same mechanism of action [134], although the chemical content and size distribution is different than that observed for traffic, dust, and biogenic particles [135,136]. Secondary inorganic aerosols (sulfate and nitrate formed from the oxidation of SO2 and NOx, respectively) have shown less toxicity in vitro; however, statistically significant associations of sulfates and nitrates with various health outcomes have been reported [137].
There have been numerous epidemiological studies linking ambient air pollution to an altered methylation status of L1 and Alu in the blood of exposed humans. For instance, occupational exposure at one of the largest steel, oil refinery, and petrochemical complexes at the Ma Ta Phut industrial estate in Southeastern Asia is associated with lower levels of L1 methylation and 3-(2-deoxy-β-D-erythropentafuranosyl) pyrimido[1,2-α]purin10(3H)-one deoxyguanosine adducts [138]. This was evident by lower levels of L1 in the blood leucocytes of 67 Ma Ta Phut industrial estate workers in comparison with 65 Ma Ta Phut residents (74.8% vs 78%; p<0.001). Similar effects were observed in 63 workers in an electric furnace steel plant in Brescia, Northern Italy, where both L1 (β= -0.19 %5-methylcytosine; p=0.04) and Alu (β= -0.34 %5-methylcytosine; p=0.04) were found to be hypomethylated in blood leukocytes after 3 days of work in comparison with those obtained on the first day of a work week [139]. Although the authors suggest that these effects were primarily caused by PM10 exposure, they could not exclude that exposures other than PM (heat, polycyclic aromatic hydrocarbons, carbon monoxide, and non-ionizing radiation) may potentially contribute to the observed effects. Short-term (130 minutes) exposure to concentrated ambient particles in healthy adult volunteers led to decreased methylation in Alu (β =0.74, adjusted-P=0.03), but not L1 elements, and was associated with increased diastolic blood pressure [140]. However, the low number (15) and wide age range (18-60) between the healthy participants do not allow for generalization to different population strats, as suggested by the authors [140].
Several studies in mice and cell culture have confirmed that exposure to ambient PM can trigger an epigenetic response, including alterations within the TEs; however, this response was not as uniform as in epidemiological studies. For instance, in contrast to a general tendency towards DNA hypomethylation observed in the majority of human studies, persistent global DNA hypermethylation was detected in the germline of C57BL/CBA mice exposed in situ for 3 to 10 weeks to ambient air near two integrated steel mills and a major highway [141].
A recent study investigated the short-term (24 hours) in vitro exposure of murine RAW264.7 macrophages (the cells that comprise the first line of defense against inhaled particles) to various concentrations of the aqueous extract of ambient PM10 (10-200 μg/ml). Although exposure did not result in changes of global DNA methylation, it did lead to redistribution of methylation patterns between L1, SINE B1, and SINE B2 [142]. This study also reported a dose-dependent loss of expression of all three DNA methyltransferases – Dnmt1, Dnmt3a, and Dnmt3b – the enzymes involved in the maintenance and de novo DNA methylation, suggesting possible global and TEs-associated hypomethylation at later timepoints. Therefore, a study that would investigate the effects of the prolonged exposure to PM would be of particular interest. At the same time, independent of methylation status, exposure to PM resulted in reactivation of SINE B2 in a dose-dependent manner, suggesting the possible role of histone modifications and non-coding RNAs in transcriptional regulation of TEs in the short-term response [142].
3.2. Traffic exhausts including diesel emissions
Traffic is one of the most important sources of urban air pollution contributing carbon mono- and dioxide, PM, and volatile organics and hydrocarbons [133]. Epidemiological studies clearly demonstrate an association between the ambient level of black carbon particles, a tracer for traffic pollution, and human diseases [143,144]. Accumulating evidence indicates that exposure to traffic-related black carbon affects the methylation of TEs. Exposure to black carbon led to decreased methylation of L1 (β= -0.11; 95% confidence interval, -0.18 to -0.04; p=0.002 [145] and Alu (β= -0.31; 95% confidence interval, 0.12- 0.50%) in 1,097 blood samples from 718 elderly participants in the Boston area Normative Aging Study [146]. Another study reports that truck drivers from Beijing, China (20 participants) exposed to trafficderived elemental carbon (exposure to elemental carbon ≥ 16.6 μg/m3) exhibited hypermethylation of one of the most youngest Alu elements – Yb8 (mean difference=0.4%, p=0.039), compared to indoor office workers (20 participants with elemental carbon exposure ≤ 16.6 μg/m3) suggesting that evolutionary age of RE subfamilies may determine differential susceptibility of DNA methylation to airborne pollutants [44].
Diesel exhaust, one of the most toxic traffic-derived pollutants, is the major contributor to fine PM in urban environments and has been classified by the United States Environmental Protection Agency (EPA) as a likely carcinogen [147,148]. Indeed, a number of cohort and case-control epidemiologic studies link diesel exhaust with lung cancer (reviewed in [148] and [149]). The mechanisms of the lung carcinogenesis are largely unknown, mainly due to the complex composition of diesel exhaust: its gaseous phase, which contains 1,3-butadiene, polycyclic aromatic hydrocarbons (PAHs), formaldehyde, benzene and acetaldehyde, and diesel exhaust particles – the largest source of emitted airborne particulate matter. Recent studies indicate that exposure to diesel also leads to altered methylation status of TEs, suggesting the epigenetic alterations may be also involved in lung carcinogenesis. Jiang and colleagues, in their controlled human crossover study, reported that short-term diesel exhaust inhalation was associated with changes in DNA methylation of circulating mononuclear cells in 16 asthmatics 6 and 30 hours postexposure [150]. Similar to the findings in RAW264.7 macrophages [142], the response to diesel exhaust exposure within L1 and Alu elements was not uniform, with sites of both hypo- and hypermethylation being identified [150]. Specifically, out of the 31 L1differentially methylated positions in the genome, 13 increased, while 18 decreased in DNA methylation after exposure. Out of the 25 Alu differentially methylated in response to short-term diesel exposure, 12 were characterized by increase and 13 by decrease in DNA methylation.
3.3. Volatile organic compounds
3.3.1. Benzene
Benzene is a known human carcinogen (classified as Group I carcinogen by IARC), causing acute myeloid and lymphoblastic leukemia and chronic myeloid leukemia [151-153]. Exposure to benzene has been also associated with other hematological disease, including myelodysplastic syndrome and aplastic anemia, bone marrow abnormalities, and neural tube defects [135,137]. Occupational exposure to lowbenzene levels in 78 gasoline filling workers and 77 urban traffic officers was associated with significant hypomethylation of L1 (-2.33%, p=0.009) and Alu (-1.00%, p=0.027) in comparsion with 58 unexposed referents in Milan, Italy [154]. Further work has reported an association of urine benzene biomarkers and hypomethylation of TEs. A study by Fustinoni et al. [155] reported a negative association between the benzene metabolite t,t-muconic acid and L1 and Alu methylation in occupationally exposed workers from the abovementioned study [154]. Weak, but significant L1 hypomethylation (-0.15%, p<0.01) was also reported with another urinary biomarker of benzene exposure, S-phenylmercapturic acid, in 158 Bulgarian petrochemical workers compared to 50 unexposed office workers [156]. Subsequent studies have demonstrated that loss of L1 methylation is associated primarily with its evolutionary older subfamilies [44].
3.3.2. 1,3-butadiene
1,3-Butadiene is a high-volume industrial chemical used in the production of synthetic rubber, resins, and plastics and is also a component of traffic-related air pollution and cigarette smoke [133,157]. It is a ubiquitous environmental pollutant and, according to IARC, known to be carcinogenic in humans [158]. The genotoxicity of 1,3-butadiene, attributed to its highly reactive metabolites, has been considered a critical event in the initiation of tumorigenesis [159]. Solid evidence of 1,3-butadiene-induced epigenetic alterations also suggests its epigenotoxicity. The latter was tightly associated with alterations in DNA methylation, primarily linked to the loss of L1 ORF1, SINE B1, and SINE B2 methylation in livers of male C57BL/6 mice after 2 weeks inhalational exposure to 1,3-butadiene [160]. Importantly, this study demonstrated the dose-dependent response, where exposure to higher concentrations of 1.3-butadiene was associated with the more pronounced loss of methylation within the TEs. Subsequent studies have shown that these effects were strain- [161] and tissue- [162] specific. Furthermore, 1,3-butadiene-induced hypomethylation of TEs was associated with their subsequent reactivation [160].
3.4. Tobacco smoke
Tobacco smoke is a potent carcinogen responsible for the vast majority of lung cancers in both men and women [163,164]. A number of known carcinogens are present in tobacco smoke, including aldehydes, polycyclic aromatic hydrocarbons (PAH), N-nitrosamines and aromatic amines, as well as 1,3- butadiene and benzene [149,165].
It is well accepted that tobacco smoke induces both genetic and epigenetic changes (reviewed in [166,167]). Among the latter, alterations in DNA methylation, associated with hypermethylation and subsequent silencing of tumor-suppressor genes, have received the most attention [168,169]. Tobacco smoke has been shown to target DNA methylation within the TEs as well. For instance, exposure to cigarette smoke condensate for up to 9 months under potentially relevant exposure conditions has led to loss of DNA methylation in L1 in human bronchial epithelial cells (HBEC) as well as in fully transformed A549 lung cancer cells [170]. This was associated with the decreased function of DNMT1 DNA methyltransferase, suggesting a possible mechanism for L1 hypomethylation.
Alterations in TEs associated with tobacco smoke are not only limited to the respiratory system. The levels of L1 in the normal esophagus mucosa from 105 patients with esophageal squamous cell carcinoma with history of cigarette smoking was inversely correlated with the Brinkman index (daily number of cigarettes × years) compared to non-smokers (p=0.037) [171]. Additionally, tobacco smoke during pregnancy has been shown to affect placental TEs. As reported by Wilhelm-Benartzi and colleagues [172], the methylation status of one of the youngest Alu elements, Yb8, was positively associated with tobacco smoke exposure (p<0.01) in 380 placental samples from full-term deliveries at the Women and Infants Hospital in Providence, Rhode Island (USA). At the same time, placental methylation of L1 was not affected by tobacco, as reported in the same studies.
3.5. Conclusions
Overall, it is without a doubt that exposure to different components of atmospheric pollution, including PM, may cause alterations in the methylation and expression of TEs, although we lack the knowledge in regard to the retrotransposition potential of such exposures. However, ambient PM is composed of different types of particles, such as soil and road dust, traffic exhausts, and biomass burning, to name a few. It is unclear, currently, whether different types of PM may cause differential responses in the methylation and expression of TEs and whether these potential differences may serve as biomarkers of exposure for certain types of ambient PM.
As indicated by epidemiological studies, exposures to atmospheric contaminants are mainly associated with hypomethylation of L1 in peripheral blood leukocytes, while some studies also indicate hypermethylation of Alu elements. Further in vitro investigations showed that the response to the same stressor is not uniform, with both hypo- and hypermethylation detected between the different TEs in the same experimental system [142]. The response to the same contaminant may vary even between the same TEs, depending on their genomic location, as shown by Jiang et al in their study on the effects of the short-term diesel exhaust inhalation in asthmatics [150]. The latter two studies provided clear evidence that alterations in the methylation status of TEs are not unidirectional, as thought before; however, the biological relevance of the TE-associated differentially methylated loci will have to be elucidated. As mentioned above, peripheral blood cells served as a material for the majority of these epidemiological studies. The authors of these studies, however, acknowledge that the observed differences in TEs methylation, although potentially representing markers of exposure or biological effects, are limited to leukocytes and, therefore, may not necessarily correlate with aberrant DNA methylation in the target tissues. Also, it has to be taken into consideration that the DNA used for the analyses of TEs comprises a pool of DNA from different types of circulating blood cells, primarily – leukocytes. Therefore, the alterations in DNA methylation may be a consequence of the shift in blood cell populations, resulting from the exposure.
3.6. Other environmental pollutants
3.6.1. Persistent organic pollutants (POPs)
Today, almost the entire general population is exposed to various POPs, such as organophosphates, carbamates, and pyrethroids through diet, inhalation, and dermal exposures. Accumulating evidence suggests that exposure to POPs, many of which are known endocrine disruptors, may be associated with increased risks of pancreatic and prostate cancer and non-Hodgkin lymphoma [173-175]. Epidemiological studies have also linked exposure to POPs and obesity [176] and have shown statistically significant correlations between pesticides exposures, preterm labor/fetal death [177], impaired semen quality [178], cryptorchidism [179], and thyroid hormones in women [180]. The Agricultural Health Study (“a prospective study of cancer and other health outcomes in a cohort of licensed pesticide applicators and their spouses from Iowa and North Carolina”) provides evidence of an association of pesticides exposures to prostate cancer in farmers [181,182] and childhood cancers [183].
Although the mechanisms of POPs-induced disease remain largely unknown, it is becoming increasingly evident that many of them may be underlined by an altered cellular epigenome [184]. A study performed on the blood samples collected from 71 Greenlandic Inuit, a population highly exposed to POPs, showed an inverse correlation between the blood percent methylcytosine (global DNA methylation) and the concentrations of a number of measured POPs [185]. Furthermore, the same study reported that the global DNA hypomethylation was associated with hypomethylation of Alu and that this was affected by increased concentrations of p,p’-DDT (β= -0.26, p=0.01), p,p’-DDE (β= -0.38, p=0.01), β-hexachlorocyclohexane (β= -0.48, p=0.01), oxychlordane (β= -0.32, p=0.01), α-chlordane (β= -0.75, p=0.05), and mirex (β= -0.27, p=0.01) in the blood, measured individually, and for the sum of all POPs (β= -0.48, p=0.01). The effects observed in L1 were not statistically significant [185].
A subsequent study in a cohort of 86 healthy adults (≥ 40 years of age) Koreans, randomly selected from 1,007 participants as controls in case-control studies on associations of POPs with diabetic or metabolic syndrome, confirmed these findings by reporting Alu hypomethylation, associated with the increased blood levels of oxychlordane (β= -0.28, p<0.05), trans-nonachlor (β= -0.28, p<0.05), and p,p’- DDE (β= -0.29, p<0.01) [186]. At the same time, exposure to POPs was not associated with the methylation status of L1. Importantly, most POPs were lower in the subjects by several orders of magnitude when compared with Greenland Inuit [185]; however, they were still able to cause significant alterations in Alu elements methylation. The most recent study in a birth cohort of Mexican-American children (n=358) reports that higher prenatal exposure to o,p’-DDT and p,p’-DDE was associated with lower Alu methylation in blood at birth, especially after adjusting for cell type composition (p=0.02) [187]. Weak associations of POPs with L1 methylation were only identified after examining the coexposure to DDT and DDE with polybrominated diphenyl ethers.
3.6.2. Benzo[a]pyrene (BaP)
BaP, a prototypical PAH, is a ubiquitous environmental pollutant and IARC group I carcinogen [164,188]. It is present in tobacco smoke and charcoal-grilled meat and is also a byproduct of fossil fuel combustion. Metabolized to reactive epoxides, BaP covalently binds to DNA causing DNA adducts and is capable of inhibiting DNA repair [189]. Accumulating evidence demonstrates that it is also a potent epigenotoxic carcinogen, and its epigenotoxicity, at least in part, is mediated by effects elicited on TEs.
Earlier studies have shown that exposure to BaP can result in reactivation and initiation of L1 retrotransposition in murine vascular smooth muscle cells vSMCs [174] and in human HeLa cells [190]. Later studies have demonstrated that these effects are tightly associated with the ability of BaP to cause L1 hypomethylation in HeLa cells by inducing early enrichment of the transcriptionally-active chromatin markers histone H3K4me3 and H3K9 acetylation and reducing DNMT1 association with the L1 promoter [191]. The effects of BaP are not limited to L1 only. Hypomethylation of SINEs and LTR elements have been reported in MCF-7 and HCC1806 breast cancer cell lines after exposure to BaP [192]. Of particular interest in this study is the identification of SINE elements hypomethylated at 16p13.3, the region of the chromosome 16 that includes the TSC2 tumor suppressor gene, in both investigated cell lines. The expression of the gene, however, was not addressed in this study, and the qualitative nature of the analysis does not allow for evaluation of the extent of the BaP-induced SINE hypomethylation.
3.6.3. Conclusions
In the limited studies, exposure to POPs was associated with significant loss of Alu methylation in the peripheral blood cells, independently of the levels of POPs, geographical location of the study, and racial and age differences. At the same time, none of the studies could identify alterations in methylation of L1 except for the study in newborns where weak L1 hypomethylation was identified after examining the co-exposure to DDT and DDE with polybrominated diphenyl ethers [187]. These findings suggest that Alu, but not L1 elements, are primarily targeted by POPs; therefore, the methylation status of Alu may serve as a surrogate biomarker for exposure to POPs.
Exposure to BaP is associated with the loss of L1, SINE, and LTR elements, suggesting global DNA hypomethylation effects; however, another study performed on C3H/10T1/2 mouse embryonic fibroblast cells, reports global DNA hypermethylation in response to BaP treatment [193]. One possible explanation of this discrepancy is that in the latter study, cells were exposed to BaP for 4 weeks, while the former studies aimed to evaluate relatively short-term effects. It has to be noted that all the investigations on BaP effects on TEs were performed in vitro, and in vivo experiments using the rodent models are clearly needed to confirm these findings.
3.7. Metals
Massive utilization of metals in industry has led to increased occupational and environmental exposure to them, many of which are considered human carcinogens [123]. Other, non-cancerous health effects of exposure to metals include pulmonary and cardiovascular disease and various neurodevelopmental deficits. Carcinogenic metals are typically weak mutagens and do not form DNA adducts; however, they are capable of causing oxidative damage to DNA. Accumulating evidence clearly indicates that exposure to metals, independent of their genotoxicity and mutagenicity, is associated with epigenetic alterations. The vast majority of investigated metals is capable of altering DNA methylation patterns, both in vitro and in vivo, and this feature is also documented in epidemiological studies [194-197].
3.7.1. Arsenic
The US EPA has listed arsenic as the number 1 chemical in its Comprehensive Environmental Response, Compensation and Liability Act Priority List of Hazardous Substances [198]. Arsenic is a ubiquitous environmental contaminant present in soil, rocks, aquatic environments, and even as airborne particles. Arsenic has been also used in insecticide production and treatment of acute promyelocytic leukemia. The most common exposure of humans to arsenic occurs via consumption of contaminated underground water and food in the form of either arsenite (AsIII) or arsenate (AsV) [199,200]. Health effects associated with arsenic exposure are various and include cardiovascular, respiratory, reproductive, and gastrointestinal disease and neurologic defects [201-203]. Exposure to inorganic arsenic is also associated with skin, liver, urinary, and lung cancers; and it has been, therefore, classified as a Group 1 human carcinogen by IARC [204,205].
Arsenic can affect a variety of cellular processes, including signal transduction pathways and redox status; it can also induce deletion mutations and chromosomal aberrations, but not point mutations (reviewed in [194]). Accumulating evidence also suggests that arsenic may promote cellular transformation via epigenetic mechanisms and DNA methylation particularly. Indeed, arsenic, in order to be detoxified and excreted from the cell, needs to be reduced and methylated. Studies indicate that chronic exposure to arsenic results in global DNA hypomethylation, decreased levels of S-adenosyl methionine (the main donor of methyl groups for DNA methylation), and reduced DNA methyltransferase activity [206,207]. In vitro and epidemiological studies suggest this global DNA hypomethylation originates from TEs, such as L1, whose methylation was negatively associated with the increasing arsenic exposure [208-211]. For instance, levels of arsenic in the 90th percentile were associated with reduced L1 methylation (p=0.04) in the blood of 459 adult participants of a population-based incident case control study in New Hampshire (USA) [208]. In utero exposure to arsenic, however, despite the significantly higher levels of arsenic in cold blood, fingernails, toenails, and hair of newborns, was not characterized by altered methylation status of L1 in their cord blood lymphocytes as reported by Intarasunanont and colleagues [209]. On the other hand, the same study reports that in vitro exposure of human lymphoblasts resulted in loss of L1 methylation after short-term (4-8 hrs) and long-term (8 weeks) exposure with arsenite [209]. Toenail levels of arsenic showed a trend of negative association with L1methylation (β= - 0.05 per one interquartile range [0.06 μg/g] increase in toenail arsenic; 95% confidence interval = -0.11 to 0.02) in 581 participants from the Normative Aging Study in Boston [210]. Interestingly, hypermethylation of Alu elements was reported in the same study (β= 0.08 per interquartile range [0.06 μg/g] increase in toenail arsenic; 95% confidence interval = 0.03 to 0.13) [210], suggesting that alterations in DNA methylation patterns upon arsenic exposure are also not unidirectional.
Arsenic can also trigger retrotransposition events. It has been shown that exposure to low levels of arsenic trioxide resulted in L1 retrotransposition in HepG2 cells [212]. Whether or not this was caused by L1 hypomethylation remains unknown since L1 methylation status was not addressed in this study. Another study reported arsenic-induced retrotransposition of viral-like 30 (VL30) LTR in mouse NIH3T3 cells that was inhibited upon treatment with antioxidant N-acetyl-cysteine [213].
3.7.2. Cadmium
Cadmium is a common environmental and occupational toxicant and is one of the most widespread environmental pollutants among metals. Battery manufacturing, tobacco usage, and consumption of certain foods are the main source of exposure to cadmium. The exposure has been associated with adverse health effects, including bone and kidney damage and developmental impairment [214-216]. Cadmium has been also classified as human carcinogen [217]. It is capable of causing damage to DNA only at high concentrations and does not form stable DNA adducts. In vitro studies clearly indicated that cadmium is a potent epigenotoxic stressor with abilities to affect both global and genespecific methylation [218-221]. An epidemiological study in 202 non-smoking Argentine women reported that DNA hypomethylation in peripheral blood was associated with dietary exposure to cadmium and indicated that this was associated with hypomethylation of L1 (β= –0.50, p = 0.0070) [197].
3.7.3. Lead
Lead is a heavy metal still considered as one of the most harmful environmental toxicants for young children, pregnant women, and other adults, despite great legislative efforts to reduce the use of lead and to control exposure pathways [222]. Sources for lead exposure include deteriorated lead based paints in older homes, drinking water, burning of fossil fuels, trades, hobbies (e.g., making stained-glass windows), ammunition, batteries involved in construction and mining, consumer products, and cultural home health remedies (e.g., azarcon, ayurvedic) [223,224].
The absorption of lead can lead to irreversible neurological damage, especially in children. Even at low exposures, over time, loss of intelligence and problems with hearing, memory, and balance have been documented [225,226]. Pregnant and lactating women are also at greater risk to low levels of lead exposure, where the weight-of-evidence is convincing that exposures can lead to lasting adverse neurotoxic outcomes on the fetus and on infant development because lead crosses the placenta and is found in breast milk [227,228].
Health outcomes of lead exposure have been related to oxidative stress and effects on the immune system based on genetic determinants, even at low levels [229,230]. Exposure to lead has also been associated with alterations in DNA methylation in peripheral blood. Specifically, L1 (β= –0.25; p< 0.01), but not Alu (β= –0.03; p= 0.4), hypomethylation was associated with patella lead levels in 2280 male participants of the Normative Aging Study [231]. In the Early Life Exposures in Mexico to Environmental Toxicants (ELEMENT) study, an inverse dose-response relationship was found in which quartiles of patella lead correlated with umbilical cord blood L1 methylation (p for trend 0.01) 103 analyzed samples [232]. At the same time, tibia lead correlated with Alu methylation (p for trend 0.05) [232]. Furthermore, maternal tibia lead was negatively associated with the methylation status of Alu elements in umbilical cord blood (β= –0.027; p= 0.01). A recent case-control study that involved 53 workers from a battery plant reported a significant decrease in blood L1 methylation (-10,3%, p<0.001), as compared to 57 healthy volunteers with matching age and gender [233]. The same study also reports a significant loss of L1 methylation in human embryonic kidney HEK293 cells 24 and 48 hrs after in vitro exposure to lead acetate [233].
3.7.4. Mercury
Mercury is among the World Health Organization (WHO)’s top 10 chemicals of major public health concern [234]. Exposure to mercury, a heavy metal pollutant, is a concern in metal and electric industries and can also occur with consumption of seafood and exposure from burning fossils. Additionally, it has been estimated that over 6,000 tons of mercury are released into the environment annually worldwide, suggesting mercury evaporation from contaminated sites as another source of exposure [235].
Biological effects elicited by mercury may be mediated via modulation of epigenetic mechanisms. Exposure to methylmercury has led to global DNA hypomethylation in neural stem cells [236]. Limited data exist on the effects of mercury on TEs. Occupational exposure to mercury among 131 dental professionals was not characterized by alterations in L1 methylation in DNA isolated from buccal mucosa; however, hypomethylation of SEPP1 promoter, the gene known to bind mercury, was detected among male participants [237]. Interestingly, a recent study reported activation of L1 in BE (2)-M17 human neuroblastoma cells, exhibited as increase in mRNA, protein expression, and genomic retrotransposition upon exposure to mercury [238]. Mercury was not able to induce similar events in three other non-neuronal cell lines, underlining the neurotoxic effects of mercury exposure.
Other environmentally ubiquitous heavy metals targeting the cellular epigenome include nickel and chromium; however, the knowledge on how they impact TEs is limited. For instance, chromium has been shown to cause global DNA hypomethylation in A549 lung cells and B lymphoblastoid cells, as well as in red blood cells of industrial chromate workers, but the methylation status of TEs was not addressed in these studies [239,240]. One study indicates that exposure to nickel chloride increased the L1 retrotransposition rates about 2.5 times in human HeLa cells [241].
3.7.5. Conclusions
The existing data on the effects of metals on TEs is derived primarily from epidemiological studies. Analysis of data obtained from human studies holds a number of advantages over the analysis of data obtained from in vitro and in vivo models. On the other hand, analysis of L1 and Alu methylation was performed mainly in peripheral blood cells, suggesting this data can be further utilized in development of biomarkers of exposure; however, it provides limited knowledge in regards to the molecular effects of metal exposure in their correspondent target organs.
Overall, exposure to metals is associated with the loss of blood L1 methylation, while the methylation status of Alu elements varies between the exposures and cohorts. Similar to the effects observed in response to PM exposure, different TEs may be differentially methylated in response to exposure to the same metal within the same experimental system. The results are also dependent on the tissue analyzed. For instance, lead patella levels were associated with L1 hypomethylation but not Alu, while tibia lead levels were associated with hypomethylation of Alu, but not L1 elements [232]. What determines such differences is currently unknown.
Accumulating evidence has also indicated that metals may induce retrotransposition events. Arsenic has been shown to be capable of causing retrotransposition of L1 and LTRs in human HepG2 and mouse NIH 3T3 cells, respectively, while mercury initiated L1 retrotransposition in human BE (2)-M17 cells, but not in three other non-neuronal cell lines. Both cell lines used for assessing of arsenic- and mercury-induced L1 retrotransposition are genomically unstable tumor cell lines; therefore, studies that will utilize the non-tumorous cell lines will be needed to confirm the L1 mobilization as an effect of metal exposure.
4. Concluding remarks
Evidence summarized in this review suggests that TEs are the sensitive endpoints for detection of effects caused by environmental stressors. Hypomethylation of L1 elements is the most frequently reported consequence of exposure observed in the studies. It was detected in numerous in vitro and in vivo, as well as in epidemiological studies; in target tissue and in peripheral or cord blood; in the tissue of exposed subjects in comparison to the tissue of control subjects and was inversely correlated with the levels of an environmental contaminant detected in the same subject. Loss of DNA methylation within the TEs, and L1 particularly, contributes to global DNA hypomethylation [91,93,160], given their abundance in mammalian genomes. Hypermethylation of TEs is a much less frequently reported effect of exposures, and the biological consequences of accumulation of methyl groups within the TEs still need to be elucidated. Limited studies that report hypermethylation of TEs link it to their transcriptional silencing [94], possibly suggesting formation of more condensed heterochromatic structures.
Accumulating evidence also demonstrates that loss of epigenetic control over TEs caused by environmental stressors may result in reactivation of TEs and initiation of retrotransposition. Numerous environmental stressors, including terrestrial, space, and UV-radiation, B[a]P, and metals were shown to cause L1 and ERV/LTR retrotransposition in experimental systems, suggesting the possible link between the exposure and development of disease.
4.1. Current challenges
Despite the significant progress in the field and rapid accumulation of data in the last ten years, some challenges exist in the comprehensive analysis and understanding of the effects exerted by the environment on TEs.
While in cancer both Alu and L1 are usually found to be hypomethylated, the early response to environmental toxicants and carcinogens, as evident from Table 2, often results in hypomethylation of L1 and hypermethylation of Alu. Furthermore, in the wealth of studies, only some classes of TEs are reported to be affected, while no significant alterations have been identified in the others. For instance, hypermethylation of Alu was frequently reported in response to exposure to POPs, while the methylation status of L1, analyzed within the same studies, remained unchanged. What underlies such differences remains unknown, but one of the possible explanations is the fact that different TEs belong to different regions of chromosomes: Alu are preferentially enriched in hypomethylated euchromatic regions, while L1 are found in hypermethylated heterochromatic and transcriptionally silent regions.
Differences in response to environmental cues may also be predetermined by differences in the tissue-specific methylation status of TEs. For instance, L1 in placenta have been reported hypomethylated in comparison with other tissues [42]. This might explain the lack of alterations in placental L1 methylation in response to in utero tobacco smoke exposure, discussed in this review earlier [172].
Furthermore, it has to be taken into consideration that certain differences in TEs methylation and expression are described between the different cell lines and even within the same tissue in different mouse strains [42,167]. Similarly, a comparison of results obtained from epidemiological studies is often challenging due to the high heterogeneity in human populations involved in these studies. Additionally, human studies are usually limited to biological fluids only, thus, often not allowing for investigations within the target organs.
Other challenges include differences in techniques and approaches for evaluation of the same endpoint that may yield incongruent results; complex structure and different evolutionary age of TEs; high variability of TEs in their retrotransposition activity; exhibited polymorphism of TEs; and extrapolation of results obtained from in vitro and in vivo studies to humans [45,49,242-244].
4.2. Future directions
4.2.1. TEs in risk assessment and as biomarkers of exposure
Epigenetic parameters hold a number of advantages over the genetic end-points and, therefore, have been proposed to be incorporated into current platforms for risk and safety assessment and as biomarkers of exposure to environmental stressors [167,245,246]. One of the particular concerns that has to be addressed before incorporation of epigenetics into regulatory practice is the large number of epigenetic alterations caused by exposures and observed in diseases. Therefore, the need for identification of specific parameters exists. Based on the growing evidence of numerous environmental stressors ability to affect the methylation of TEs and the persistent nature of these effects, TEs may serve as a unique platform for risk and safety assessment, as well as serve as biomarkers of exposure to environmental stressors with potential epi-mutagenic effects. However, it has to be determined which specific TEs and their families are the most sensitive targets for environmental stressors, as it has been shown on the example of L1 element that exposure to PM affects the methylation status of specific L1 families [44]. Furthermore, there is a need to determine a generally available methodology for TEs methylation assessment that will yield reproducible and comparable results across different laboratories.
4.2.2. TEs: role in cancer and biomarkers in cancer diagnostics
Global genomic hypomethylation that is usually originated from TEs is associated with decondensation of the chromatin structure, may affect the expression of genes that acquired TE insertions during evolution and lead to genomic instability. Furthermore, hypomethylation itself may predispose to tumor development, as evident from the mouse models of erythroleukemia and lymphoma [247,248].
Alterations in the methylation status of TEs observed after exposure may also result in the loss of proper control over the TEs expression and may subsequently lead to new somatic retrotransposition events. Up to date, a number of L1 retrotranspositions have been reported in human cancers [64,65,249]. Although retrotransposition rates in cancer are relatively low, insertional mutagenesis that results in inactivation of tumor-suppressor gene may have a detrimental effect on cancer initiation and promotion, as has been shown in the case of APC gene and colorectal cancer [62]. A recent study indicating that the TE-associated insertional mutagenesis in cancerous tissues preferentially occurs at genes commonly mutated in cancer and substantially disrupts their expression [64] further supports this hypothesis.
As has been shown above, environmental stressors, many of which are carcinogens or suspected carcinogens, are capable of causing persistent alterations in methylation of TEs and initiate retrotransposition. Furthermore, accumulating evidence demonstrates that L1 becomes a useful tool in cancer diagnostics, including utilization of L1 as a surrogate biomarker in identifying the cancer stage, tumor response to chemo- and radiotherapy and determination of tumor metastatic potential (reviewed in [45]). Similarly, L1 and Alu/SINEs present a promising platform for the development of surrogate biomarkers of exposures to environmental stressors with carcinogenic potential.
In conclusion, exposures to environmental stressors contribute to over 13 million deaths each year due to cancer and non-cancerous diseases [249]. Although the etiology and pathogenesis of these diseases vary, they are all characterized by alterations in TEs, including their hypo- and hypermethylation, reactivation, and retrotransposition. Accumulating evidence clearly demonstrates that these alterations are not the simple consequence of the disease, but often may drive the pathogenesis, as they can be detected early during disease development and even shortly after exposure to a given environmental stressor. These findings suggest that TEs may potentially be introduced into safety and risk assessment and serve as biomarkers of exposure to environmental stressors, as well as surrogate biomarkers in clinic and possible targets for therapeutic modalities for disease treatment and prevention.
Acknowledgments
We are thankful to Drs. Frederick A. Beland, Yuri Dubrova, Kristy Kutanzi, and Luisa Camacho for critical reading and Rebecca Helm for editing this manuscript. We apologize to our colleagues whose research was not mentioned here owing to the focus on specific models and space limitations. The research in Koturbash’ Lab is supported, in part, by the NIH/UAMS Clinical and Translational Science Award [UL1TR000039 and KL2TR000063], National Space Biomedical Research Institute (RE03701) through National Aeronautics and Space Administration NCC 9-58, and the Arkansas Biosciences Institute. The research in Fergusons’ Lab is supported, in part, by EPA NE-00F65601-0 and X8-00F10401-1 grants.
Abbreviations
- AML
Acute Myeloid Leukemia
- BaP
Benzo[a]pyrene
- CDC
Center for Disease Control and Prevention
- cGy
centiGrey
- COBRA
Combined Bisulfite Restriction Analysis
- DDE
Dichlorodiphenyldichloroethylene
- DDT
dichlorodiphenyltrichloroethane
- DNMT1
DNA Methyltransferase 1
- EPA
Environmental Protection Agency
- ERV
Endogenous Retrovirus
- H3K4me3
Histone 3, Lysine 4 trimethylation
- HBEC
Human Bronchial Epithelial Cells
- Gy
Grey
- IAP
Intracysternal A Particle
- IARC
International Agency for Research on Cancer
- IR
Ionizing Radiation
- LET
Linear Energy Transfer
- LINE
Long Interspersed Nucleotide Element
- LTR
Long Terminal Repeat
- MeV
Megaelectron Volt
- mRNA
messenger RNA
- miRNA
microRNA
- MS qPCR
Methylation-Sensitive quantitative Polymerase Chain Reaction
- ORF
Open Reading Frame
- PAH
polycyclic aromatic hydrocarbon
- PCR
Polymerase Chain Reaction
- PIWI
P-element Induced Wimpy testis
- piRNA
PIWI-interacting RNA
- PM
Particulate Matter
- POP
Persistent Organic Pollutant
- RNAi
RNA interference
- RUNX
Runt-related Transcription Factor
- SEPP1
Selenoprotein P1
- siRNA
small interfering RNA
- SINE
Short Interspersed Nucleotide Element
- SOCS1
Suppressor Of Cytokine Signaling 1
- SRY
Sex-determining Region Y
- SVA
SINE-R VNTR and Alu
- TE
Transposable Element
- UTR
Untranslated Region
- UV
Ultraviolet radiation
- VL30
Viral-Like 30
- WHO
World Health Organization
- YY1
Ying Yang 1
Footnotes
The views expressed in this manuscript do not necessarily represent those of the US Food and Drug Administration
Conflict of interests
The authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Isabelle R. Miousse, Email: IRacinemiousse@uams.edu.
Marie-Cecile G. Chalbot, Email: MGChalbot@uams.edu.
Annie Lumen, Email: Annie.Lumen@fda.hhs.gov.
Alesia Ferguson, Email: AFerguson@uams.edu.
Ilias G. Kavouras, Email: IKavouras@uams.edu.
References
- 1.de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7:e1002384. doi: 10.1371/journal.pgen.1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chenais B, Caruso A, Hiard S, Casse N. The impact of transposable elements on eukaryotic genomes: From genome size increase to genetic adaptation to stressful environments. Gene. 2012;509:7–15. doi: 10.1016/j.gene.2012.07.042. [DOI] [PubMed] [Google Scholar]
- 3.Ostertag EM, Kazazian HH., Jr Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001;35:501–538. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- 4.Burns KH, Boeke JD. Human transposon tectonics. Cell. 2012;149:740–752. doi: 10.1016/j.cell.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rebollo R, Romanish MT, Mager DL. Transposable elements: An abundant and natural source of regulatory sequences for host genes. Annu Rev Genet. 2012;46:21–42. doi: 10.1146/annurev-genet-110711-155621. [DOI] [PubMed] [Google Scholar]
- 6.Cowley M, Oakey RJ. Transposable elements re-wire and fine-tune the transcriptome. PLoS Genet. 2013;9:e1003234. doi: 10.1371/journal.pgen.1003234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belan E. LINEs of evidence: Noncanonical DNA replication as an epigenetic determinant. Biol Direct. 2013;8:22. doi: 10.1186/1745-6150-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jurka J, Kapitonov VV, Kohany O, Jurka MV. Repetitive sequences in complex genomes: Structure and evolution. Annu Rev Genomics Hum Genet. 2007;8:241–259. doi: 10.1146/annurev.genom.8.080706.092416. [DOI] [PubMed] [Google Scholar]
- 9.Faulkner GJ. Retrotransposons: Mobile and mutagenic from conception to death. FEBS Lett. 2011;585:1589–1594. doi: 10.1016/j.febslet.2011.03.061. [DOI] [PubMed] [Google Scholar]
- 10.Kaer K, Speek M. Retroelements in human disease. Gene. 2013;518:231–241. doi: 10.1016/j.gene.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Feng Q, Moran JV, Kazazian HH, Jr, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87:905–916. doi: 10.1016/s0092-8674(00)81997-2. [DOI] [PubMed] [Google Scholar]
- 12.Cost GJ, Feng Q, Jacquier A, Boeke JD. Human L1 element target-primed reverse transcription in vitro. EMBO J. 2002;21:5899–5910. doi: 10.1093/emboj/cdf592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tchenio T, Casella JF, Heidmann T. Members of the SRY family regulate the human LINE retrotransposons. Nucleic Acids Res. 2000;28:411–415. doi: 10.1093/nar/28.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang N, Zhang L, Zhang Y, Kazazian HH., Jr An important role for RUNX3 in human L1 transcription and retrotransposition. Nucleic Acids Res. 2003;31:4929–4940. doi: 10.1093/nar/gkg663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris CR, Dewan A, Zupnick A, Normart R, Gabriel A, Prives C, Levine AJ, Hoh J. p53 responsive elements in human retrotransposons. Oncogene. 2009;28:3857–3865. doi: 10.1038/onc.2009.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matlik K, Redik K, Speek M. L1 antisense promoter drives tissue-specific transcription of human genes. J Biomed Biotechnol. 2006;2006:71753. doi: 10.1155/JBB/2006/71753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cruickshanks HA, Vafadar-Isfahani N, Dunican DS, Lee A, Sproul D, Lund JN, Meehan RR, Tufarelli C. Expression of a large LINE-1-driven antisense RNA is linked to epigenetic silencing of the metastasis suppressor gene TFPI-2 in cancer. Nucleic Acids Res. 2013;41:6857–6869. doi: 10.1093/nar/gkt438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang N, Kazazian HH., Jr L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat Struct Mol Biol. 2006;13:763–771. doi: 10.1038/nsmb1141. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Kannan M, Trivett AL, Liao H, Wu X, Akagi K, Symer DE. An antisense promoter in mouse L1 retrotransposon open reading frame-1 initiates expression of diverse fusion transcripts and limits retrotransposition. Nucleic Acids Res. 2014;42:4546–4562. doi: 10.1093/nar/gku091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang CR, Burns KH, Boeke JD. Active transposition in genomes. Annu Rev Genet. 2012;46:651–675. doi: 10.1146/annurev-genet-110711-155616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexandrova EA, Olovnikov IA, Malakhova GV, Zabolotneva AA, Suntsova MV, Dmitriev SE, Buzdin AA. Sense transcripts originated from an internal part of the human retrotransposon LINE-1 5’ UTR. Gene. 2012;511:46–53. doi: 10.1016/j.gene.2012.09.026. [DOI] [PubMed] [Google Scholar]
- 22.Kulpa DA, Moran JV. Cis-preferential LINE-1 reverse transcriptase activity in ribonucleoprotein particles. Nat Struct Mol Biol. 2006;13:655–660. doi: 10.1038/nsmb1107. [DOI] [PubMed] [Google Scholar]
- 23.Kazazian HH., Jr Mobile elements: Drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 24.Goodier JL, Kazazian HH., Jr Retrotransposons revisited: The restraint and rehabilitation of parasites. Cell. 2008;135:23–35. doi: 10.1016/j.cell.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 25.Trelogan SA, Martin SL. Tightly regulated, developmentally specific expression of the first open reading frame from LINE-1 during mouse embryogenesis. Proc Natl Acad Sci U S A. 1995;92:1520–1524. doi: 10.1073/pnas.92.5.1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kano H, Godoy I, Courtney C, Vetter MR, Gerton GL, Ostertag EM, Kazazian HH., Jr L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes Dev. 2009;23:1303–1312. doi: 10.1101/gad.1803909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin SL, Li WL, Furano AV, Boissinot S. The structures of mouse and human L1 elements reflect their insertion mechanism. Cytogenet Genome Res. 2005;110:223–228. doi: 10.1159/000084956. [DOI] [PubMed] [Google Scholar]
- 28.McCarthy EM, McDonald JF. Long terminal repeat retrotransposons of mus musculus. Genome Biol. 2004;5:R14. doi: 10.1186/gb-2004-5-3-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mills RE, Bennett EA, Iskow RC, Devine SE. Which transposable elements are active in the human genome? Trends Genet. 2007;23:183–191. doi: 10.1016/j.tig.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Maksakova IA, Gagnier L, van dL, Mager DL. Genome-wide assessments reveal extremely high levels of polymorphism of two active families of mouse endogenous retroviral elements. PLoS Genet. 2008;4:e1000007. doi: 10.1371/journal.pgen.1000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malik HS, Burke WD, Eickbush TH. The age and evolution of non-LTR retrotransposable elements. Mol Biol Evol. 1999;16:793–805. doi: 10.1093/oxfordjournals.molbev.a026164. [DOI] [PubMed] [Google Scholar]
- 32.Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev. 1999;9:657–663. doi: 10.1016/s0959-437x(99)00031-3. [DOI] [PubMed] [Google Scholar]
- 33.Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- 34.Cordaux R, Hedges DJ, Herke SW, Batzer MA. Estimating the retrotransposition rate of human alu elements. Gene. 2006;373:134–137. doi: 10.1016/j.gene.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 35.Luo Y, Lu X, Xie H. Dynamic alu methylation during normal development, aging, and tumorigenesis. Biomed Res Int. 2014;2014:784706. doi: 10.1155/2014/784706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked alu sequences. Nat Genet. 2003;35:41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- 37.Walters RD, Kugel JF, Goodrich JA. InvAluable junk: The cellular impact and function of alu and B2 RNAs. IUBMB Life. 2009;61:831–837. doi: 10.1002/iub.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yakovchuk P, Goodrich JA, Kugel JF. B2 RNA and alu RNA repress transcription by disrupting contacts between RNA polymerase II and promoter DNA within assembled complexes. Proc Natl Acad Sci U S A. 2009;106:5569–5574. doi: 10.1073/pnas.0810738106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roman AC, Gonzalez-Rico FJ, Fernandez-Salguero PM. B1-SINE retrotransposons: Establishing genomic insulatory networks. Mob Genet Elements. 2011;1:66–70. doi: 10.4161/mge.1.1.15455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Estecio MR, Gallegos J, Dekmezian M, Lu Y, Liang S, Issa JP. SINE retrotransposons cause epigenetic reprogramming of adjacent gene promoters. Mol Cancer Res. 2012;10:1332–1342. doi: 10.1158/1541-7786.MCR-12-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schläppi M, Raina R, Fedoroff N. Epigenetic regulation of the maize spm transposable element: Novel activation of a methylated promoter by TnpA. Cell. 1994;77:427–437. doi: 10.1016/0092-8674(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 42.Ross JP, Rand KN, Molloy PL. Hypomethylation of repeated DNA sequences in cancer. Epigenomics. 2010;2:245–269. doi: 10.2217/epi.10.2. [DOI] [PubMed] [Google Scholar]
- 43.Jones PA. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 44.Byun HM, Motta V, Panni T, Bertazzi PA, Apostoli P, Hou L, Baccarelli AA. Evolutionary age of repetitive element subfamilies and sensitivity of DNA methylation to airborne pollutants. Part Fibre Toxicol. 2013;10:28. doi: 10.1186/1743-8977-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miousse IR, Koturbash I. The fine LINE: Methylation drawing the cancer landscape. BioMed Research International. 2015 doi: 10.1155/2015/131547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335–340. doi: 10.1016/s0168-9525(97)01181-5. [DOI] [PubMed] [Google Scholar]
- 47.Bourc’his D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431:96–99. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- 48.Leung DC, Lorincz MC. Silencing of endogenous retroviruses: When and why do histone marks predominate? Trends Biochem Sci. 2012;37:127–133. doi: 10.1016/j.tibs.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 49.Macia A, Muñoz-Lopez M, Cortes JL, Hastings RK, Morell S, Lucena-Aguilar G, Marchal JA, Badge RM, Garcia-Perez JL. Epigenetic control of retrotransposon expression in human embryonic stem cells. Mol Cell Biol. 2011;31:300–316. doi: 10.1128/MCB.00561-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martens JH, O’Sullivan RJ, Braunschweig U, Opravil S, Radolf M, Steinlein P, Jenuwein T. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J. 2005;24:800–812. doi: 10.1038/sj.emboj.7600545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, Shinkai Y, Mager DL, Jones S, Hirst M, Lorincz MC. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 2011;8:676–687. doi: 10.1016/j.stem.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rangasamy D. Distinctive patterns of epigenetic marks are associated with promoter regions of mouse LINE-1 and LTR retrotransposons. Mob DNA. 2013;4:27. doi: 10.1186/1759-8753-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carmell MA, Girard A, van dK, Bourc’his D, Bestor TH, de Rooij DG, Hannon GJ. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell. 2007;12:503–514. doi: 10.1016/j.devcel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 54.De Fazio S, Bartonicek N, Di GM, Abreu-Goodger C, Sankar A, Funaya C, Antony C, Moreira PN, Enright AJ, O’Carroll D. The endonuclease activity of mili fuels piRNA amplification that silences LINE1 elements. Nature. 2011;480:259–263. doi: 10.1038/nature10547. [DOI] [PubMed] [Google Scholar]
- 55.Rodic N, Sharma R, Sharma R, Zampella J, Dai L, Taylor MS, Hruban RH, Iacobuzio-Donahue CA, Maitra A, Torbenson MS, Goggins M, Shih I, Duffield AS, Montgomery EA, Gabrielson E, Netto GJ, Lotan TL, De Marzo AM, Westra W, Binder ZA, Orr BA, Gallia GL, Eberhart CG, Boeke JD, Harris CR, Burns KH. Long interspersed element-1 protein expression is a hallmark of many human cancers. Am J Pathol. 2014;184:1280–1286. doi: 10.1016/j.ajpath.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boyle AL, Ballard SG, Ward DC. Differential distribution of long and short interspersed element sequences in the mouse genome: Chromosome karyotyping by fluorescence in situ hybridization. Proc Natl Acad Sci U S A. 1990;87:7757–7761. doi: 10.1073/pnas.87.19.7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han JS, Szak ST, Boeke JD. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature. 2004;429:268–274. doi: 10.1038/nature02536. [DOI] [PubMed] [Google Scholar]
- 58.Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435:903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- 59.Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, Schroder K, Cloonan N, Steptoe AL, Lassmann T, Waki K, Hornig N, Arakawa T, Takahashi H, Kawai J, Forrest AR, Suzuki H, Hayashizaki Y, Hume DA, Orlando V, Grimmond SM, Carninci P. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009;41:563–571. doi: 10.1038/ng.368. [DOI] [PubMed] [Google Scholar]
- 60.Wolff EM, Byun HM, Han HF, Sharma S, Nichols PW, Siegmund KD, Yang AS, Jones PA, Liang G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010;6:e1000917. doi: 10.1371/journal.pgen.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hur K, Cejas P, Feliu J, Moreno-Rubio J, Burgos E, Boland CR, Goel A. Hypomethylation of long interspersed nuclear element-1 (LINE-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut. 2014;63:635–646. doi: 10.1136/gutjnl-2012-304219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, Vogelstein B, Nakamura Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992;52:643–645. [PubMed] [Google Scholar]
- 63.Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, Van Meir EG, Vertino PM, Devine SE. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010;141:1253–1261. doi: 10.1016/j.cell.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ, III, Lohr JG, Harris CC, Ding L, Wilson RK, Wheeler DA, Gibbs RA, Kucherlapati R, Lee C, Kharchenko PV, Park PJ. Landscape of somatic retrotransposition in human cancers. Science. 2012;337:967–971. doi: 10.1126/science.1222077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Solyom S, Ewing AD, Rahrmann EP, Doucet T, Nelson HH, Burns MB, Harris RS, Sigmon DF, Casella A, Erlanger B, Wheelan S, Upton KR, Shukla R, Faulkner GJ, Largaespada DA, Kazazian HH., Jr Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res. 2012;22:2328–2338. doi: 10.1101/gr.145235.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kazazian HH, Jr, Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature. 1988;332:164–166. doi: 10.1038/332164a0. [DOI] [PubMed] [Google Scholar]
- 67.Beck CR, Garcia-Perez JL, Badge RM, Moran JV. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. 2011;12:187–215. doi: 10.1146/annurev-genom-082509-141802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hancks DC, Kazazian HH., Jr Active human retrotransposons: Variation and disease. Curr Opin Genet Dev. 2012;22:191–203. doi: 10.1016/j.gde.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rangwala SH, Kazazian HH., Jr The L1 retrotransposition assay: A retrospective and toolkit. Methods. 2009;49:219–226. doi: 10.1016/j.ymeth.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ewing AD, Kazazian HH., Jr High-throughput sequencing reveals extensive variation in humanspecific L1 content in individual human genomes. Genome Res. 2010;20:1262–1270. doi: 10.1101/gr.106419.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stewart C, Kural D, Strömberg MP, Walker JA, Konkel MK, Stütz AM, Urban AE, Grubert F, Lam HY, Lee WP, Busby M, Indap AR, Garrison E, Huff C, Xing J, Snyder MP, Jorde LB, Batzer MA, Korbel JO, Marth GT. A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet. 2011;7:e1002236. doi: 10.1371/journal.pgen.1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Witherspoon DJ, Xing J, Zhang Y, Watkins WS, Batzer MA, Jorde LB. Mobile element scanning (ME-scan) by targeted high-throughput sequencing. BMC Genomics. 2010;11:410. doi: 10.1186/1471-2164-11-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.White TB, McCoy AM, Streva VA, Fenrich J, Deininger PL. A droplet digital PCR detection method for rare L1 insertions in tumors. Mob DNA. 2014;5:30. doi: 10.1186/s13100-014-0030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Helman E, Lawrence MS, Stewart C, Sougnez C, Getz G, Meyerson M. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res. 2014;24:1053–1063. doi: 10.1101/gr.163659.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Criscione SW, Zhang Y, Thompson W, Sedivy JM, Neretti N. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics. 2014;15:583. doi: 10.1186/1471-2164-15-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mir AA, Philippe C, Cristofari G. euL1db: The european database of L1HS retrotransposon insertions in humans. Nucleic Acids Res. 2015;43:D43–D47. doi: 10.1093/nar/gku1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brenner DJ, Mossman KL. Do radiation doses below 1 cGy increase cancer risks? Radiat Res. 2005;163:692–693. [PubMed] [Google Scholar]
- 78.Hall EJ, Brenner DJ. Cancer risks from diagnostic radiology: The impact of new epidemiological data. Br J Radiol. 2012;85:e1316–e1317. doi: 10.1259/bjr/13739950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.American Cancer Society. Cancer facts and figures. American Cancer Society. 2003:1–52. [Google Scholar]
- 80.Brenner DJ. Slowing the increase in the population dose resulting from CT scans. Radiat Res. 2010;174:809–815. doi: 10.1667/RR1859.1. [DOI] [PubMed] [Google Scholar]
- 81.Malone J, Guleria R, Craven C, Horton P, Järvinen H, Mayo J, O’reilly G, Picano E, Remedios D, Le Heron J, Rehani M, Holmberg O, Czarwinski R. Justification of diagnostic medical exposures: Some practical issues. report of an international atomic energy agency consultation. Br J Radiol. 2012;85:523–538. doi: 10.1259/bjr/42893576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lyon CM, Klinge DM, Liechty KC, Gentry FD, March TH, Kang T, Gilliland FD, Adamova G, Rusinova G, Telnov V, Belinsky SA. Radiation-induced lung adenocarcinoma is associated with increased frequency of genes inactivated by promoter hypermethylation. Radiat Res. 2007;168:409–414. doi: 10.1667/RR0825.1. [DOI] [PubMed] [Google Scholar]
- 83.Tawa R, Kimura Y, Komura J, Miyamura Y, Kurishita A, Sasaki MS, Sakurai H, Ono T. Effects of X-ray irradiation on genomic DNA methylation levels in mouse tissues. J Radiat Res. 1998;39:271–278. doi: 10.1269/jrr.39.271. [DOI] [PubMed] [Google Scholar]
- 84.Pogribny I, Raiche J, Slovack M, Kovalchuk O. Dose-dependence, sex- and tissue-specificity, and persistence of radiation-induced genomic DNA methylation changes. Biochem Biophys Res Commun. 2004;320:1253–1261. doi: 10.1016/j.bbrc.2004.06.081. [DOI] [PubMed] [Google Scholar]
- 85.Koturbash I, Pogribny I, Kovalchuk O. Stable loss of global DNA methylation in the radiationtarget tissue--a possible mechanism contributing to radiation carcinogenesis? Biochem Biophys Res Commun. 2005;337:526–533. doi: 10.1016/j.bbrc.2005.09.084. [DOI] [PubMed] [Google Scholar]
- 86.Pogribny I, Koturbash I, Tryndyak V, Hudson D, Stevenson SM, Sedelnikova O, Bonner W, Kovalchuk O. Fractionated low-dose radiation exposure leads to accumulation of DNA damage and profound alterations in DNA and histone methylation in the murine thymus. Mol Cancer Res. 2005;3:553–561. doi: 10.1158/1541-7786.MCR-05-0074. [DOI] [PubMed] [Google Scholar]
- 87.Loree J, Koturbash I, Kutanzi K, Baker M, Pogribny I, Kovalchuk O. Radiation-induced molecular changes in rat mammary tissue: Possible implications for radiation-induced carcinogenesis. Int J Radiat Biol. 2006;82:805–815. doi: 10.1080/09553000600960027. [DOI] [PubMed] [Google Scholar]
- 88.Giotopoulos G, McCormick C, Cole C, Zanker A, Jawad M, Brown R, Plumb M. DNA methylation during mouse hemopoietic differentiation and radiation-induced leukemia. Exp Hematol. 2006;34:1462–1470. doi: 10.1016/j.exphem.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 89.Koturbash I, Kutanzi K, Hendrickson K, Rodriguez-Juarez R, Kogosov D, Kovalchuk O. Radiation-induced bystander effects in vivo are sex specific. Mutat Res. 2008;642:28–36. doi: 10.1016/j.mrfmmm.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 90.Wang J, Zhang Y, Xu K, Mao X, Xue L, Liu X, Yu H, Chen L, Chu X. Genome-wide screen of DNA methylation changes induced by low dose X-ray radiation in mice. PLoS One. 2014;9:e90804. doi: 10.1371/journal.pone.0090804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koturbash I, Boyko A, Rodriguez-Juarez R, McDonald RJ, Tryndyak VP, Kovalchuk I, Pogribny IP, Kovalchuk O. Role of epigenetic effectors in maintenance of the long-term persistent bystander effect in spleen in vivo. Carcinogenesis. 2007;28:1831–1838. doi: 10.1093/carcin/bgm053. [DOI] [PubMed] [Google Scholar]
- 92.Lima F, Ding D, Goetz W, Yang AJ, Baulch JE. High LET 56Fe ion irradiation induces tissue-specific changes in DNA methylation in the mouse. Environ Mol Mutagen. 2014;55:266–277. doi: 10.1002/em.21832. [DOI] [PubMed] [Google Scholar]
- 93.Miousse IR, Shao L, Chang J, Feng W, Wang Y, Allen AR, Turner J, Stewart B, Raber J, Zhou D, Koturbash I. Exposure to low-dose (56)Fe-ion radiation induces long-term epigenetic alterations in mouse bone marrow hematopoietic progenitor and stem cells. Radiat Res. 2014;182:92–101. doi: 10.1667/RR13580.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nzabarushimana E, Miousse IR, Shao L, Chang J, Allen AR, Turner J, Stewart B, Raber J, Koturbash I. Long-term epigenetic effects of exposure to low doses of 56Fe in the mouse lung. J Radiat Res. 2014;55:823–828. doi: 10.1093/jrr/rru010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aypar U, Morgan WF, Baulch JE. Radiation-induced epigenetic alterations after low and high LET irradiations. Mutat Res. 2011;707:24–33. doi: 10.1016/j.mrfmmm.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 96.Goetz W, Morgan MN, Baulch JE. The effect of radiation quality on genomic DNA methylation profiles in irradiated human cell lines. Radiat Res. 2011;175:575–587. doi: 10.1667/RR2390.1. [DOI] [PubMed] [Google Scholar]
- 97.Lahtz C, Bates SE, Jiang Y, Li AX, Wu X, Hahn MA, Pfeifer GP. Gamma irradiation does not induce detectable changes in DNA methylation directly following exposure of human cells. PLoS One. 2012;7:e44858. doi: 10.1371/journal.pone.0044858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Newman MR, Sykes PJ, Blyth BJ, Bezak E, Lawrence MD, Morel KL, Ormsby RJ. A single whole-body low dose X-irradiation does not affect L1, B1 and IAP repeat element DNA methylation longitudinally. PLoS One. 2014;9:e93016. doi: 10.1371/journal.pone.0093016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Newman MR, Sykes PJ, Blyth BJ, Bezak E, Lawrence MD, Morel KL, Ormsby RJ. The methylation of DNA repeat elements is sex-dependent and temporally different in response to X radiation in radiosensitive and radioresistant mouse strains. Radiat Res. 2014;181:65–75. doi: 10.1667/RR13460.1. [DOI] [PubMed] [Google Scholar]
- 100.Blakely EA, Kronenberg A. Heavy-ion radiobiology: New approaches to delineate mechanisms underlying enhanced biological effectiveness. Radiat Res. 1998;150:S126–S145. [PubMed] [Google Scholar]
- 101.Weil MM, Bedford JS, Bielefeldt-Ohmann H, Ray FA, Genik PC, Ehrhart EJ, Fallgren CM, Hailu F, Battaglia CL, Charles B, Callan MA, Ullrich RL. Incidence of acute myeloid leukemia and hepatocellular carcinoma in mice irradiated with 1 GeV/nucleon (56)fe ions. Radiat Res. 2009;172:213–219. doi: 10.1667/RR1648.1. [DOI] [PubMed] [Google Scholar]
- 102.Weil MM, Ray FA, Genik PC, Yu Y, McCarthy M, Fallgren CM, Ullrich RL. Effects of 28Si ions, 56Fe ions, and protons on the induction of murine acute myeloid leukemia and hepatocellular carcinoma. PLoS One. 2014;9:e104819. doi: 10.1371/journal.pone.0104819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Herceg Z, Paliwal A. Epigenetic mechanisms in hepatocellular carcinoma: How environmental factors influence the epigenome. Mutat Res. 2011;727:55–61. doi: 10.1016/j.mrrev.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 104.Christofidou-Solomidou M, Pietrofesa RA, Arguiri E, Schweitzer KS, Berdyshev EV, McCarthy M, Corbitt A, Alwood JS, Yu Y, Globus RK, Solomides CC, Ullrich RL, Petrache I. Space radiation associated lung injury in a murine model. Am J Physiol Lung Cell Mol Physiol. 2014 doi: 10.1152/ajplung.00260.2014. ajplung. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kroeger H, Jelinek J, Estecio MR, He R, Kondo K, Chung W, Zhang L, Shen L, Kantarjian HM, Bueso-Ramos CE, Issa JP. Aberrant CpG island methylation in acute myeloid leukemia is accentuated at relapse. Blood. 2008;112:1366–1373. doi: 10.1182/blood-2007-11-126227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bujko M, Musialik E, Olbromski R, Przestrzelska M, Libura M, Pastwinska A, Juszczynski P, Zwierzchowski L, Baranowski P, Siedlecki JA. Repetitive genomic elements and overall DNA methylation changes in acute myeloid and childhood B-cell lymphoblastic leukemia patients. Int J Hematol. 2014;100:79–87. doi: 10.1007/s12185-014-1592-0. [DOI] [PubMed] [Google Scholar]
- 107.Tanaka A, Nakatani Y, Hamada N, Jinno-Oue A, Shimizu N, Wada S, Funayama T, Mori T, Islam S, Hoque SA, Shinagawa M, Ohtsuki T, Kobayashi Y, Hoshino H. Ionising irradiation alters the dynamics of human long interspersed nuclear elements 1 (LINE1) retrotransposon. Mutagenesis. 2012;27:599–607. doi: 10.1093/mutage/ges025. [DOI] [PubMed] [Google Scholar]
- 108.Banaz-Yasar F, Gedik N, Karahan S, Diaz-Carballo D, Bongartz BM, Ergün S. LINE-1 retrotransposition events regulate gene expression after X-ray irradiation. DNA Cell Biol. 2012;31:1458–1467. doi: 10.1089/dna.2012.1676. [DOI] [PubMed] [Google Scholar]
- 109.Jonason AS, Kunala S, Price GJ, Restifo RJ, Spinelli HM, Persing JA, Leffell DJ, Tarone RE, Brash DE. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci U S A. 1996;93:14025–14029. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ichihashi M, Ueda M, Budiyanto A, Bito T, Oka M, Fukunaga M, Tsuru K, Horikawa T. UV-induced skin damage. Toxicology. 2003;189:21–39. doi: 10.1016/s0300-483x(03)00150-1. [DOI] [PubMed] [Google Scholar]
- 111.Zhang W, Hanks AN, Boucher K, Florell SR, Allen SM, Alexander A, Brash DE, Grossman D. UVB-induced apoptosis drives clonal expansion during skin tumor development. Carcinogenesis. 2005;26:249–257. doi: 10.1093/carcin/bgh300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Banerjee G, Gupta N, Tiwari J, Raman G. Ultraviolet-induced transformation of keratinocytes: Possible involvement of long interspersed element-1 reverse transcriptase. Photodermatol Photoimmunol Photomed. 2005;21:32–39. doi: 10.1111/j.1600-0781.2005.00136.x. [DOI] [PubMed] [Google Scholar]
- 113.Teneng I, Stribinskis V, Ramos KS. Context-specific regulation of LINE-1. Genes Cells. 2007;12:1101–1110. doi: 10.1111/j.1365-2443.2007.01117.x. [DOI] [PubMed] [Google Scholar]
- 114.Nair-Shalliker V, Dhillon V, Clements M, Armstrong BK, Fenech M. The association between personal sun exposure, serum vitamin D and global methylation in human lymphocytes in a population of healthy adults in south australia. Mutat Res Fundam Mol Mech Mutagen. 2014;765C:6–10. doi: 10.1016/j.mrfmmm.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 115.Serafino A, Balestrieri E, Pierimarchi P, Matteucci C, Moroni G, Oricchio E, Rasi G, Mastino A, Spadafora C, Garaci E, Vallebona PS. The activation of human endogenous retrovirus K (HERV-K) is implicated in melanoma cell malignant transformation. Exp Cell Res. 2009;315:849–862. doi: 10.1016/j.yexcr.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 116.Stengel S, Fiebig U, Kurth R, Denner J. Regulation of human endogenous retrovirus-K expression in melanomas by CpG methylation. Genes Chromosomes Cancer. 2010;49:401–411. doi: 10.1002/gcc.20751. [DOI] [PubMed] [Google Scholar]
- 117.Muster T, Waltenberger A, Grassauer A, Hirschl S, Caucig P, Romirer I, Födinger D, Seppele H, Schanab O, Magin-Lachmann C, Löwer R, Jansen B, Pehamberger H, Wolff K. An endogenous retrovirus derived from human melanoma cells. Cancer Res. 2003;63:8735–8741. [PubMed] [Google Scholar]
- 118.Schmitt K, Reichrath J, Roesch A, Meese E, Mayer J. Transcriptional profiling of human endogenous retrovirus group HERV-K(HML-2) loci in melanoma. Genome Biol Evol. 2013;5:307–328. doi: 10.1093/gbe/evt010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Reiche J, Pauli G, Ellerbrok H. Differential expression of human endogenous retrovirus K transcripts in primary human melanocytes and melanoma cell lines after UV irradiation. Melanoma Res. 2010;20:435–440. doi: 10.1097/CMR.0b013e32833c1b5d. [DOI] [PubMed] [Google Scholar]
- 120.Schanab O, Humer J, Gleiss A, Mikula M, Sturlan S, Grunt S, Okamoto I, Muster T, Pehamberger H, Waltenberger A. Expression of human endogenous retrovirus K is stimulated by ultraviolet radiation in melanoma. Pigment Cell Melanoma Res. 2011;24:656–665. doi: 10.1111/j.1755-148X.2011.00860.x. [DOI] [PubMed] [Google Scholar]
- 121.Brunekreef B, Holgate ST. Air pollution and health. Lancet. 2002;360:1233–1242. doi: 10.1016/S0140-6736(02)11274-8. [DOI] [PubMed] [Google Scholar]
- 122.Dockery DW, Pope CA, III, Xu X, Spengler JD, Ware JH, Fay ME, Ferris BG, Jr, Speizer FE. An association between air pollution and mortality in six U.S. cities. N Engl J Med. 1993;329:1753–1759. doi: 10.1056/NEJM199312093292401. [DOI] [PubMed] [Google Scholar]
- 123.Jarup L. Hazards of heavy metal contamination. Br Med Bull. 2003;68:167–182. doi: 10.1093/bmb/ldg032. [DOI] [PubMed] [Google Scholar]
- 124.Pope CA., III Epidemiology of fine particulate air pollution and human health: Biologic mechanisms and who’s at risk? Environ Health Perspect. 2000;108(Suppl 4):713–723. doi: 10.1289/ehp.108-1637679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Demerjian KL, Mohnen VA. Synopsis of the temporal variation of particulate matter composition and size. J Air Waste Manag Assoc. 2008;58:216–233. doi: 10.3155/1047-3289.58.2.216. [DOI] [PubMed] [Google Scholar]
- 126.Brunekreef B, Forsberg B. Epidemiological evidence of effects of coarse airborne particles on health. Eur Respir J. 2005;26:309–318. doi: 10.1183/09031936.05.00001805. [DOI] [PubMed] [Google Scholar]
- 127.Delfino RJ, Sioutas C, Malik S. Potential role of ultrafine particles in associations between airborne particle mass and cardiovascular health. Environ Health Perspect. 2005;113:934–946. doi: 10.1289/ehp.7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sioutas C, Delfino RJ, Singh M. Exposure assessment for atmospheric ultrafine particles (UFPs) and implications in epidemiologic research. Environ Health Perspect. 2005;113:947–955. doi: 10.1289/ehp.7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.de Hartog JJ, Ayres JG, Karakatsani A, Analitis A, Brink HT, Hameri K, Harrison R, Katsouyanni K, Kotronarou A, Kavouras I, Meddings C, Pekkanen J, Hoek G. Lung function and indicators of exposure to indoor and outdoor particulate matter among asthma and COPD patients. Occup Environ Med. 2010;67:2–10. doi: 10.1136/oem.2008.040857. [DOI] [PubMed] [Google Scholar]
- 130.Loomis D, Grosse Y, Lauby-Secretan BA, El Ghissassi F, Bouvard VA, Benbrahim-Tallaa L, Guha N, Baan R, Mattock H, Straif K. The carcinogenicity of outdoor air pollution. The Lancet Oncology. 2013;14:1262–1263. doi: 10.1016/s1470-2045(13)70487-x. [DOI] [PubMed] [Google Scholar]
- 131.Valavanidis A, Fiotakis K, Vlachogianni T. Airborne particulate matter and human health: Toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2008;26:339–362. doi: 10.1080/10590500802494538. [DOI] [PubMed] [Google Scholar]
- 132.Akhtar US, McWhinney RD, Rastogi N, Abbatt JP, Evans GJ, Scott JA. Cytotoxic and proinflammatory effects of ambient and source-related particulate matter (PM) in relation to the production of reactive oxygen species (ROS) and cytokine adsorption by particles. Inhal Toxicol. 2010;22(Suppl 2):37–47. doi: 10.3109/08958378.2010.518377. [DOI] [PubMed] [Google Scholar]
- 133.DeMarini DM. Genotoxicity biomarkers associated with exposure to traffic and near-road atmospheres: A review. Mutagenesis. 2013;28:485–505. doi: 10.1093/mutage/get042. [DOI] [PubMed] [Google Scholar]
- 134.Franzi LM, Bratt JM, Williams KM, Last JA. Why is particulate matter produced by wildfires toxic to lung macrophages? Toxicol Appl Pharmacol. 2011;257:182–188. doi: 10.1016/j.taap.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nelson PF, Tibbett AR, Day SJ. Effects of vehicle type and fuel quality on real world toxic emissions from diesel vehicles. Atmospheric Environment. 2008;42:5291–5303. [Google Scholar]
- 136.Li R, Ning Z, Majumdar R, Cui J, Takabe W, Jen N, Sioutas C, Hsiai T. Ultrafine particles from diesel vehicle emissions at different driving cycles induce differential vascular pro-inflammatory responses: Implication of chemical components and NF-kappaB signaling. Part Fibre Toxicol. 2010;7:6. doi: 10.1186/1743-8977-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Reiss R, Anderson EL, Cross CE, Hidy G, Hoel D, McClellan R, Moolgavkar S. Evidence of health impacts of sulfate-and nitrate-containing particles in ambient air. Inhal Toxicol. 2007;19:419–449. doi: 10.1080/08958370601174941. [DOI] [PubMed] [Google Scholar]
- 138.Peluso M, Bollati V, Munnia A, Srivatanakul P, Jedpiyawongse A, Sangrajrang S, Piro S, Ceppi M, Bertazzi PA, Boffetta P, Baccarelli AA. DNA methylation differences in exposed workers and nearby residents of the ma ta phut industrial estate, rayong, thailand. Int J Epidemiol. 2012;41:1753–1760. doi: 10.1093/ije/dys129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tarantini L, Bonzini M, Apostoli P, Pegoraro V, Bollati V, Marinelli B, Cantone L, Rizzo G, Hou L, Schwartz J. Effects of particulate matter on genomic DNA methylation content and iNOS promoter methylation. Environmental Health Perspectives. 2009;117:217. doi: 10.1289/ehp.11898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bellavia A, Urch B, Speck M, Brook RD, Scott JA, Albetti B, Behbod B, North M, Valeri L, Bertazzi PA, Silverman F, Gold D, Baccarelli AA. DNA hypomethylation, ambient particulate matter, and increased blood pressure: Findings from controlled human exposure experiments. J Am Heart Assoc. 2013;2:e000212. doi: 10.1161/JAHA.113.000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yauk C, Polyzos A, Rowan-Carroll A, Somers CM, Godschalk RW, Van Schooten FJ, Berndt ML, Pogribny IP, Koturbash I, Williams A, Douglas GR, Kovalchuk O. Germ-line mutations, DNA damage, and global hypermethylation in mice exposed to particulate air pollution in an urban/industrial location. Proc Natl Acad Sci U S A. 2008;105:605–610. doi: 10.1073/pnas.0705896105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Miousse IR, Chalbot MC, Aykin-Burns N, Wang X, Basnakian A, Kavouras IG, Koturbash I. Epigenetic alterations induced by ambient particulate matter in mouse macrophages. Environ Mol Mutagen. 2014;55:428–435. doi: 10.1002/em.21855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Adar SD, Gold DR, Coull BA, Schwartz J, Stone PH, Suh H. Focused exposures to airborne traffic particles and heart rate variability in the elderly. Epidemiology. 2007;18:95–103. doi: 10.1097/01.ede.0000249409.81050.46. [DOI] [PubMed] [Google Scholar]
- 144.Park SK, O’Neill MS, Vokonas PS, Sparrow D, Spiro A, III, Tucker KL, Suh H, Hu H, Schwartz J. Traffic-related particles are associated with elevated homocysteine: The VA normative aging study. Am J Respir Crit Care Med. 2008;178:283–289. doi: 10.1164/rccm.200708-1286OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, Zanobetti A, Sparrow D, Vokonas PS, Schwartz J. Rapid DNA methylation changes after exposure to traffic particles. American Journal of Respiratory and Critical Care Medicine. 2009;179:572–578. doi: 10.1164/rccm.200807-1097OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Madrigano J, Baccarelli A, Mittleman MA, Wright RO, Sparrow D, Vokonas PS, Tarantini L, Schwartz J. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men. Environ Health Perspect. 2011;119:977–982. doi: 10.1289/ehp.1002773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Laden F, Neas LM, Dockery DW, Schwartz J. Association of fine particulate matter from different sources with daily mortality in six U.S. cities. Environ Health Perspect. 2000;108:941–947. doi: 10.1289/ehp.00108941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ris C. U.S. EPA health assessment for diesel engine exhaust: A review. Inhal Toxicol. 2007;19(Suppl 1):229–239. doi: 10.1080/08958370701497960. [DOI] [PubMed] [Google Scholar]
- 149.IARC. Diesel and gasoline engine exhausts and some nitroarenes. IARC Monograph on the Evaluation of Carcinogenic Risks to Humans. 2012;105:1–714. [PMC free article] [PubMed] [Google Scholar]
- 150.Jiang R, Jones MJ, Sava F, Kobor MS, Carlsten C. Short-term diesel exhaust inhalation in a controlled human crossover study is associated with changes in DNA methylation of circulating mononuclear cells in asthmatics. Part Fibre Toxicol. 2014;11:71. doi: 10.1186/s12989-014-0071-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.IARC. IARC monographs programme on the evaluation of the carcinogenic risk of chemicals to humans. preamble. IARC Monographs Programme on the Evaluation of the Carcinogenic Risk of Chemicals to Humans. 1986;39:13–32. [PubMed] [Google Scholar]
- 152.Smith MT. Advances in understanding benzene health effects and susceptibility. Annu Rev Public Health. 2010;31:133–148. doi: 10.1146/annurev.publhealth.012809.103646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Khalade A, Jaakkola MS, Pukkala E, Jaakkola JJ. Exposure to benzene at work and the risk of leukemia: A systematic review and meta-analysis. Environ Health. 2010;9:31. doi: 10.1186/1476-069X-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, Byun HM, Jiang J, Marinelli B, Pesatori AC, Bertazzi PA, Yang AS. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67:876–880. doi: 10.1158/0008-5472.CAN-06-2995. [DOI] [PubMed] [Google Scholar]
- 155.Fustinoni S, Rossella F, Polledri E, Bollati V, Campo L, Byun HM, Agnello L, Consonni D, Pesatori AC, Baccarelli A, Bertazzi PA. Global DNA methylation and low-level exposure to benzene. Med Lav. 2012;103:84–95. [PubMed] [Google Scholar]
- 156.Seow WJ, Pesatori AC, Dimont E, Farmer PB, Albetti B, Ettinger AS, Bollati V, Bolognesi C, Roggieri P, Panev TI, Georgieva T, Merlo DF, Bertazzi PA, Baccarelli AA. Urinary benzene biomarkers and DNA methylation in bulgarian petrochemical workers: Study findings and comparison of linear and beta regression models. PLoS One. 2012;7:e50471. doi: 10.1371/journal.pone.0050471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.White WC. Butadiene production process overview. Chem Biol Interact. 2007;166:10–14. doi: 10.1016/j.cbi.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 158.IARC. 1,3-butadiene, ethylene oxide and vinyl halides (vinyl fluoride, vinyl chloride and vinyl bromide) IARC Monograph on the Evaluation of Carcinogenic Risks to Humans. 2008;97:3–471. [PMC free article] [PubMed] [Google Scholar]
- 159.Cochrane JE, Skopek TR. Mutagenicity of butadiene and its epoxide metabolites: I. mutagenic potential of 1,2-epoxybutene, 1,2,3,4-diepoxybutane and 3,4-epoxy-1,2-butanediol in cultured human lymphoblasts. Carcinogenesis. 1994;15:713–717. doi: 10.1093/carcin/15.4.713. [DOI] [PubMed] [Google Scholar]
- 160.Koturbash I, Scherhag A, Sorrentino J, Sexton K, Bodnar W, Tryndyak V, Latendresse JR, Swenberg JA, Beland FA, Pogribny IP, Rusyn I. Epigenetic alterations in liver of C57BL/6J mice after short-term inhalational exposure to 1,3-butadiene. Environ Health Perspect. 2011;119:635–640. doi: 10.1289/ehp.1002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Koturbash I, Scherhag A, Sorrentino J, Sexton K, Bodnar W, Swenberg JA, Beland FA, Pardo-Manuel DF, Rusyn I, Pogribny IP. Epigenetic mechanisms of mouse interstrain variability in genotoxicity of the environmental toxicant 1,3-butadiene. Toxicol Sci. 2011;122:448–456. doi: 10.1093/toxsci/kfr133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chappell G, Kobets T, O’Brien B, Tretyakova N, Sangaraju D, Kosyk O, Sexton KG, Bodnar W, Pogribny IP, Rusyn I. Epigenetic events determine tissue-specific toxicity of inhalational exposure to the genotoxic chemical 1,3-butadiene in male C57BL/6J mice. Toxicol Sci. 2014;142:375–384. doi: 10.1093/toxsci/kfu191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Walser T, Cui X, Yanagawa J, Lee JM, Heinrich E, Lee G, Sharma S, Dubinett SM. Smoking and lung cancer: The role of inflammation. Proc Am Thorac Soc. 2008;5:811–815. doi: 10.1513/pats.200809-100TH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.IARC. Personal habits and indoor combustions. IARC Monograph on the Evaluation of Carcinogenic Risks to Humans. 2012;100E:1–598. [PMC free article] [PubMed] [Google Scholar]
- 165.Hecht SS. Cigarette smoking and lung cancer: Chemical mechanisms and approaches to prevention. Lancet Oncol. 2002;3:461–469. doi: 10.1016/s1470-2045(02)00815-x. [DOI] [PubMed] [Google Scholar]
- 166.DeMarini DM. Genotoxicity of tobacco smoke and tobacco smoke condensate: A review. Mutat Res. 2004;567:447–474. doi: 10.1016/j.mrrev.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 167.Koturbash I, Beland FA, Pogribny IP. Role of epigenetic events in chemical carcinogenesis--a justification for incorporating epigenetic evaluations in cancer risk assessment. Toxicol Mech Methods. 2011;21:289–297. doi: 10.3109/15376516.2011.557881. [DOI] [PubMed] [Google Scholar]
- 168.Belinsky SA. Silencing of genes by promoter hypermethylation: Key event in rodent and human lung cancer. Carcinogenesis. 2005;26:1481–1487. doi: 10.1093/carcin/bgi020. [DOI] [PubMed] [Google Scholar]
- 169.Kim JS, Kim H, Shim YM, Han J, Park J, Kim DH. Aberrant methylation of the FHIT gene in chronic smokers with early stage squamous cell carcinoma of the lung. Carcinogenesis. 2004;25:2165–2171. doi: 10.1093/carcin/bgh217. [DOI] [PubMed] [Google Scholar]
- 170.Liu F, Killian JK, Yang M, Walker RL, Hong JA, Zhang M, Davis S, Zhang Y, Hussain M, Xi S, Rao M, Meltzer PA, Schrump DS. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene. 2010;29:3650–3664. doi: 10.1038/onc.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kawano H, Saeki H, Kitao H, Tsuda Y, Otsu H, Ando K, Ito S, Egashira A, Oki E, Morita M, Oda Y, Maehara Y. Chromosomal instability associated with global DNA hypomethylation is associated with the initiation and progression of esophageal squamous cell carcinoma. Ann Surg Oncol. 2014;21(Suppl 4):S696–S702. doi: 10.1245/s10434-014-3818-z. [DOI] [PubMed] [Google Scholar]
- 172.Wilhelm-Benartzi CS, Houseman EA, Maccani MA, Poage GM, Koestler DC, Langevin SM, Gagne LA, Banister CE, Padbury JF, Marsit CJ. In utero exposures, infant growth, and DNA methylation of repetitive elements and developmentally related genes in human placenta. Environ Health Perspect. 2012;120:296–302. doi: 10.1289/ehp.1103927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Engel LS, Lan Q, Rothman N. Polychlorinated biphenyls and non-hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev. 2007;16:373–376. doi: 10.1158/1055-9965.EPI-07-0055. [DOI] [PubMed] [Google Scholar]
- 174.Hardell L, Carlberg M, Hardell K, Björnfoth H, Wickbom G, Ionescu M, van BB, Lindström G. Decreased survival in pancreatic cancer patients with high concentrations of organochlorines in adipose tissue. Biomed Pharmacother. 2007;61:659–664. doi: 10.1016/j.biopha.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 175.Ritchie JM, Vial SL, Fuortes LJ, Guo H, Reedy VE, Smith EM. Organochlorines and risk of prostate cancer. J Occup Environ Med. 2003;45:692–702. doi: 10.1097/01.jom.0000071510.96740.0b. [DOI] [PubMed] [Google Scholar]
- 176.Lee DH, Lee IK, Porta M, Steffes M, Jacobs DR., Jr Relationship between serum concentrations of persistent organic pollutants and the prevalence of metabolic syndrome among nondiabetic adults: Results from the national health and nutrition examination survey 1999-2002. Diabetologia. 2007;50:1841–1851. doi: 10.1007/s00125-007-0755-4. [DOI] [PubMed] [Google Scholar]
- 177.Pathak R, Ahmed RS, Tripathi AK, Guleria K, Sharma CS, Makhijani SD, Banerjee BD. Maternal and cord blood levels of organochlorine pesticides: Association with preterm labor. Clin Biochem. 2009;42:746–749. doi: 10.1016/j.clinbiochem.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 178.Cohn BA, Wolff MS, Cirillo PM, Sholtz RI. DDT and breast cancer in young women: New data on the significance of age at exposure. Environ Health Perspect. 2007;115:1406–1414. doi: 10.1289/ehp.10260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Weidner IS, Moller H, Jensen TK, Skakkebaek NE. Cryptorchidism and hypospadias in sons of gardeners and farmers. Environ Health Perspect. 1998;106:793–796. doi: 10.1289/ehp.98106793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rathore M, Bhatnagar P, Mathur D, Saxena GN. Burden of organochlorine pesticides in blood and its effect on thyroid hormones in women. Sci Total Environ. 2002;295:207–215. doi: 10.1016/s0048-9697(02)00094-3. [DOI] [PubMed] [Google Scholar]
- 181.Mahajan R, Bonner MR, Hoppin JA, Alavanja MC. Phorate exposure and incidence of cancer in the agricultural health study. Environ Health Perspect. 2006;114:1205–1209. doi: 10.1289/ehp.8911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Van Maele-Fabry G, Libotte V, Willems J, Lison D. Review and meta-analysis of risk estimates for prostate cancer in pesticide manufacturing workers. Cancer Causes Control. 2006;17:353–373. doi: 10.1007/s10552-005-0443-y. [DOI] [PubMed] [Google Scholar]
- 183.Reynolds P, Von BJ, Gunier RB, Goldberg DE, Hertz A, Harnly ME. Childhood cancer and agricultural pesticide use: An ecologic study in california. Environ Health Perspect. 2002;110:319–324. doi: 10.1289/ehp.02110319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Collotta M, Bertazzi PA, Bollati V. Epigenetics and pesticides. Toxicology. 2013;307:35–41. doi: 10.1016/j.tox.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 185.Rusiecki JA, Baccarelli A, Bollati V, Tarantini L, Moore LE, Bonefeld-Jorgensen EC. Global DNA hypomethylation is associated with high serum-persistent organic pollutants in greenlandic inuit. Environ Health Perspect. 2008;116:1547–1552. doi: 10.1289/ehp.11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kim KY, Kim DS, Lee SK, Lee IK, Kang JH, Chang YS, Jacobs DR, Steffes M, Lee DH. Association of low-dose exposure to persistent organic pollutants with global DNA hypomethylation in healthy Koreans. Environ Health Perspect. 2010;118:370–374. doi: 10.1289/ehp.0901131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Huen K, Yousefi P, Bradman A, Yan L, Harley KG, Kogut K, Eskenazi B, Holland N. Effects of age, sex, and persistent organic pollutants on DNA methylation in children. Environ Mol Mutagen. 2014;55:209–222. doi: 10.1002/em.21845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Benford D, Dinovi M, Setzer RW. Application of the margin-of-exposure (MoE) approach to substances in food that are genotoxic and carcinogenic e.g.: Benzo[a]pyrene and polycyclic aromatic hydrocarbons. Food Chem Toxicol. 2010;48(Suppl 1):S42–S48. doi: 10.1016/j.fct.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 189.Pelkonen O, Nebert DW. Metabolism of polycyclic aromatic hydrocarbons: Etiologic role in carcinogenesis. Pharmacol Rev. 1982;34:189–222. [PubMed] [Google Scholar]
- 190.Stribinskis V, Ramos KS. Activation of human long interspersed nuclear element 1 retrotransposition by benzo(a)pyrene, an ubiquitous environmental carcinogen. Cancer Res. 2006;66:2616–2620. doi: 10.1158/0008-5472.CAN-05-3478. [DOI] [PubMed] [Google Scholar]
- 191.Teneng I, Montoya-Durango DE, Quertermous JL, Lacy ME, Ramos KS. Reactivation of L1 retrotransposon by benzo(a)pyrene involves complex genetic and epigenetic regulation. Epigenetics. 2011;6:355–367. doi: 10.4161/epi.6.3.14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sadikovic B, Rodenhiser DI. Benzopyrene exposure disrupts DNA methylation and growth dynamics in breast cancer cells. Toxicol Appl Pharmacol. 2006;216:458–468. doi: 10.1016/j.taap.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 193.Yauk CL, Polyzos A, Rowan-Carroll A, Kortubash I, Williams A, Kovalchuk O. Tandem repeat mutation, global DNA methylation, and regulation of DNA methyltransferases in cultured mouse embryonic fibroblast cells chronically exposed to chemicals with different modes of action. Environ Mol Mutagen. 2008;49:26–35. doi: 10.1002/em.20359. [DOI] [PubMed] [Google Scholar]
- 194.Salnikow K, Zhitkovich A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: Nickel, arsenic, and chromium. Chem Res Toxicol. 2008;21:28–44. doi: 10.1021/tx700198a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Martinez-Zamudio R, Ha HC. Environmental epigenetics in metal exposure. Epigenetics. 2011;6:820–827. doi: 10.4161/epi.6.7.16250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chervona Y, Arita A, Costa M. Carcinogenic metals and the epigenome: Understanding the effect of nickel, arsenic, and chromium. Metallomics. 2012;4:619–627. doi: 10.1039/c2mt20033c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hossain MB, Vahter M, Concha G, Broberg K. Low-level environmental cadmium exposure is associated with DNA hypomethylation in argentinean women. Environ Health Perspect. 2012;120:879–884. doi: 10.1289/ehp.1104600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.US EPA. Comprehensive environmental response, compensation, and liability act (CERCLA) priority list of hazardous substances. Agency for Toxic Substances and Disease Registry. 2010 [Google Scholar]
- 199.Bertolero F, Pozzi G, Sabbioni E, Saffiotti U. Cellular uptake and metabolic reduction of pentavalent to trivalent arsenic as determinants of cytotoxicity and morphological transformation. Carcinogenesis. 1987;8:803–808. doi: 10.1093/carcin/8.6.803. [DOI] [PubMed] [Google Scholar]
- 200.Ahsan H, Chen Y, Parvez F, Zablotska L, Argos M, Hussain I, Momotaj H, Levy D, Cheng Z, Slavkovich V, van GA, Howe GR, Graziano JH. Arsenic exposure from drinking water and risk of premalignant skin lesions in bangladesh: Baseline results from the health effects of arsenic longitudinal study. Am J Epidemiol. 2006;163:1138–1148. doi: 10.1093/aje/kwj154. [DOI] [PubMed] [Google Scholar]
- 201.Schuhmacher-Wolz U, Dieter HH, Klein D, Schneider K. Oral exposure to inorganic arsenic: Evaluation of its carcinogenic and non-carcinogenic effects. Crit Rev Toxicol. 2009;39:271–298. doi: 10.1080/10408440802291505. [DOI] [PubMed] [Google Scholar]
- 202.Wang CH, Hsiao CK, Chen CL, Hsu LI, Chiou HY, Chen SY, Hsueh YM, Wu MM, Chen CJ. A review of the epidemiologic literature on the role of environmental arsenic exposure and cardiovascular diseases. Toxicol Appl Pharmacol. 2007;222:315–326. doi: 10.1016/j.taap.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 203.Ray PD, Yosim A, Fry RC. Incorporating epigenetic data into the risk assessment process for the toxic metals arsenic, cadmium, chromium, lead, and mercury: Strategies and challenges. Front Genet. 2014;5:201. doi: 10.3389/fgene.2014.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.IARC. Some drinking-water disinfectants and contaminants, including arsenic. IARC Monograph on the Evaluation of Carcinogenic Risks to Humans. 2004;84:1–477. [PMC free article] [PubMed] [Google Scholar]
- 205.Straif K, Benbrahim-Tallaa L, Baan R, Grosse Y, Secretan B, El GF, Bouvard V, Guha N, Freeman C, Galichet L, Cogliano V. A review of human carcinogens--part C: Metals, arsenic, dusts, and fibres. Lancet Oncol. 2009;10:453–454. doi: 10.1016/s1470-2045(09)70134-2. [DOI] [PubMed] [Google Scholar]
- 206.Zhao CQ, Young MR, Diwan BA, Coogan TP, Waalkes MP. Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc Natl Acad Sci U S A. 1997;94:10907–10912. doi: 10.1073/pnas.94.20.10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Xie Y, Liu J, Benbrahim-Tallaa L, Ward JM, Logsdon D, Diwan BA, Waalkes MP. Aberrant DNA methylation and gene expression in livers of newborn mice transplacentally exposed to a hepatocarcinogenic dose of inorganic arsenic. Toxicology. 2007;236:7–15. doi: 10.1016/j.tox.2007.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wilhelm CS, Kelsey KT, Butler R, Plaza S, Gagne L, Zens MS, Andrew AS, Morris S, Nelson HH, Schned AR, Karagas MR, Marsit CJ. Implications of LINE1 methylation for bladder cancer risk in women. Clin Cancer Res. 2010;16:1682–1689. doi: 10.1158/1078-0432.CCR-09-2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Intarasunanont P, Navasumrit P, Waraprasit S, Chaisatra K, Suk WA, Mahidol C, Ruchirawat M. Effects of arsenic exposure on DNA methylation in cord blood samples from newborn babies and in a human lymphoblast cell line. Environ Health. 2012;11:31. doi: 10.1186/1476-069X-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lambrou A, Baccarelli A, Wright RO, Weisskopf M, Bollati V, Amarasiriwardena C, Vokonas P, Schwartz J. Arsenic exposure and DNA methylation among elderly men. Epidemiology. 2012;23:668–676. doi: 10.1097/EDE.0b013e31825afb0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tajuddin SM, Amaral AF, Fernandez AF, Rodriguez-Rodero S, Rodriguez RM, Moore LE, Tardon A, Carrato A, Garcia-Closas M, Silverman DT, Jackson BP, Garcia-Closas R, Cook AL, Cantor KP, Chanock S, Kogevinas M, Rothman N, Real FX, Fraga MF, Malats N. Genetic and non-genetic predictors of LINE-1 methylation in leukocyte DNA. Environ Health Perspect. 2013;121:650–656. doi: 10.1289/ehp.1206068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Karimi A, Madjd Z, Habibi L, Akrami SM. Exposure of hepatocellular carcinoma cells to lowlevel As2O3 causes an extra toxicity pathway via L1 retrotransposition induction. Toxicol Lett. 2014;229:111–117. doi: 10.1016/j.toxlet.2014.05.025. [DOI] [PubMed] [Google Scholar]
- 213.Markopoulos G, Noutsopoulos D, Mantziou S, Vartholomatos G, Monokrousos N, Angelidis C, Tzavaras T. Arsenic induces VL30 retrotransposition: The involvement of oxidative stress and heatshock protein 70. Toxicol Sci. 2013;134:312–322. doi: 10.1093/toxsci/kft118. [DOI] [PubMed] [Google Scholar]
- 214.Engström A, Michaëlsson K, Vahter M, Julin B, Wolk A, Åkesson A. Associations between dietary cadmium exposure and bone mineral density and risk of osteoporosis and fractures among women. Bone. 2012;50:1372–1378. doi: 10.1016/j.bone.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 215.Kippler M, Tofail F, Hamadani JD, Gardner RM, Grantham-McGregor SM, Bottai M, Vahter M. Early-life cadmium exposure and child development in 5-year-old girls and boys: A cohort study in rural bangladesh. Environ Health Perspect. 2012;120:1462–1468. doi: 10.1289/ehp.1104431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Sommar JN, Svensson MK, Björ BM, Elmståhl SI, Hallmans G, Lundh T, Schön SM, Skerfving S, Bergdahl IA. End-stage renal disease and low level exposure to lead, cadmium and mercury; a population-based, prospective nested case-referent study in sweden. Environ Health. 2013;12:9. doi: 10.1186/1476-069X-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.National Toxicology Program. Ninth report on carcinogens. 2000 [Google Scholar]
- 218.Takiguchi M, Achanzar WE, Qu W, Li G, Waalkes MP. Effects of cadmium on DNA- (cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp Cell Res. 2003;286:355–365. doi: 10.1016/s0014-4827(03)00062-4. [DOI] [PubMed] [Google Scholar]
- 219.Huang D, Zhang Y, Qi Y, Chen C, Ji W. Global DNA hypomethylation, rather than reactive oxygen species (ROS), a potential facilitator of cadmium-stimulated K562 cell proliferation. Toxicol Lett. 2008;179:43–47. doi: 10.1016/j.toxlet.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 220.Jiang G, Xu L, Song S, Zhu C, Wu Q, Zhang L, Wu L. Effects of long-term low-dose cadmium exposure on genomic DNA methylation in human embryo lung fibroblast cells. Toxicology. 2008;244:49–55. doi: 10.1016/j.tox.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 221.Yuan D, Ye S, Pan Y, Bao Y, Chen H, Shao C. Long-term cadmium exposure leads to the enhancement of lymphocyte proliferation via down-regulating p16 by DNA hypermethylation. Mutat Res. 2013;757:125–131. doi: 10.1016/j.mrgentox.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 222.Ferguson A, Rimmer T. Navigating the lead laws/rules/standards for renovation and repair activities: A guide for contractors and laborers. Journal of Environmental Protection. 2012;2012 [Google Scholar]
- 223.Levin R, Brown MJ, Kashtock ME, Jacobs DE, Whelan EA, Rodman J, Schock MR, Padilla A, Sinks T. Lead exposures in U.S. children, 2008: Implications for prevention. Environ Health Perspect. 2008;116:1285–1293. doi: 10.1289/ehp.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Datta-Mitra A, Ahmed O., Jr Ayurvedic medicine use and lead poisoning in a child: A continued concern in the united states. Clin Pediatr (Phila) 2014 doi: 10.1177/0009922814553397. [DOI] [PubMed] [Google Scholar]
- 225.Bellinger DC. Lead neurotoxicity and socioeconomic status: Conceptual and analytical issues. Neurotoxicology. 2008;29:828–832. doi: 10.1016/j.neuro.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Advisory Committee on Childhood Lead, Poisoning Prevention. ACCLPP; Atlanta, GA, USA: 2012. Low level lead exposure harms children: A renewed call for primary prevention: Report to the CDCP; pp. 1–54. [Google Scholar]
- 227.Hernandez-Avila M, Peterson KE, Gonzalez-Cossio T, Sanin LH, Aro A, Schnaas L, Hu H. Effect of maternal bone lead on length and head circumference of newborns and 1-month-old infants. Arch Environ Health. 2002;57:482–488. doi: 10.1080/00039890209601441. [DOI] [PubMed] [Google Scholar]
- 228.Centers for Disease Control and Prevention. Guidelines for the identification and management of lead exposure in pregnant and lactating women. Atlanta, GA: US Dept of Health and Human Services; 2010. [Google Scholar]
- 229.Gundacker C, Wittmann KJ, Kukuckova M, Komarnicki G, Hikkel I, Gencik M. Genetic background of lead and mercury metabolism in a group of medical students in austria. Environ Res. 2009;109:786–796. doi: 10.1016/j.envres.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 230.Sirivarasai J, Wananukul W, Kaojarern S, Chanprasertyothin S, Thongmung N, Ratanachaiwong W, Sura T, Sritara P. Association between inflammatory marker, environmental lead exposure, and glutathione S-transferase gene. Biomed Res Int. 2013;2013 doi: 10.1155/2013/474963. 474963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Wright RO, Schwartz J, Wright RJ, Bollati V, Tarantini L, Park SK, Hu H, Sparrow D, Vokonas P, Baccarelli A. Biomarkers of lead exposure and DNA methylation within retrotransposons. Environ Health Perspect. 2010;118:790–795. doi: 10.1289/ehp.0901429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Pilsner JR, Hu H, Ettinger A, Sánchez BN, Wright RO, Cantonwine D, Lazarus A, Lamadrid-Figueroa H, Mercado-García A, Téllez-Rojo MM, Hernández-Avila M. Influence of prenatal lead exposure on genomic methylation of cord blood DNA. Environ Health Perspect. 2009;117:1466–1471. doi: 10.1289/ehp.0800497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Li C, Yang X, Xu M, Zhang J, Sun N. Epigenetic marker (LINE-1 promoter) methylation level was associated with occupational lead exposure. Clin Toxicol (Phila) 2013;51:225–229. doi: 10.3109/15563650.2013.782410. [DOI] [PubMed] [Google Scholar]
- 234.Risher JF. Elemental mercury and inorganic mercury compounds: Human health aspects. World Health Organization; 2003. Concise International Chemical Assessment Document. [Google Scholar]
- 235.Swain EB, Jakus PM, Rice G, Lupi F, Maxson PA, Pacyna JM, Penn A, Spiegel SJ, Veiga MM. Socioeconomic consequences of mercury use and pollution. Ambio. 2007;36:45–61. doi: 10.1579/0044-7447(2007)36[45:scomua]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 236.Bose R, Onishchenko N, Edoff K, Janson Lang AM, Ceccatelli S. Inherited effects of low-dose exposure to methylmercury in neural stem cells. Toxicol Sci. 2012;130:383–390. doi: 10.1093/toxsci/kfs257. [DOI] [PubMed] [Google Scholar]
- 237.Goodrich JM, Basu N, Franzblau A, Dolinoy DC. Mercury biomarkers and DNA methylation among michigan dental professionals. Environ Mol Mutagen. 2013;54:195–203. doi: 10.1002/em.21763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Habibi L, Shokrgozar MA, Tabrizi M, Modarressi MH, Akrami SM. Mercury specifically induces LINE-1 activity in a human neuroblastoma cell line. Mutat Res Genet Toxicol Environ Mutagen. 2014;759:9–20. doi: 10.1016/j.mrgentox.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 239.Wang TC, Song YS, Wang H, Zhang J, Yu SF, Gu YE, Chen T, Wang Y, Shen HQ, Jia G. Oxidative DNA damage and global DNA hypomethylation are related to folate deficiency in chromate manufacturing workers. J Hazard Mater. 2012;213-214:440–446. doi: 10.1016/j.jhazmat.2012.02.024. [DOI] [PubMed] [Google Scholar]
- 240.Lou J, Wang Y, Yao C, Jin L, Wang X, Xiao Y, Wu N, Song P, Song Y, Tan Y, Gao M, Liu K, Zhang X. Role of DNA methylation in cell cycle arrest induced by cr (VI) in two cell lines. PLoS One. 2013;8:e71031. doi: 10.1371/journal.pone.0071031. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 241.El-Sawy M, Kale SP, Dugan C, Nguyen TQ, Belancio V, Bruch H, Roy-Engel AM, Deininger PL. Nickel stimulates L1 retrotransposition by a post-transcriptional mechanism. J Mol Biol. 2005;354:246–257. doi: 10.1016/j.jmb.2005.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH., Jr Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci U S A. 2003;100:5280–5285. doi: 10.1073/pnas.0831042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Lutz SM, Vincent BJ, Kazazian HH, Jr, Batzer MA, Moran JV. Allelic heterogeneity in LINE-1 retrotransposition activity. Am J Hum Genet. 2003;73:1431–1437. doi: 10.1086/379744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Boissinot S, Entezam A, Young L, Munson PJ, Furano AV. The insertional history of an active family of L1 retrotransposons in humans. Genome Res. 2004;14:1221–1231. doi: 10.1101/gr.2326704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Goodman JI, Augustine KA, Cunnningham ML, Dixon D, Dragan YP, Falls JG, Rasoulpour RJ, Sills RC, Storer RD, Wolf DC, Pettit SD. What do we need to know prior to thinking about incorporating an epigenetic evaluation into safety assessments? Toxicol Sci. 2010;116:375–381. doi: 10.1093/toxsci/kfq133. [DOI] [PubMed] [Google Scholar]
- 246.LeBaron MJ, Rasoulpour RJ, Klapacz J, Ellis-Hutchings RG, Hollnagel HM, Gollapudi BB. Epigenetics and chemical safety assessment. Mutat Res. 2010;705:83–95. doi: 10.1016/j.mrrev.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 247.Fan T, Schmidtmann A, Xi S, Briones V, Zhu H, Suh HC, Gooya J, Keller JR, Xu H, Roayaei J, Anver M, Ruscetti S, Muegge K. DNA hypomethylation caused by lsh deletion promotes erythroleukemia development. Epigenetics. 2008;3:134–142. doi: 10.4161/epi.3.3.6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 249.Prüss-Ürsün A, Corvalàn C. Preventing disease through healthy environments. towards an estimate of the environmental burden of disease. World Health Organization; 2006. [Google Scholar]