

Abstract
Delineation of a cell’s ultrastructure is important for understanding its function. This can be a daunting project for rare cell types diffused throughout tissues made of diverse cell types, such as enteroendocrine cells of the intestinal epithelium. These gastrointestinal sensors of food and bacteria have been difficult to study because they are dispersed among other epithelial cells at a ratio of 1:1,000. Recently, transgenic reporter mice have been generated to identify enteroendocrine cells by means of fluorescence. One of those is the peptide YY-GFP mouse. Using this mouse, we developed a method to correlate confocal and serial block-face scanning electron microscopy. We named the method cocem3D and applied it to identify a specific enteroendocrine cell in tissue and unveil the cell’s ultrastructure in 3D. The resolution of cocem3D is sufficient to identify organelles as small as secretory vesicles and to distinguish cell membranes for volume rendering. Cocem3D can be easily adapted to study the 3D ultrastructure of other specific cell types in their native tissue.
Keywords: Cellular Biology, Issue 101, Gut-Brain neurobiology, 3D electron microscopy, serial block-face scanning electron microscopy, chemosensory cell, neuropod, nutrient-sensing, gastrointestinal chemosensation
Introduction
Life inside a cell takes place in time and space. Changes over time are often studied using time-lapse microscopy combined with fluorescence imaging techniques, like super-resolution microscopy. Space, in particular the arrangement of organelles inside a cell or cell-to-cell interactions, can only be derived by a complete account of the cell’s fine structure. A comprehensive account of a cell’s fine structure can also bring clarity to genomic function in cases where the genome is available, like the C. elegans nematode1 or the flat placozoa tricoplax adherens2. Serial sectioning electron microscopy is now a reproducible, time efficient, and less expensive task thanks to the development of automated 3D electron microscopy technologies, like serial block-face scanning electron microscopy3 (SBEM).
The need for structural information to elucidate function is very evident in certain cell types where function depends on physical cell-to-cell interactions, such as neurons, glia, or sensory epithelial cells. We are particularly interested in elucidating how sensory signals from nutrients in the lumen of the gut are transduced into an electrical signal that ultimately modulates appetitive behaviors. The circuit is complex but begins at the wall of the intestine where nutrients come into contact with sensory epithelial cells, called enteroendocrine cells. Unlike other sensory epithelial cells, such as taste cells, enteroendocrine cells are dispersed throughout the gut epithelium at a ratio of one to one thousand4-7. Consequently, they have been difficult to identify and study, and for long time they were viewed only as a source of gut hormones. But with the development of cell-specific fluorescence reporter mice, the complex sensory function of these cells is emerging. Using one of these reporter mice, a peptide YY-GFP (PyyGFP) mouse, we found that enteroendocrine cells have a prominent cytoplasmic tail that we named neuropod. The appearance of neuropods suggested a conserved function in cell-to-cell communication. Thus, we reasoned that by documenting the ultrastructure of an enteroendocrine cell, the function of neuropods could be derived.
The need to understand the structure of an appendage in a dispersed cell which is hard to identify was the main rationale for developing a method to combine confocal microscopy and SBEM. The cell of interest was identified using PyyGFP enteroendocrine cell-specific reporter mice. The method allowed us to document the entire ultrastructure of an enteroendocrine cell and its neuropod. Within neuropods, we found structural features of neuronal axons, and outside neuropods, we found a physical relationship to enteric glia8. Indeed, neuropods contain about 70% of all secretory vesicles suggesting an essential role in the secretory function of these cells. Building on the structural data, more recently we found that through these neuropods, enteroendocrine cells and neurons innervating the gut form a neuroepithelial circuit, similar to that of taste cells in the tongue8,9.
Uncovering such features of gastrointestinal chemosensory mechanisms stemmed from structural data gathered using this correlative microscopy method. We believe this method could be useful in other areas of cell biology, particularly where cells are dispersed within the tissues at a very low ratio. We referred to the method as correlative confocal and serial block face scanning electron microscopy in 3D (Cocem3D). The method is comprised of the following main steps: tissue dissection, confocal microscopy, SBEM imaging, SBEM and confocal image correlation, and manual segmentation. Compared to other correlative methods, the concept is rather simple because the correlation is physical rather than chemical.
Protocol
All animal care and experiments were done in accordance with a protocol approved by the Duke University Institutional Animal Care and Use Committee.
Cocem3D stemmed from a combination of previously published protocols10,11 and was applied here to study a specific enteroendocrine cell using a PyyGFP reporter mouse12. This method can readily be applied to other cells of interest using commercially available transgenic reporter mice. The method is described in three sections: Correlative confocal microscopy, serial block-face scanning electron microscopy (SBEM), and data rendering in 3D.
Correlative Confocal Microscopy
The objective of this section is to harvest a tissue segment from the distal colon of dimensions that allow correlative confocal and SBEM imaging. The protocol is as follows:
1. Harvesting the Tissue
Prepare 10 ml of 50 µg/ml heparin solution in PBS.
- Prepare xylazine/ketamine anesthetic stock by mixing 10 ml of Ketamine (100 mg/ml) and 1 ml of xylazine (20 mg/ml).
- Dilute xylazine/ketamine stock 1:4 in 0.1M PBS.
Prepare a 100 ml of fixative solution containing 4% paraformaldehyde and 0.1% glutaraldehyde in PBS.
Anesthetize a PyyGFP mouse, 6 to 10 weeks old, with a lethal dose of xylazine/ketamine anesthetic. Inject the anesthetic intraperitoneally at 0.15 ml per 20 g of mouse weight.
Confirm proper mouse anesthetization by pinching the tail or toes, and use ointment on eyes to prevent dryness.
Use a variable flow peristaltic pump to perfuse fixative intracardially10. Using surgical scissors (13 cm length) cut open the abdominal cavity to expose the intestines, heart, and lungs. Hold heart with narrow-pattern curved forceps (12 cm length) and insert butterfly needle (19 G) from peristaltic pump into left ventricle. Immediately, cut right atrium with spring straight scissors (length 4 mm). Perfuse at a rate of 2 ml/min first with heparin solution for 1 min, and then with ice-cold fixative solution for 15 min until mouse’s tail is completely rigid. Note: It is very important to perfuse at a slow rate to prevent bursting of small vessels in the intestinal mucosa. Bursting of vessels will compromise the tissue’s ultrastructure. If perfusion is adequate, the liver should turn pale in color within 3-5 min.
Using small scissors open the abdominal cavity and excise the entire colon from the junction with the cecum to the distal rectum. Place the tissue in ice-cold PBS. While submerged in PBS, cut open with small spring scissors the colon along the mesentery.
2. Dissecting and Fixing Tissue Segments
Using a scalpel, cut about 6 small tissue segments (2 mm2) from the distal colon. Do this on a sheet of dental wax and submerged in few drops of PBS.
Post-fix the tissue segments in fixative solution for 3 hr at 4 °C.
3. Micro-dissecting Tissue Blocks
Prepare 5% low-melting agarose in PBS and keep it in a water bath at 45 °C.
Embed the tissue segments in 5 % low-melting agarose using a small plastic container, like the standard size Tissue-Tek Cryomold.
Mount the embedded sections on a vibrating blade microtome and fill the buffer tray with ice-cold PBS.
Cut 300 µm tissue strips at 0.8 amplitude and 0.04 mm/sec speed. Note: At this point, the tissue strips will be dislodged from the agarose.
Re-embed 300 µm tissue strips in agarose and mount them on microtome perpendicularly to the vibratome’s blade.
Using a vibratome, cut tissue strips at a thickness of 50 µm. Note: The final tissue blocks should be about 300 µm wide x 50 µm thick. These dimensions are optimized from pilot confocal experiments that showed that the thickness of enteroendocrine cell is between 15-20 µm and the neuropod’s length 30-70 nm. Therefore, an entire cell could be contained within a block of tissue 300 µm wide by 50 µm thick if the cell’s orientation is parallel to the face of the tissue block. Vary these dimensions depending on the tissue and cell type of interest.
Store the tissue blocks in PBS at 4 °C.
4. Confocal Imaging
Prepare a nuclear stain solution by diluting DAPI in PBS at 1:4,000.
Incubate tissue blocks in DAPI nuclear stain for 5 min. Note: DAPI staining facilitates distinguishing individual cells by their nuclei, which is helpful for correlating the confocal and SBEM images.
Mount tissue blocks on charged glass slide, add a few drops of PBS, and cover them with a coverslip.
- Using a confocal microscope, identify blocks with intact villi and PyyGFP cells of interest and imaged them to obtain z-stacks optical sections.
- Use a 20X/0.8 Zeiss Plan Apochromat objective to obtain z-stacks of 1 µm optical sections.
- For the z-stack, use the channels for 405 nm (DAPI) to determine the relationship to other cells, 488 nm endogenous GFP to localize the cell of interest, and differential interference contrast (DIC) to determine location of the cell relative to the lumen.
- Use image resolution of 1,024 pixels or higher.
5. Embedding Blocks in Agarose
Prepare 10 ml of fixative containing 4% paraformaldehyde and 2.5% glutaraldehyde in PBS.
Remove the tissue blocks from the glass slide by carefully adding PBS in the edges of the cover slip to slide the coverslip away from the glass slide without damaging the tissue block. Then, use a fine art paintbrush to transfer the tissue block from the slide to a 1.5 ml microcentrifuge tube containing 4% paraformaldehyde and 2.5% glutaraldehyde fixative.
Post-fix tissue blocks O/N at 4 °C.
Transfer tissue blocks to PBS. Then, embed blocks flat in 5% low-melting agarose by sandwiching them between two glass slides. Note: Embedding the blocks in a thin layer of agarose eases posterior manipulation during staining.
Cut the agarose in a square and make a notch on the upper hand side. Note: This step is necessary to maintain the orientation in subsequent steps.
Store blocks in a 1.5 ml microcentrifuge tube with PBS at 4 °C until further processing.
Serial Block-face Scanning Electron Microscopy (SBEM)
In this section, the tissue block is prepared and imaged with SBEM at low-magnification. The survey image of the block face is then correlated with the confocal data to identify the region containing the cell of interest. Once the region is identified, the tissue is imaged at a resolution of 7 nm/pixel and slices of 70 nm. This was sufficient to resolve and distinguish large-dense core secretory vesicles from other organelles. In enteroendocrine cells, this type of vesicles varies between 100 and 150 nm in diameter13. The protocol is as follows:
6. Staining Tissue Sections
Remove tissue from PBS and rinse three times, 5 min each, in 0.1 M cacodylate buffer.
Stain blocks for 1 hr in 0.1 % tannic acid dissolved in 0.1 M cacodylate buffer to enhance contrast of cell membranes14.
Carry out subsequent staining and dehydration of tissue according to the published protocol11. Following Deerinck et al. protocol, prepare these solutions: 1) 1% (w/v) thiocarbonhydrazide (THC); 2) lead aspartate solution; 3) 1% uranyl acetate, and 4) osmium tetraoxyde/ potassium ferrocyanide (OsO4/K Ferrocyanide). Mix 4% OsO4 and 2x K Ferrocyanide stock [0.3g K Ferrocyanide and 0.86g Na Cacodylate in 10 ml H2O] at 1:1 ratio to make OsO4/K Ferrocyanide. Note: Having the tissue embedded in agarose facilitates handling during staining. Carefully remove agarose for subsequent resin infiltration.
Rinse samples three times, 5 min each, in 0.1 M cacodylate buffer and then stain with OsO4/K Ferrocyanide solution for 2 hr at 4 °C.
Rinse in ultra pure water three times, 5 min each, and stain with TCH for 30 min at 60 °C.
Rinse in ultra pure water three times, 5 min each and stain with 2% OsO4 (no K Ferrocyanide) for 60 min at RT. Rinse again in ultra pure water three times, 5 min each.
Stain with 1% uranyl acetate O/N at 4 °C. Rinse in ultra pure water three times, 5 min each.
Stain with lead aspartate for 30 min at 60 °C. Rinse in ultra pure water three times, 5 min each.
Dehydrate tissues by rinsing in solutions with increasing concentrations of ethanol. Rinse 2 times, 5 min each, with 50%, 75%, 85%, 95%, and absolute 100% ethanol. At the end, rinse sections with propylene oxide two times, 10 min each.
7. Infiltrating and Embedding Tissue Sections in Resin
Infiltrate the blocks with resin using the commercially avaialble kit. Embed tissues in resin using an epon embedding kit. Prepare a resin mix of 48% Epoxy, 20% ml DDSA, 30% ml NMA, and 2% DMP30. Agitate resin mixture vigorously for 5 min and then place mix in vacuum for 30 min to allow bubbles to come to the surface.
Mix resin with propylene oxide at 1:1 ratio and shake vigorously. Remove the final dehydration rinse from tissues, and add resin mixed with propylene oxide. Place vial with samples in a rotator O/N and leave vial uncapped. The next day, add freshly made epon resin and mix for 90 min in rotator.
Embed blocks in resin as flat as possible by sandwiching them in between glass slides that have been cured with liquid release agent to prevent them from sticking to each other15.
Once the blocks are embedded flat, cure the blocks for an addition 48 hr at 60 °C.
Pull slides apart to release the blocks.
8. Mounting and Trimming the Tissue Block for Imaging
Under a dissecting scope, match the orientation of the tissue blocks embedded in resin with that of the confocal micrographs to facilitate identifying the regions containing the cells of interest before trimming the block.
Trim the resin embedded block manually down to a ~500 x 500 µm block-face.
Mount the block flat on a pin containing conductive epoxy and dry for 30 min. Then, lay the block flat on a surface and dry O/N at 60 °C.
Coat the block with colloidal silver liquid. Maintain the tissue sections flat to ensure slicing of the block at a right angle to facilitate correlating the serial block-face and confocal micrographs.
9. SBEM
Image the block using a Scanning Electron Microscope equipped with a SBEM system (e.g., 3View).
Set initial thickness section at ~2 µm increments and cut until the face of tissue emerges from the block.
Acquire a surver image of the entire block face.
Using Fiji software, take measurements of both the serial block-face and confocal micrographs to generate a common denominator and account for sample warping during fixation and staining16.
Locate the area of interest on the block face by multiplying the denominator to the coordinates in the confocal images. Use also other structural features, like the position of microvilli, goblet cells, or lamina propria, as a reference.
Image region of interest at a 2.25 kV and 7 nm/pixel (or 15,147X magnification) in high vacuum mode. Slice increments should be set at 70 nm or less.
Collect SBEM data in raw 16-bit .dm3 format.
10. Optimizing SBEM Images for Surface Segmentation
Convert SBEM images from raw .dm3 format to 8-bit .tiff.
Filter .tiff images using a 0.8 gaussian blur filter in Fiji.
Scale down the data set to 25 % of the original size and save it as a .tiff stack to minimize the amount of RAM memory necessary to handle the set.
Align the stack of SBEM images using the Fiji plugin “linear stack alignment with SIFT” in translation mode and cropped using the “crop 3D” plugin. Note: Cropping helps to further reduced the amount of RAM memory necessary for segmenting and volume rendering.
Data Rendering in 3D
11. Confocal Microscopy
Reconstruct z-stacks using the automatic surpass mode of the “surfaces” tool. Figure 1D, shows a reconstruction of each channel using the smooth option on, the surfaces area detail at 0.126 µm, and thresholding as absolute intensity.
12. Serial Block-face SEM
Note: Manual data segmentation is a very time consuming procedure and depending on the features to be rendered the procedure can take several weeks. Segmentation for the videos and figures presented in Bohórquez et al. 20148 was done manually and took about 500 hr of labor. It is recommended to prioritize the essential features needing rendering before beginning the segmentation process. The procedure is as follows:
Use the “surfaces” tool in draw mode and the “contour” option at drawing mode of 50 msec to segment manually and volume rendered the cell of interest.
Trace contours on every slice of the cell for smoother rendering or every 5 slices for faster rendering.
Export final images using the “snapshot” tool at a resolution of 300 dpi or more.
Representative Results
The method presented here was used to study the ultrastructure of a specific cell within a layer of epithelium. The cell of interest in this case is the elusive enteroendocrine cell. Their identification in situ has been possible only in the last few years with the development of cell-specific fluorescence reporter mice. In 2011, we developed a mouse in which the promoter of the hormone PYY drives the expression of green fluorescent protein17. In this PyyGFP mouse, enteroendocrine cells of the distal part of the small intestine and colon are easily distinguished under UV light. This mouse model was a foundation to identify a tissue block containing an enteroendocrine cell and process it for SBEM. The correlation method is referred here as cocem3D and was built on previously published protocols10,11.
Optimizing the dimensions of the tissue block is critical for the correlation. In preliminary experiments, it was determined that in a tissue block 300 µm long x 50 µm thick the number of SBEM images is reduced to a minimum while covering the entire body of the cell of interest. Figure 1A shows an overlook of the dissection using a vibrating blade microtrome to obtain the tissue block with the right dimensions. At least 100 blocks are obtained before sorting through them to pick those with intact cells of interest. Selected blocks are imaged by confocal microscopy and image z-stacks are optically spaced every 1 µm (Figure 1B and C). Figure 1D shows a volume rendered view of an enteroendocrine cell in the ileum of the mouse. Its prominent neuropod extends underneath epithelial cells for ~60 µm.
The cell of interest is found by correlating the confocal z-stacks with a SBEM image of the entire block face. An example is presented here of a block from the distal colon of a PyyGFP mouse. After obtaining confocal z-stacks (Figure 2A), the block is embedded in a thin layer of low-melting agarose (Figure 2B). This is necessary for subsequent manipulation of the block. It is important to maintain the orientation of the block as flat as possible to match the optical slices with the SBEM slices. In Figure 2C, a representative image is shown of the entire face of the tissue block. Parts of this figure were previously published in Bohorquez et al., 20148. This was subsequently correlated with the confocal image by taking into account tissue landmarks and dimensions. Figure 2D shows an overlay of the confocal and SBEM image of the block, revealing the precise location of our cell of interest.
Once the cell has been identified, SBEM imaging is done at a resolution high enough to identify secretory vesicles and other cell organelles. The data set presented here was imaged at 7 nm per pixel as images of the block were taken every 70 nm. The data set contained 643 images that span 45 µm of the tissue depth. A few microns are shaved during initial imaging of the entire block face at low magnification. The raw SBEM data set was acquired in .dm3 format and transformed to .tiff for subsequent handling. The SBEM stack was cropped using the Fiji software plugin “crop (3D)” block to contain only the region of interest. This reduces the amount of RAM memory needed for volume rendering. The cell was identified by its position and secretory vesicles, and segmented using Imaris software. Figure 3 contains a rendering in 3D of the enteroendocrine cell.
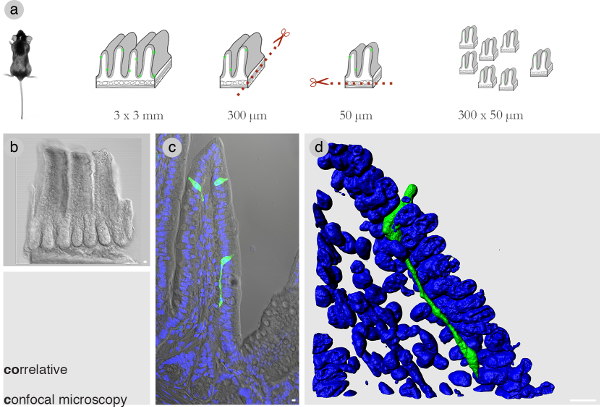
Figure 1: Correlative confocal microscopy. (A) Workflow to dissect a piece of intestinal tissue into a tissue block 300 x 50 µm thick. (B) A representative tissue block of the right dimensions. Note that a tissue block of these dimensions from the mouse small intestine spans about 3 villi or, in the case of the colon about 8 crypts. (C) A villus in the small intestine containing a cell of interest. (D) A confocal z-stack rendered in 3D using Imaris software shows an enteroendocrine cell with a prominent neuropod running underneath the intestinal epithelium. Blue = DAPI nuclear stain. Bars = 10 µm. Please click here to view a larger version of this figure.
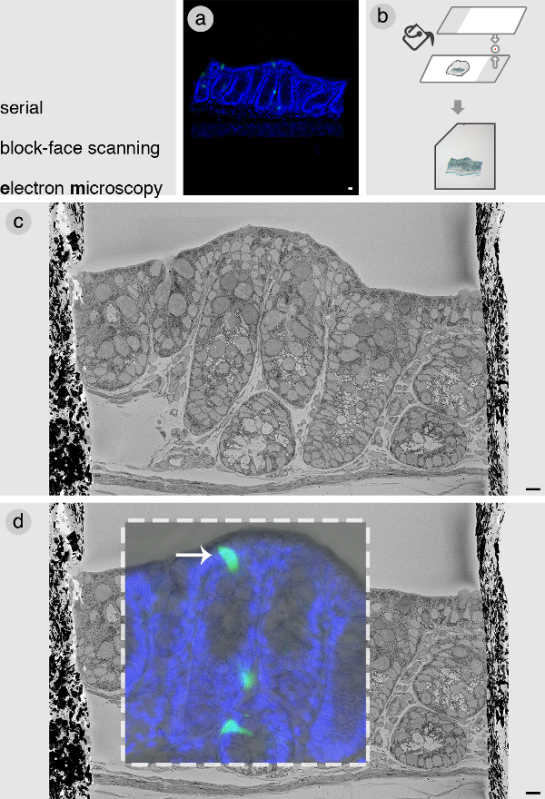 Figure 2: Serial block-face scanning electron microscopy. (A) Confocal image of a selected tissue block from the colon containing PyyGFP enteroendocrine cells (green) of interest. (B) Once the block is imaged by confocal microscopy, it is then embedded flat in low-melting agarose using glass slides. Embedding the tissue block in agarose facilitates subsequent manipulation, and the notch on the left corner helps to mount the block within the SBEM in the right orientation. (C) SBEM image of the face of the block. (D) Correlation of confocal data with SBEM to identify cell of interest (white arrow). Images in c and d of this figure are modified from Bohorquez et al., 20148. Blue = DAPI nuclear stain. Bars = 10 µm. Please click here to view a larger version of this figure.
Figure 2: Serial block-face scanning electron microscopy. (A) Confocal image of a selected tissue block from the colon containing PyyGFP enteroendocrine cells (green) of interest. (B) Once the block is imaged by confocal microscopy, it is then embedded flat in low-melting agarose using glass slides. Embedding the tissue block in agarose facilitates subsequent manipulation, and the notch on the left corner helps to mount the block within the SBEM in the right orientation. (C) SBEM image of the face of the block. (D) Correlation of confocal data with SBEM to identify cell of interest (white arrow). Images in c and d of this figure are modified from Bohorquez et al., 20148. Blue = DAPI nuclear stain. Bars = 10 µm. Please click here to view a larger version of this figure.
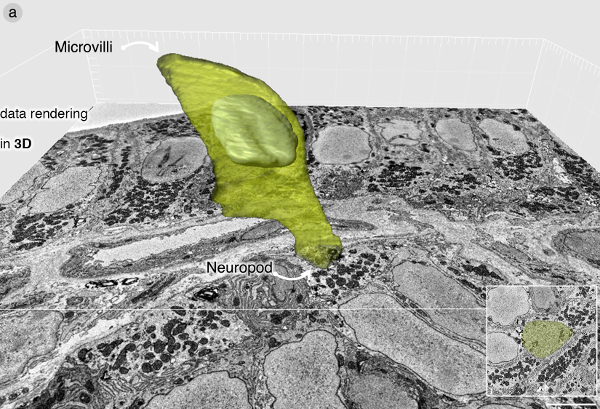 Figure 3: Data rendering in 3D. (A) Volume rendering of enteroendocrine cell using Imaris software. The cell contains a neuropod packed with secretory vesicles. For a complete description of the ultrastructure of an enteroendocrine cell, please refer to Bohórquez et al., 20148. Please click here to view a larger version of this figure.
Figure 3: Data rendering in 3D. (A) Volume rendering of enteroendocrine cell using Imaris software. The cell contains a neuropod packed with secretory vesicles. For a complete description of the ultrastructure of an enteroendocrine cell, please refer to Bohórquez et al., 20148. Please click here to view a larger version of this figure.
Discussion
Gastrointestinal chemosensation is emerging as an exciting new field in biomedical research. This is in great part due to the discovery of functional taste receptors in enteroendocrine cells18. Subsequent studies have shown that enteroendocrine cells express specific receptors for nutrients, including carbohydrates, lipids, and aminoacids5,6,19,20. The catalytic factor for these discoveries has been the development of reporter mice, in which enteroendocrine cells are fluorescently labeled20. We developed one of these mice, the PyyGFP mouse, to study enteroendocrine cells of the distal small intestine and colon17,21. These cells are of interest because they secrete PYY and glucagon-like peptide 1, both of which are inducers of satiety22,23. At the time, a complete ultra-structural account of these cells was missing, which we believed was essential to understand their signaling mechanisms.
Here, we described a visually a method to bridge confocal microscopy with SBEM. The method is referred as Cocem3D, which allowed us to document the complete ultrastructure of enteroendocrine cells. We have reported that these cells have a neuropod that contains three-quarters of all of the secretory vesicles8. Within the neuropod, there are neurofilaments and much like neuronal axons, neuropods are nurtured by enteric glial cells8. More important, it is though these neuropods that enteroendocrine cells physically connect to neurons innervating the intestine and colon9.
One of the strengths of cocem3D is its simplicity. Reducing the tissue sample into a block that can be imaged by confocal and SBEM facilitates the identification of a specific cell within a tissue. Secretory vesicles and other organelles can be identified with ease at a resolution of 7 nm/pixel. Because the sections in SBEM are discarded, further processing of tissues to identify specific proteins is not an option at this point. However, the development of methods such as ATUM24, in which sections from the tissue block are preserved, are likely to enable the identification of specific proteins in the cell. Determining the specific location of chemosensory receptors on enteroendocrine cells is essential information for the development of drug therapies for obesity, because enteroendocrine cells are a sensory interface between food in the gut and satiety in the brain.
Disclosures
Dr. Satish Medicetty is an employee of and receives 100% salary from Renovo Neural Inc; however, this does not alter the author’s adherence to JoVE policies on sharing data and materials. No conflict of interest is declared for remaining authors.
Acknowledgments
Our sincere appreciation is expressed to the following people: Drs. Sam Johnson and Benjamin Carlson of the Duke Light Microscopy Core Facility for their assistance with data visualization software, and Ms. Valerie Lapham and Dr. John M. Mackenzie, Jr. of the Center for Electron Microscopy at North Carolina State University for their advice on electron microscopy. We thank Dr. Elaine B. Bohórquez for her editorial assistance. Authors contributed in the following manner: DVB, SM, and RAL designed experiments and analyzed data. DVB performed experiments and FH performed manual rendering of data. SM is director of Renovo Neural, where SBEM data was acquired. DVB wrote the manuscript and all authors reviewed and edited the final manuscript. This work was supported by NIH grants R01DK091946 and Veterans Affairs grant I01BX002230 to RAL, and F32DK094704, to DVB.
References
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philosophical transactions of the Royal Society of London. Series B, Biological. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Smith CL, et al. Novel cell types, neurosecretory cells, and body plan of the early-diverging metazoan Trichoplax adhaerens. Current biology : CB. 2014;24:1565–1572. doi: 10.1016/j.cub.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk W, Horstmann H. Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS biology. 2004;2:e329. doi: 10.1371/journal.pbio.0020329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelstoft MS, et al. Seven transmembrane G protein-coupled receptor repertoire of gastric ghrelin cells. Molecular metabolism. 2013;2:376–392. doi: 10.1016/j.molmet.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra R, et al. Immunoglobulin-like domain containing receptor 1 mediates fat-stimulated cholecystokinin secretion. The Journal of clinical investigation. 2013;123:3343–3352. doi: 10.1172/JCI68587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. Amino acids stimulate cholecystokinin release through the Ca2+-sensing receptor. Am J Physiol Gastrointest Liver Physiol. 2011;300:G528–G537. doi: 10.1152/ajpgi.00387.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GJ, et al. Electrical activity-triggered glucagon-like peptide-1 secretion from primary murine L-cells. The Journal of physiology. 2011;589:1081–1093. doi: 10.1113/jphysiol.2010.198069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohórquez DV, et al. An enteroendocrine cell-enteric glia connection revealed by 3D electron microscopy. PloS one. 2014;9:e89881. doi: 10.1371/journal.pone.0089881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohórquez DV, et al. Neuroepithelial circuit formed by innervation of sensory enteroendocrine cells. The Journal of clinical investigation. 2015. [DOI] [PMC free article] [PubMed]
- Knott GW, Holtmaat A, Trachtenberg JT, Svoboda K, Welker E. A protocol for preparing GFP-labeled neurons previously imaged in vivo and in slice preparations for light and electron microscopic analysis. Nature protocols. 2009;4:1145–1156. doi: 10.1038/nprot.2009.114. [DOI] [PubMed] [Google Scholar]
- Deerinck T, Bushong E, Thor A, Ellisman M. NCMIR methods for 3D EM: a new protocol for preparation of biological specimens for serial block face scanning electron microscopy. San Diego San Diego, CA: University of California; 2010. [Google Scholar]
- Bohórquez DV, Chandra R, Samsa LA, Vigna SR, Liddle RA. Characterization of basal pseudopod-like processes in ileal and colonic PYY cells. Journal of molecular histology. 2011;42:3–13. doi: 10.1007/s10735-010-9302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson O, et al. Distribution and immunocytochemical colocalization of peptide YY and enteroglucagon in endocrine cells of the rabbit colon. Endocrinology. 1991;129:139–148. doi: 10.1210/endo-129-1-139. [DOI] [PubMed] [Google Scholar]
- Mizuhira V, Futaesaku Y. New fixation method for biological membranes using tannic acids. Acta Histochem Cytochem. 1972;5:233–236. [Google Scholar]
- Reymond OL, Pickett-Heaps JD. A routine flat embedding method for electron microscopy of microorganisms allowing selection and precisely orientated sectioning of single cells by light microscopy. Journal of microscopy. 1983;130:79–84. doi: 10.1111/j.1365-2818.1983.tb04200.x. [DOI] [PubMed] [Google Scholar]
- Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nature. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohórquez D, Chandra R, Samsa L, Vigna S, Liddle R. Characterization of basal pseudopod-like processes in ileal and colonic PYY cells. J Mol Histol. 2011;42:3–13. doi: 10.1007/s10735-010-9302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang HJ, et al. Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15069–15074. doi: 10.1073/pnas.0706890104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou AP, et al. The G-protein-coupled receptor GPR40 directly mediates long-chain fatty acid-induced secretion of cholecystokinin. Gastroenterology. 2011;140:903–912. doi: 10.1053/j.gastro.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann F, et al. Glucose sensing in L cells: a primary cell study. Cell metabolism. 2008;8:532–539. doi: 10.1016/j.cmet.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohorquez DV, Liddle RA. Axon-like basal processes in enteroendocrine cells: characteristics and potential targets. Clinical and translational science. 2011;4:387–391. doi: 10.1111/j.1752-8062.2011.00299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batterham RL, et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature. 2002;418:650–654. doi: 10.1038/nature00887. [DOI] [PubMed] [Google Scholar]
- Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. The Journal of clinical investigation. 1998;101:515–520. doi: 10.1172/JCI990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayworth KJ, et al. Imaging ATUM ultrathin section libraries with WaferMapper: a multi-scale approach to EM reconstruction of neural circuits. Frontiers in neural circuits. 2014;8:68. doi: 10.3389/fncir.2014.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]