

Abstract
Mineral supplements are often included in multivitamin preparations because of their beneficial effects on metabolism. In this study, we used an animal model of light-induced retinal degeneration to test for photoreceptor cell protection by the essential trace element zinc. Rats were treated with various doses of zinc oxide and then exposed to intense visible light for as long as 8 h. Zinc treatment effectively prevented retinal light damage as determined by rhodopsin and retinal DNA recovery, histology and electrophoretic analysis of DNA damage and oxidized retinal proteins. Zinc oxide was particularly effective when given before light exposure and at doses two- to four-fold higher than recommended by the age-related eye disease study group. Treated rats exhibited higher serum and retinal pigment epithelial zinc levels and an altered retinal gene expression profile. Using an Ingenuity database, 512 genes with known functional annotations were found to be responsive to zinc supplementation, with 45% of these falling into a network related to cellular growth, proliferation, cell cycle and death. Although these data suggest an integrated and extensive regulatory response, zinc induced changes in gene expression also appear to enhance antioxidative capacity in retina and reduce oxidative damage arising from intense light exposure.
INTRODUCTION
By its action on rhodopsin light triggers the physiological process of visual transduction. Intense or prolonged light exposure, however, can initiate pathological processes within visual cells commonly referred to as retinal light damage. Light-induced retinal damage can give rise to a series of apoptotic reactions, leading to photoreceptor cell death (1,2), and to repair processes resulting in recovery of photoreceptor function (3). In animal models, previous light rearing history, age and diet are all known to impact the extent of retinal cell loss from light exposure, whereas the inherent susceptibility to light damage is influenced by genetics and environmental circadian factors (4). Originally described in 1966 (5), retinal light damage has long served as an end point model of retinal degenerations arising from genetic inheritance and age-related visual cell loss. For example, there are remarkable morphological similarities between end stage retinal remodeling in the light-damaged rat retina and in advanced atrophic age-related macular degeneration (AMD; 6).
Oxidative stress has been implicated in retinal light damage, as numerous antioxidants are known to prevent photoreceptor cell damage following exposure to intense visible light (4,7). Likewise, the progression of AMD appears to depend to some extent on oxidative stress and chronic light levels. Crabb et al. (8) and Nakata et al. (9) found that drusen from AMD patients contained a variety of oxidized-lipid protein adducts, present in quantities greater than found in age-matched unaffected individuals. Epidemiological evidence indicates that micronutrient antioxidants reduce the risk of neovascular AMD (10) and that supplementation with antioxidants plus zinc reduces the rate of disease progression to advanced AMD (11). The age-related eye disease study (AREDS) also demonstrated beneficial effects with zinc or antioxidants alone (11), confirming earlier findings in a smaller cohort of patients receiving a high-zinc dietary supplement (12).
Among ocular tissues zinc levels are relatively high in the retina and retinal pigment epithelium (RPE; 13,14). Although zinc is present in all retinal cells it appears to be concentrated in photoreceptor rod outer segments (ROS), the outer nuclear layer (ONL) and in the photoreceptor cell synaptic region (14). Visual transduction may be affected by zinc binding to rhodopsin or phosphodiesterase within ROS, as well as to disk membranes (15). Its distribution in ROS, and more generally its nuclear to cytoplasmic ratio has been reported to change during, or in response to, light exposure (16). During light adaptation zinc may also migrate from the ONL to the rod inner segment (RIS), and changes in the ratio of free to protein-bound zinc also can occur (17). Zinc modulates synaptic transmission in rod and cone photoreceptors and blocks the depolarizing effects of GABA in horizontal cells (18), further suggesting an important role in the generation of electrogenic potentials (13). Zinc also binds cysteine residues in metallothionein, a protein reservoir for zinc with important regulatory functions in homeostasis. Metallothionein, presumably bound to zinc, is known to contribute to the translocation and consequent activation of protein kinase C, which has two zinc-binding motifs at its N-terminus (19). The tight regulation of cellular zinc levels appears to be required not only for controlling protein structure, function and transcription but also its potential for toxicity, which can emerge in the retina at higher levels (14).
In the RPE, zinc is known to serve as a cofactor for enzymes involved in degrading the shed tips of ROS (20) and for matrix metalloproteinases involved in the remodeling and turnover of Bruch’s membrane (21). Incubation of RPE cells with zinc in vitro leads to an induction of metallothionein expression (22) and increased glutathione synthesis (23). Dietary zinc also increases RPE metallothionein and reduces peroxide formation (24). Zinc ions are known to inhibit voltage-gated proton channels (25), which are linked to superoxide production by NADPH oxidase (26). This suggests that in retinal tissues zinc can regulate superoxide formation specifically. These protective effects of zinc, however, appear to be concentration dependent. At low levels zinc promotes RPE cell viability, whereas higher zinc concentrations lead to RPE cell death (27,28). Neurotrophic factors control expression of zinc transmembrane influx and efflux transport proteins and thus, zinc uptake into the RPE (28). At micromolar concentrations zinc can enhance the binding of complement factor H (CFH) to factor C3b (29,30) leading to an inhibition of lysis in intact host cells, whereas at higher levels the protective effect of zinc appears to be lost (29). Recently, serum CFH polymorphism H402 has been found to reduce binding strength for malondialdehyde (MDA)-modified protein epitopes, modifications generated by oxidative stress and proinflammatory reactions (31). This extent of binding also may be modulated by zinc.
Herein, we describe a concentration dependent protective effect of zinc oxide observed in the rat model of light-induced retinal degeneration. As determined by biochemical measures and histology, zinc, at a level 2–4 times higher by body mass than found with the AREDS formulation, prevented photoreceptor cell damage from light. The protective effect of zinc oxide was greatest when administered 1–4 h before light exposure and did not appear to depend on the form of zinc salt used. Other divalent cations were ineffective in preventing retinal photoreceptor cell degeneration. Zinc treatment also altered the protein profile and genetic signature of the rat retina, but no dramatic increase in retinal zinc was found. We suggest that zinc uptake from blood by the RPE has an effect, from changes in retinal gene expression and protein abundance or structure, on visual cell recovery following intense light treatment.
MATERIALS AND METHODS
Animals and rearing conditions
Male weanling Sprague–Dawley rats from Harlan Inc. (Indianapolis, IN) were maintained in darkness or in a dim light / dark environment for 40 days. The cyclic light environment consisted of 12 h of white light (20–40 lux) per day, while routine animal maintenance in the dark environment was carried out under dim red illumination (>600 nm) for less than 30 min each day. All animals received food and water ad libitum. The use of rats in this investigation conformed to the ARVO statement for the Use of Animals in Ophthalmic and Vision Research and with Laboratory Animal Resource Committee guidelines at Wright State University.
Intense light exposure and zinc treatment
At P60-65 dark reared rats were exposed to intense visible light (1200–1400 lux), beginning at 9:00 A.M. Cyclic light reared rats were dark adapted for 16 h before 8 h of intense light treatment starting at 1:00 A.M. (4). Light exposures were conducted in green Plexiglas cylinders (#2092), which transmit 490–580 nm light (5), surrounded by seven circular fluorescent bulbs (32). Light treatments normally lasted for 4 h, with two dark reared rats per chamber. For western protein analysis light exposures were either 4 or 24 h. Normal core body temperature was maintained by a gentle flow of air through the cylinders. Water and food were available during light treatment. Following light exposure most animals were returned to darkness for 14 days, to allow for the removal of necrotic photoreceptor cell material and repair and recovery in still surviving photoreceptors (5). Some rats were sacrificed immediately after light treatment, while others were killed 24 or 48 h later. Zinc oxide (Alfa Aesar, Ward Hill, MA) was dissolved in acidified water (pH ~2) and given 1× IP to rats, normally 1 h before the start of light treatment. Vehicle-treated rats received an equivalent volume of acidified water. In some experiments other zinc salts, at equal molar concentrations, or other divalent cations were used. Figure 1 contains a diagram outlining the time line for zinc injection and light treatment of rats, along with sample times and tissue analysis.
Figure 1.

Time line for zinc injections and intense light treatment of rats reared in dim cyclic light or darkness. Following intense light treatment all rats were placed into the dark environment for various periods of time, depending on the measurements to be performed.
Measurements of photoreceptor cell survival
Whole eye rhodopsin and retinal DNA levels were used as end point estimates of visual cell survival, essentially as described (33). These determinations were made 2 weeks after intense light exposure, using the two eyes of each animal. Survival was calculated from the average rhodopsin and DNA levels in experimental animals, compared with those found in rats unexposed to damaging light. Briefly, rats were killed in a CO2 saturated chamber under dim red illumination. The eyes were then either enucleated for rhodopsin determinations, or retinas excised for DNA measurements. Rhodopsin was determined using Emulphogene BC-720 detergent (Sigma, St. Louis, MO) extraction and measuring absorbance at 500 nm, before and after bleaching in vitro (34). Photoreceptor cell DNA was determined by the Hoechst (Calbiochem-Behring, La Jolla, CA) dye-binding assay (35). For both experimental and control eyes, photoreceptor DNA was estimated by subtracting the DNA content in the inner retinal layers from the total retinal DNA (35).
Efficacy of drug treatment
Average visual cell recovery was determined from the respective averages for rhodopsin and DNA for each concentration of zinc used or for each time point studied. Protective efficacy was determined by the following formula:
Retinal histology
Prior to enucleation, the superior aspect of the eye was marked to insure proper orientation during processing. Enucleated eyes had the lens removed and the eyecups were fixed in 50% Karnovsky’s solution for 24 h. The eyes were then transferred to 0.1 M sodium cacodylate buffer, pH 7.4, and left at 4°C until processed. Eyes were paraffin embedded using a Tissue-Tek Vacuum infiltration processor (Sakura Finetek, Torrance, CA). Eyes were sectioned vertically with a Shandon Finesse microtome (Thermo Fisher Scientific, Rockford, IL) at 4 μm and stained with hematoxylin and eosin. The midsuperior aspect of the retina was examined.
DNA extraction and electrophoresis
Retinal DNA was extracted as described in Maniatis et al. (36) and separated by neutral pH gel electrophoresis on 1.5% agarose gels (37). DNA fragmentation was visualized by ethidium bromide staining and photographed under UV light (37).
Western analysis of retinal proteins
Tissues were homogenized (2 retina per mL) in PBS supplemented with protease inhibitors (Sigma), DTPA (2 mM), BHT (100 μM) and EDTA (1 mM). Homogenates were mixed with sample buffer and heated at 37°C for 1 h before being loaded onto the gel. SDS-PAGE gels consisting of a 4% stacking gel and a 12.5% denaturing gel were run. Each lane contained 20 μg of protein as determined by the Bradford method. The proteins were either visualized with GelCode® blue stain reagent (Thermo Fisher Scientific) or transferred to a PVDF membrane overnight at 200 mA using a CAPS (10 mM, pH 11.0) 20% methanol transfer buffer. Nonspecific binding sites were blocked with 2% nonfat dry milk in PBS / Tween for 1 h at 37°C; blots were then incubated with primary antibody for 1 h at 37°C. Following three washes of 5 min each with blocking buffer diluted 1 to 10, the appropriate HRP-conjugated secondary antibody was incubated for 1 h at 37°C. Again, the blots were washed 3X, and then reactive bands visualized by chemiluminescence on X-ray film. Table 1 contains a list of the primary antibodies studied and their dilutions. The appropriate secondary antibody was used for each primary, either goat anti-rabbit or goat anti-mouse IgG HRP conjugate (Bio-Rad, Hercules, CA).
Table 1.
Primary antibodies used for western analysis of proteins.
| Protein | Dilution | Supplier |
|---|---|---|
| Glyceraldehyde-P-dehydrogenase (GAPDH) | 1 / 1000 | Enzo Life Sciences, Plymouth Meeting, PA |
| Heme oxygenase (HO-1) | 1 / 1000 | Enzo Life Sciences, Plymouth Meeting, PA |
| Carboxyethylpyrrole (CEP) | 1 / 500 | Dr. J. Crabb, Cole Eye Inst., Cleveland, OH |
| Transducin (Tα), | 1 / 1000 | Cytosignal, Irvine, CA |
| S-antigen (S-ag; C10C10) | 1 / 2000 | L. Donoso, Wills Eye Hosp., Philadelphia, PA |
Retinal gene array analysis
The four treatment conditions were described elsewhere, and briefly were with / without zinc oxide injection (5.2 mg kg−1) and with / without 4 h of intense light exposure. In each case, retinas were harvested 5 h after vehicle or zinc oxide treatment and then flash frozen. For each treatment condition three biological replicates, each consisting of retinas from three different animals, were analyzed. That is, tissues from nine different animals were evaluated for each condition. Total RNA was extracted from the tissues with Trizol reagent and the supplier’s protocol (Invitrogen, Carlsbad, CA) and further purified through an RNAeasy column (Qiagen, Germantown, MD). The integrity of the RNA was assayed using an Agilent 2100 BioAnalyzer NanoChip (Agilent Technologies, Santa Clara, CA). High quality RNA samples were screened using an Illumina bead chip rat gene expression array following the supplier’s protocol (Ratref-12 expression bead chip; Illumina, San Diego, CA). Each bead chip allows the gene profiling of 22 525 mRNA transcripts. Three array chips were screened per treatment condition, each screen representing a biological replicate. The relative levels of gene expression data were measured using Illumina Genome / Bead Studio software (http://www.illumina.com/documents/products/datasheets/datasheet_genomestudio_software.pdf) and quality confirmed, and normalized using lumi (38,39). Comparison of pooled data sets defining different treatment groups (two at a time) were made using limma (40). A threshold P-value of 0.05 (adjusted for multiple testing) was used as the cut-off to define the threshold at which a transcript was considered statistically significantly differentially expressed relative to control. Additional informatic analyses of gene lists derived from this analysis were performed with IPA software (Ingenuity Systems, Redwood City, CA).
Tissue zinc analysis
Rats were injected (IP) with 5.2 mg kg−1 of zinc oxide and tissue samples taken at 0, 1 or 5 h after injection. Serum was collected from trunk blood immediately after respiratory arrest and then frozen on dry ice before being sent for analysis. Other rats were subjected to trans-cardiac perfusion with saline prior to retina removal. Retinas and eye cups (RPE / choroid / sclera) were excised and immediately frozen. An additional set of rats was injected with zinc and retinas collected without perfusion. All samples were sent to Columbia Analytical Services (Kelso, WA) for analysis of zinc content by inductively coupled plasma mass spectroscopy (ICP-MS). N = 4–5 rats for each time point.
RESULTS
Photoreceptor cell survival in zinc-treated rats
To assess the protective efficacy of zinc in our animal model of light-induced retinal degeneration, we treated dark reared rats with various doses of zinc oxide and then exposed them to intense visible light for 4 h. Figure 2 contains rhodopsin and DNA levels, measures of visual cell survival, determined 2 weeks after light treatment along with the protective efficacy of zinc, calculated from the average recovery of rhodopsin and DNA. As shown in Fig. 2A, the control rats unexposed to intense light had an average rhodopsin value of 2.1 nmol per eye, while photoreceptor cell DNA was 187 μg per retina. Zinc treatment of the unexposed, dark maintained, control animals did not affect these values. At a dose of 1.3 mg kg−1, which approximates the AREDS recommended level of zinc (11) a modest improvement in visual cell survival was found after light treatment. Rhodopsin was about 50% of the dark control value, whereas photoreceptor cell DNA was 43% of control. At doses of 2.6 mg kg−1 and above, the protective effect of zinc was greater. Rhodopsin levels were 1.3–1.5 nmol per eye at the higher zinc concentrations and DNA recovery was more than 125 μg per retina. These values are 62–71% and over 67%, respectively, of the rhodopsin and DNA levels found in unexposed dark maintained rats. Using the averages for rhodopsin and DNA recovery, overall efficacy was then calculated. When zinc treatment was compared with the average values for vehicle-treated light exposed rats its protective efficacy was approximately 25% at 1.3 mg kg−1 and 60–70% at the higher zinc levels (Fig. 2B).
Figure 2.

Dose response curve for zinc oxide treatment of rats 1 h before intense light exposure. P60 dark reared rats were given zinc oxide, or the aqueous vehicle (IP) and then exposed to light for 4 h. Visual cell recovery was determined by rhodopsin and retinal DNA measurements, performed after a 14 day dark recovery period (* = P < 0.05; A). Data were presented as the mean ± S.D. for n = 8 rats with one eye used for rhodopsin and the fellow eye for DNA measurements. (B) Percent protective efficacy, calculated from the average rhodopsin and retinal DNA values in zinc-treated rats in comparison to vehicle-treated animals.
Retinal histology and DNA gel electrophoresis
Biochemical measures of visual cell survival were confirmed by retinal histology in sections from the superior hemisphere of rat eyes (Fig. 3). As shown in Fig. 3B, 5.2 mg kg−1 of zinc had no apparent effect on retinal morphology in dark maintained rats. The ONL contained 8–9 rows of photoreceptor cell nuclei (41), the INL was unaffected, and both layers resembled the retinal morphology found in dark maintained-vehicle treated rats (Fig. 3A). In contrast to its respective dark control, intense light caused considerable loss of photoreceptor cell nuclei in vehicle-treated rats (Fig. 3C). The ONL was reduced to approximately 2–3 rows of nuclei. ROS/ RIS were absent and the RPE appears to be damaged or is missing. The INL contained several vacuoles or cysts, typical of severely light-damaged rat retina (5). Overall retinal thickness was also considerably less than found in the unexposed rat retina (Fig. 3A). However, zinc treatment 1 h before light exposure resulted in significantly better retinal morphology (Fig. 3D). The ONL was intact, the ROS/ RIS region retained much of its overall length and RPE morphology appeared normal. As expected, the inner retinal layers also exhibited relatively normal morphology (compare Fig. 3D with 3B).
Figure 3.
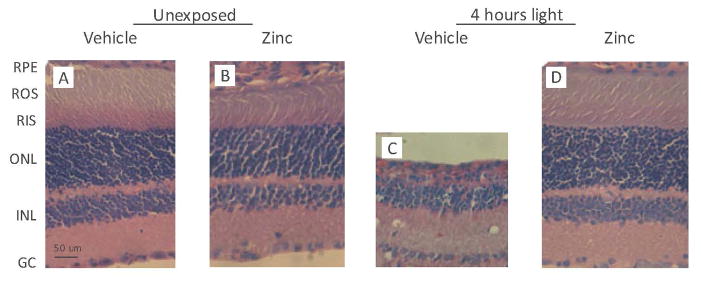
Representative retinal sections from rats treated with zinc oxide before intense light exposure. Rats were given zinc oxide, at a dose of 5.2 mg kg−1, or vehicle and then kept in darkness (panels A and B), or exposed to light for 4 h (panels C and D). After a 14 day dark recovery period rats were euthanized in a CO2 atmosphere and their eyes enucleated and placed in Karnovsky’s fixative for 24 h. Paraffin-embedded sections were stained with hematoxylin / eosin. The vehicle-treated rat retina (panel C), exhibits distinct losses of photoreceptor nuclei, RPE cell damage and overall thinning compared with unexposed retinas. Zinc treatment (panel D) appears to ameliorate the light-induced changes seen in panel (C). All sections are from the superior hemisphere along the vertical meridian. Abbreviations: RPE, retinal pigment epithelium; ROS, rod outer segments; RIS, rod inner segments; ONL, outer nuclear layer; INL, inner nuclear layer; GC, ganglion cell. Magnification: Bar shown in panel (A) represents 50 μm.
Further evidence of the protective effect of zinc was found by neutral gel electrophoresis of retinal DNA extracted 2 days after light exposure. As shown in Fig. 4 a pattern of DNA degradation products, consisting of an array of DNA fragments of various sizes and a DNA ladder of lower molecular weight bands, characteristic of endonuclease mediated apoptosis (1,2), was present in the extracts from vehicle-treated, light-exposed rats. The retinal extract from light-exposed rats treated with zinc oxide contained primarily high-molecular weight DNA fragments. This pattern resembles that found for the undegraded DNA present in retinal extracts from control animals unexposed to intense light.
Figure 4.

Gel electrophoresis of retinal DNA 48 h after intense light exposure. DNA was extracted and pooled from the retinas of two rats treated with zinc oxide (5.2 mg kg−1), or vehicle, before light exposure. DNA fragmentation was visualized by ethidium bromide staining under UV light. A ladder of lower molecular weight DNA fragments and a variety of higher molecular weight fragments are present in the sample from vehicle-treated rats, whereas both the zinc-treated and dark control rat retinal DNA is retained as a high-molecular weight band near the top of the gel.
Time course of zinc uptake and relative efficacy
Tissue zinc levels were measured to determine relative tissue concentrations and whether zinc oxide treatment led to increased ocular tissue levels. Table 2 contains the results of ICP-MS analysis for serum zinc and for excised retina and RPE tissues, 1 and 5 h after zinc oxide administration. In untreated rats, base line serum zinc was 2.2 μg mL−1 and in good agreement with previously published values (13,14,42). One hour after an IP dose of 5.2 mg zinc oxide per kg body weight, serum zinc levels increased to 7.6 μg mL−1. Four hours later the concentration of serum zinc was 6.9 μg mL−1. These serum zinc levels were significantly higher than found in untreated rats. Table 2 also shows that zinc administration did not result in an increase in retinal zinc. We found 0.11–0.12 μg zinc / retina, which also compared favorably to earlier work (24), but which is much lower than found in serum. In addition, there were no detectable differences in retinal zinc levels dependent on whether the rats were perfused with saline or not. Because blood would be expected to cross-contaminate the RPE (RPE / choroid / sclera), these tissues were excised from rats previously perfused with saline. In untreated control rats, RPE zinc levels were 0.18 μg, approximately 1.5 fold higher than in retina. Compared with the untreated controls, zinc treatment led to a significant increase in the RPE level of zinc 1 h after administration (0.23 μg vs 0.18 μg; P < 0.02). Although the RPE level of zinc was also higher 5 h after zinc oxide treatment, the difference between its levels in control and treated animals was not significant (P > 0.1).
Table 2.
Tissue zinc levels in rats given 5.2 mg kg−1 zinc oxide IP.
| Serum (μg mL−1; n = 4) | Retina (μg per retina; n = 8–9) | RPE (μg per eye cup; n = 8–10) | |
|---|---|---|---|
| 0 h | 2.2 ± 0.4 | 0.11 ± 0.02 | 0.18 ± 0.03 |
| 1 h | 7.6 ± 0.4*** | 0.11 ± 0.01 | 0.23 ± 0.05* |
| 5 h | 6.9 ± 1.7** | 0.12 ± 0.02 | 0.21 ± 0.04 |
P < 0.001;
P < 0.01;
P < 0.02 compared with 0 h.
The time course of zinc’s protective effect was determined by giving a single dose of zinc oxide to rats at various times before or after the onset of light (Fig. 5). As determined by visual cell recovery, zinc was very effective at a dose of 5.2 mg kg−1 when given up to 4 h before onset of light exposure, but largely ineffective when administered after light exposure had begun (Fig. 5A). Compared to the average values in vehicle-treated rats, rhodopsin and retinal DNA levels were significantly higher (P < 0.001) in rats pretreated with zinc 1–4 h before light. At those time points, the efficacy for zinc treatment ranged between 60% and 70% (Fig. 5B). Zinc administration 5 h before light resulted in an intermediate level of photoreceptor protection (P < 0.01) and about 30% efficacy, whereas zinc was ineffective when given 6–24 h before lights on. Similarly, when given 1–4 h after light onset the efficacy of zinc treatment was only about 20% and the differences between zinc and vehicle-treated rats were not significant (P > 0.10).
Figure 5.

Time course of zinc oxide protection in rats exposed to intense visible light. Rats were given a single dose of zinc oxide (5.2 mg kg−1) at various times before or after the onset of light. Two weeks after a 4 h light exposure rhodopsin and retinal DNA were measured to determine the effect of zinc on visual cell recovery. Zinc oxide was effective for the recovery of visual cell markers when given 1–4 h before light and less effective, or ineffective, at earlier times and after light exposure had started. (A) Results represent the mean ± SD for n = 8 rats per time point; * = P < 0.001. (B) Protective efficacy for zinc treatment calculated from the average values for rhodopsin and retinal DNA.
To estimate the duration of its effectiveness, we treated rats previously reared in dim cyclic light with zinc oxide 1 h before an 8 h light exposure starting at 1:00 A.M. This start time has been shown to result in about 50% photoreceptor cell loss after 8 h of intense light (33). As expected and as shown in Fig. 6, rhodopsin and DNA in the vehicle-treated animals were about 50% of the levels present in unexposed rats. However, in the zinc-treated animals rhodopsin and DNA recoveries were 67% and 83%, respectively, of the values found in unexposed control animals. Thus, using a dose of 5.2 mg zinc oxide per kg, zinc may provide protection from exposure to intense illumination for durations as long as 8 h.
Figure 6.

Visual cell recovery in rats previously reared in dim cyclic light. Weanling rats were maintained in a dim cyclic light environment for 40 days and then exposed to intense visible light for 8 h starting at 1:00 A.M. Prior to light treatment rats were given zinc oxide (5.2 mg kg−1) or the aqueous vehicle. Rhodopsin and retinal DNA were measured 2 weeks after light exposure. Zinc treatment was effective in reducing visual cell loss for the 8 h period studied (* = P < 0.01). Results are for a total n = 12–16 animals from two separate experiments and shown as the mean ± SD.
Different zinc compounds and other divalent cations
To demonstrate that zinc ion was effective in reducing the extent of retinal light damage, we gave equal molar concentrations of zinc oxide and other zinc salts to rats and compared their protective efficacy. In addition, magnesium (Mg) and calcium (Ca) chloride were tested in our light damage model. Figure 7 contains the results of these findings. Zinc oxide and zinc gluconate were equally effective in reducing retinal light damage. Rhodopsin and photoreceptor cell DNA levels were 70–80% of the values found in unexposed, dark maintained rats. These values were significantly higher than found in light-exposed vehicle-treated animals (P < 0.001). Zinc chloride (ZnCl2) was also significantly more effective, with recoveries for rhodopsin and DNA that were 80–90% of the dark control values and over two times higher than in vehicle-treated light exposed rats (P < 0.001). Because acidification of zinc oxide with HCl would be expected to result in the formation of ZnCl2, we determined the protective efficacy of MgCl2 and CaCl2. At equal molar concentrations to that of ZnCl2 neither MgCl2 nor CaCl2 protected against light-induced retinal degeneration. The levels of rhodopsin and retinal DNA were essentially the same as found in vehicle injected animals exposed to intense visible light.
Figure 7.
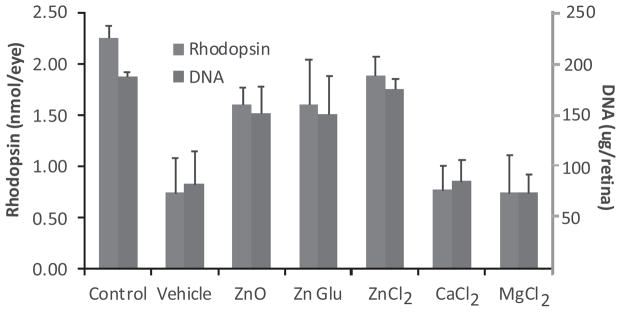
The effects of various zinc salts on visual cell recovery after light exposure. Dark reared rats were given zinc oxide at 5.2 mg kg−1, or an equivalent molar amount of zinc in the form of its gluconate (Glu) 29.1 mg kg−1, or chloride (Cl) salt, 8.7 mg kg−1. One hour later, the rats were exposed to intense visible light for 4 h and rhodopsin and retinal DNA measured 2 weeks later. All forms of zinc were significantly effective in preventing retinal light damage (P < 0.001). Equal molar amounts of the divalent cations MgCl2 (6.1 mg kg−1) and CaCl2 (7.1 mg kg−1) were ineffective in preventing the loss of retinal photoreceptors. Data represents the mean ± SD for n = 6 animals for each condition.
Western analysis of retinal protein expression
To assess the effects of light and zinc on retinal proteins, we treated dark reared rats with 4 h or 24 h of intense visible light and then extracted the proteins for western analysis 48 h after the start of light exposure. Figure 8 contains data showing that zinc treatment reduces oxidative stress in the retina. Whether light treatment was for 4 h or for 24 h, the level of carboxyethylpyrrole (CEP) adducted retinal protein was markedly reduced in extracts from rats previously treated with zinc. CEP reactivity has been found to be elevated in drusen from patients affected with AMD (8). In unexposed vehicle-treated rats CEP-protein molecular weights ranged from 25 to 64 kDa (lanes 1 and 2). More intense labeling was found in the vehicle-treated rat retinal extract after 4 h of light (lane 3), while zinc-treated rats exhibited a dramatic reduction in CEP reactivity (lane 4). Following 24 h of light, relative CEP staining in the vehicle-treated rat retinal extract was similar to that found in zinc-treated rats after 4 h of light (lane 5 vs 4). However, the staining of CEP proteins was greatly reduced in the zinc-treated retinal extract (lane 6 vs 5). To demonstrate that the reduced CEP staining was specific for zinc we treated other rats with MgCl2 or CaCl2 (lanes 7, 8). As shown, CEP staining was either the same, or somewhat greater, than found in vehicle-treated rats.
Figure 8.

Western analysis of oxidative protein markers and transduction proteins in retinas from light-exposed rats. Dark reared rats were given zinc oxide or vehicle and then exposed to intense light for 4 or 24 h. Retinas were excised 48 h after the onset of light and proteins extracted for gel electrophoresis and western analysis. Each lane contains 20 μg protein extracted from the pooled retinas of two rats. Abbreviations: CEP, carboxyethylpyrrole; HO-1, heme oxygenase-1; S-ag, S-antigen (arrestin); t-α, transducin alpha; GAPDH, glyceraldehyde- 3-phosphate dehydrogenase. Lanes 1 and 2, unexposed rat retinal extracts; lanes 3 and 5, vehicle-treated rat retinal extracts; lanes 4 and 6, zinc oxide treatment (5.2 mg kg−1); lanes 7 and 8, MgCl2 and CaCl2 at equal molar concentration to ZnCl2 (see Fig. 7).
Hemeoxygenase-1 (HO-1) is an inducible protein marker of oxidative stress (43), which is produced in the inner retinal layers and in Muller cells. HO-1 was absent in retinal extracts from rats treated with light for only 4 h, but staining was clearly present in the vehicle-treated retinal extract after 24 h of light (lane 5). HO-1 reactivity was significantly reduced by zinc, magnesium or calcium treatment (lanes 6–8) suggesting that retinal stress in the inner retinal layers is decreased by all three divalent cations. Staining of the retinal photoreceptor cell markers, S-antigen (S-ag; arrestin) and transducin (T-α) was reduced after 4 h of light (lanes 3, 4 vs lanes 1, 2). However, following 24 h of light the staining of retinal S-ag was absent and T-α staining was greatly reduced. This indicates that the light-induced loss of photoreceptor cells 1 day after 24 h of intense light treatment was greater than found in rats after only 4 h of light exposure.
Zinc changes the genetic signature of the rat retina
To examine the effects of zinc treatment on the genetic signature of the retina with and without a 4 h light treatment we screened three series of Ratref-12 expression array chips (four chips per series) with independent sets of c-RNA representing retinas from animals treated with: (1) 1 h of a zinc oxide treatment (ZN) followed by a 4 h light treatment (LT); (2) ZN, followed by a 4 h dark period (NOLT); (3) 1 h of vehicle treatment (VEH) followed by 4 h LT; and (4) VEH followed by a 4 h dark period (NOLT). A screen of the VEH NOLT gene profile with ZN NOLT gene profile revealed very little difference in gene expression profiles between the two NOLT treatments, with only 9 of the 22 523 mRNA transcripts being differentially expressed (Table 3, Column [A]). A differentially expressed gene is one which shows a significant difference in mRNA expression for one treatment condition as compared with another and is the index we used to define changes in the genetic signature of the retina.
Table 3.
Comparisons of gene screens consequential to nutritional and physical interventions.
| * | Entrez gene name | Gene symbol | Gene ID | (A) ZN NOLT X VEH NOLT: fold change | Adjusted P-value | (B) ZN LTX ZN NOLT: fold change | Adjusted P-value | (C) VEH LT X VEH NOLT: fold change | Adjusted P-value | (D) Effect |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Rhodopsin | Rho | 24717 | NA | P > 0.05 | NA | P > 0.05 | NA | P > 0.05 | No detectable effects |
| 1 | Guanine nucleotide binding protein (G protein), alpha inhibiting 1 (transducin, alpha subunit) | Gnatl | 363143 | NA | P > 0.05 | NA | P > 0.05 | NA | P > 0.05 | No detectable effects |
| 1 | Protein kinase C, gamma | Prkcg | 24681 | NA | P > 0.05 | NA | P > 0.05 | NA | P > 0.05 | No detectable effects |
| 1 | S-antigen; retina and pineal gland (arrestin) | Sag | 25539 | NA | P > 0.05 | 1.29 | 0.0051989 | NA | P > 0.05 | Zinc-modified light response |
| 1 | Phosducin | Pdc | 25343 | NA | P > 0.05 | NA | P > 0.05 | 0.71 | 0.0181533 | Zinc-repressed light response |
| 2 | Hexokinase 2 | Hk2 | 25059 | 1.26 | 0.0024618 | 0.54 | 7.02E-11 | 0.51 | 1.53E-11 | Light-modified zinc response |
| 2 | Spermine oxidase | Smox | 308652 | 1.26 | 0.0131823 | NA | P > 0.05 | NA | P > 0.05 | Light-repressed zinc response |
| 2 | Heme oxygenase (decycling) 1 | Hmoxl | 24451 | NA | P > 0.05 | NA | P > 0.05 | NA | P > 0.05 | No detectable effects |
| 2 | Superoxide dismutase 1, soluble | Sod1 | 24786 | NA | P > 0.05 | NA | P > 0.05 | NA | P > 0.05 | No detectable effects |
| 2 | NADPH oxidase 4 | Nox4 | 85431 | NA | P > 0.05 | NA | P > 0.05 | NA | P > 0.05 | No detectable effects |
| 3 | Metallothionein la | Mtla | 24567 | 2.66 | 0.0001353 | 1.85 | 0.0010279 | 2.5 | 1.60E-05 | Light and zinc response |
| 3 | Metal-regulatory transcription factor 1 | Mtfl | 362591 | NA | P > 0.05 | NA | P > 0.05 | NA | P > 0.05 | No detectable effects |
| 3 | CCHC-type zinc finger, nucleic acid binding protein | Cnbp | 64530 | NA | P > 0.05 | 1.15 | 0.0445115 | NA | P > 0.05 | Zinc-modified light response |
| 3 | Solute carrier family 30 (zinc transporter), member 1 | Slc30al | 58976 | NA | P > 0.05 | NA | P > 0.05 | 1.11 | 0.0472235 | Zinc-repressed light response |
| 3 | Transferrin | Tf | 24825 | NA | P > 0.05 | 0.5 | 0.015603 | 1.92 | 0.0273489 | Zinc-repressed light response |
| 4 | Lysophosphatidylcholine acyltransferase 1 | Lpcatl | 361467 | 0.78 | 0.0408628 | 1.79 | 2.64E-08 | 1.53 | 3.54E-06 | Light-modified zinc response |
| 4 | Acyl-CoA synthetase long-chain family member 3 | Acsl3 | 114024 | 1.4 | 0.0024618 | NA | P > 0.05 | NA | P > 0.05 | Light-repressed zinc response |
| 4 | Phospholipase C, delta 1 | Plcdl | 24655 | NA | P > 0.05 | 1.22 | 0.0225638 | NA | P > 0.05 | Zinc-modified light response |
| 5 | Bestrophin 2 | Best2 | 364973 | 1.64 | 0.0108329 | NA | P > 0.05 | NA | Light-repressed zinc response | |
| 6 | FK506 binding protein 5 | Fkbp5 | 361810 | 1.66 | 5.27E-05 | NA | P > 0.05 | NA | P > 0.05 | Light-repressed zinc response |
| 6 | Calcium/calmodulin-dependent protein kinase ID-like | RGD1560691 | 307124 | 1.44 | 0.0007777 | NA | P > 0.05 | NA | P > 0.05 | Light-repressed zinc response |
| 6 | Ubiquitin specific peptidase 2 | Usp2 | 115771 | 1.58 | 0.0108329 | NA | P > 0.05 | NA | P > 0.05 | Light-repressed zinc response |
| 6 | Caspase 7, apoptosis-related cysteine peptidase | Casp7 | 64026 | NA | P > 0.05 | 1.45 | 0.000114 | 1.4 | 0.0004618 | Light response |
| 6 | Caspase 3, apoptosis-related cysteine peptidase | Casp3 | 25402 | NA | P > 0.05 | NA | P > 0.05 | 1.19 | 0.0356835 | Zinc-repressed light response |
NA, not applicable; *functional annotation: (1) retinal cell biology, (2) stress response, (3) metal binding regulation, (4) fatty acid metabolism, (5) ion channel, (6) cell death; modifications:
 ; increase or
; increase or
 decrease in fold change.
decrease in fold change.
Differentially expressed gene markers from a VEH LT X VEH NOLT comparison and a ZN LT X ZN NOLT comparison identified 764 genes and 988 genes, respectively (Fig. 9). Three broad categories of differential genes were determined (Fig. 9A): (1) differential genes unique to the (VEH LT X VEH NOLT) screen; (2) differential genes that respond to light in both screens; and (3) differential genes unique to the (ZN LT X ZN NOLT) screen. A dissection of this collection of genes allowed the identification of 461 gene marker loci that defined light-induced changes (genes that show the same response to light treatment regardless of a ZN or VEH pretreatment) and 831 gene marker loci that defined zinc influenced changes (genes that show a different response to light treatment depending on whether a ZN or VEH pretreatment had been administered, Fig. 9B). Comparison of the three different screens is provided in Table 3 for a subset consisting of some of the major ocular related genes. Including all three screens in a single comparison allows differentiation in types of response, designated in Table 3, column D. The major categories consisted of light-modified zinc responses (Hk2 and Lpcat1), light-repressed zinc responses (Smox, Acsl3, Best2, Fkbp5, RGD1560691 and Usp2), zinc-modified light responses (Sag, Cnbp and Plcd1), zinc-repressed light response (Pdc, Slc30a1 and Casp3), and others for which the response was non-detectable (Rho, Gnat1, Prkcg, Hmox1, Sod1, Nox4 and Mtf1). Because so few differential gene changes are observed in the ZN NO LT X VEH NOLT screen most zinc-mediated changes detected in the comparison of the VEH LT X VEH NOLT screen with the ZN LT X ZN NOLT screen will represent zinc-modified light responses (e.g. Sag, Cnbp and Plcd1), zinc repressed light responses (Pdc, Slc30a1 and Casp3) or zinc reversed light responses (TF). In these cases, zinc treatment alone does not mediate a direct change in gene expression, but rather in the presence of a light stress either directly or indirectly mediates a response that results in specific changes in the expression of specific genes. In contrast, the expression profile of Mt1a defines a common response to either zinc or light, and the expression profile of Caspase 7 defines a light response.
Figure 9.

Light- and zinc-mediated effects on the genetic signature of the retina. Dark reared rats were given zinc oxide, at a dose of 5.2 mg kg−1 (ZN), or vehicle (VEH) at 8:00 A.M. and then kept in darkness for 5 h (NOLT), or exposed to light for 4 h (LT) starting at 9:00 A.M. Four treatment groups were generated for gene array analysis using Illumina Genome / Bead Studio software (ZN LT, ZN NOLT, VEH LT and VEH NOLT) and two differential comparisons of pooled data for each treatment group under consideration were made using limma (VEH LT X VEH NOLT and ZN LT X ZN NOLT). A threshold P-value of 0.05 or less (adjusted for multiple testing) was used to define a differentially expressed transcript relative to its control. (A) Venn diagram showing the distribution of differentially expressed genes with respect to VEH LT (relative to VEH NOLT) and ZN LT (relative to ZN NOLT) that are unique to each condition, and shared between the two comparisons. (B) Graph showing the distribution of the differentially expressed genes identified into light-mediated and zinc-mediated changes to the genetic signature of the retina. DN: differentially expressed genes showing a decrease in mRNA expression levels after LT (as compared with NOLT). UP: differentially expressed genes that show an increase in expression levels after LT (as compared with NOLT). X: genes that did not show a change in expression levels after LT (as compared with NOLT).
To identify the most prevalent biological effect of the zinc-mediated gene changes, we analyzed the list of 831 gene markers using Ingenuity systems pathway analysis (Fig. 10). Of the 831 gene markers, only 512 were annotated in the Ingenuity Knowledge Database in terms of abbreviated gene ontology. Seven biofunction categories each harbored more than 10% of the genes examined. The two largest biofunction categories were “Cell death” and “Cellular growth and proliferation.” Integral to cell death and growth / proliferation is the cell cycle, one of the other categories harboring greater than 10% of the gene list examined. These three categories are combined in the area designated as “Other cellular processes” in Fig. 10, in which there are now only five categories assigned. Note: genes can be in more than one category. Because most proteins / genes are multifunctional and because related biological processes are often intertwined we looked for the overlap between the genes in these three ontology groups (Fig. 11). In total, 232 of the 512 (45%) Zn responsive genes that are annotated in the Ingenuity database fall into a network of cell death, cellular growth and proliferation and cell cycle related genes.
Figure 10.

Most significant biological functions associated with zinc-mediated gene changes. Five hundred and twelve of the 831 gene marker loci defining zinc-mediated gene changes are annotated in the Ingenuity Knowledge Base. Functional analysis of these 512 zinc-mediated differential genes was performed using Ingenuity Pathways Analysis (Ingenuity® Systems, http://www.ingenuity.com). Right-tailed Fisher’s exact test was used to calculate a P-value determining the probability that each biological function assigned to the data set is due to chance alone, the lower the P-value the higher the probability that the gene list is associated with the biological function indicated.
Figure 11.
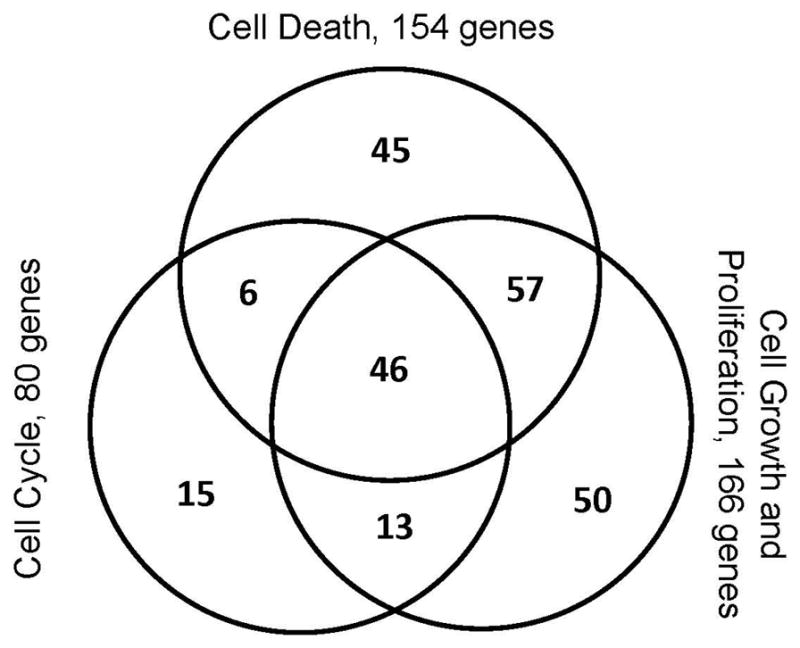
Venn diagram showing the overlap of cell death, cell cycle, cell growth and proliferation associated zinc-mediated differential genes. In total, 232 genes, representing 45% of the 512 genes considered, belong in this gene subset.
DISCUSSION
Animal models of light-induced retinal degeneration allow for testing drug treatments and dietary components in a preclinical setting under defined environmental conditions. In this regard, our data show that zinc reduces the extent of retinal photoreceptor damage caused by intense visible light. By either biochemical end point determinations or retinal histology, we found enhanced visual cell recovery in zinc-treated rats compared with vehicle-treated animals (Figs. 2 and 3). The efficacy of pretreatment was 60–70% at zinc oxide concentrations above 5 mg kg−1 and the protective effect lasted for at least 8 h of intense light exposure (Fig. 6). Zinc was particularly effective when given 1–4 h before light exposure, but was ineffective 6–24 h before and 1–4 h after the onset of light (Fig. 5). This window of protective efficacy also correlated with an increase in serum zinc levels and with its uptake by the RPE (Table 2). Zinc gluconate and its chloride salt were as effective as zinc oxide, indicating that zinc, and not the counter ion, was the effective agent. Two other divalent metal cations were ineffective in preventing retinal light damage (Fig. 7).
Gel electrophoresis of retinal DNA and western analysis of retinal proteins revealed that zinc effectively prevented DNA degradation and reduced oxidative stress. Zinc oxide treatment also altered the genetic signature of the rat retina (Figs. 9 and 10) but, unexpectedly, its concentration in the retina was unchanged by IP injection of a 5.2 mg kg−1 dose. The reasons for this finding are not yet known; both a systemic effect of zinc and an RPE mediated effect are possible. Incubation of RPE cells with zinc in vitro results in neuroprotection and reduced oxidative stress (27). A reduction in apoptotic photoreceptor cell death in vitro also has been demonstrated with exposure to zinc (44). In one of the few in vivo studies to date, Ugarte and Osborne (14) found a beneficial effect of acute zinc treatment on the ERG b-wave in a rabbit model of ischemia / reperfusion injury. Our findings support these earlier studies as both apoptotic retinal DNA ladders (Fig. 4) and oxidative CEP-protein adducts (Fig. 8) were greatly reduced by zinc treatment. Unknown at this time is whether a change in translocation of zinc between nucleoplasm and cytoplasm, or in the ratio of free vs bound forms of this critical metal can explain these findings. Additional work, perhaps using higher zinc concentrations, will be required to determine if retinal zinc levels can be increased by acute treatment, or long-term diet and whether its potential toxic effects (14,27) can be prevented, for example by coadministration of selective antioxidants.
Light and zinc change the genetic signature of the retina
Effects on the genetic signature of the retina will vary due to changes in the duration, intensity, wave-length of light, total photon energy, and likely the amount and time of zinc administered. A 4 h light exposure with respect to our gene profiling studies affords an opportunity to look at changes in gene expression relatively early in the light-induced retinal degeneration process. A (VEH LT X VEH NOLT) screen identified 764 gene markers that define the difference in the retinal genetic signature between a light exposed and unexposed state. A (ZN LT X ZN NOLT) screen identified 988 retinal expressed gene markers that differentiate zinc pretreatment with or without a subsequent light treatment (Fig. 9). A pairwise comparison of the differential gene markers in these two screens identified: (1) a subset of 303 gene markers that are involved in light-induced retinal degeneration, (2) a subset of 527 zinc inducible genes that appear to be essential for retinal response to or protection from light treatment, and (3) a gene, transferrin, that shows an interesting polar opposite response to light treatment in the two screens (Fig. 9). Ingenuity systems pathway analysis was used to analyze the list of zinc responsive genes to generate a cellular profile of how these genes might impact retinal biology (Fig. 10). In total, 45% of the zinc responsive genes that are also annotated in the Ingenuity Knowledge Database fall into a network of cell death, cellular growth and proliferation and cell cycle related genes (Figs. 10 and 11).
An alternative way to interrogate the data is by examining small clusters of genes known to be involved in a specific cellular function or trait. An analysis of the effects of zinc treatment on genes known to be involved in retinal cell biology (Rho, Gnat1, Sag, Pdc and Prkcg) indicates that zinc treatment alone is not likely to alter normal photoreceptor gene expression. In contrast, zinc treatment with light treatment results in specific changes in select genes of this pathway indicating a differential response to light exposure in the presence or absence of zinc. Moreover, a 4 h light exposure does not immediately result in a strong oxidative stress response. Although Hmox1, Sod1 and Nox4 mRNA levels (biomarkers for high level of oxidative stress) are not altered with a 4 h light exposure and / or zinc treatment, other markers indicative of a stress response (Hk2 and Smox) and metal ion regulation (Mt1a) are differentially affected by light and zinc treatment. In contrast, a differential change in gene expression is noted for Cnbp, Slc30a1 and TF (markers of metal ion regulation) in the presence of light with or without zinc treatment. Lastly, a 4 h light exposure does not result in the detection of an immediate DNA fragmentation pattern, suggesting that an apoptotic state has not yet been fully realized. Zinc treatment alone increased the relative mRNA levels of Fkbp5, RGD1560691 and Usp2 (three markers associated with an apoptotic molecular environment). Light exposure (with or without zinc treatment) resulted in an increase in Caspase 7 levels. Light exposure alone resulted in an increase in Caspase 3 levels, suggesting that the presence of zinc represses the normal light driven response in Caspase 3. The cell death microenvironment is clearly different in the absence or presence of zinc, although both are likely to be caspase-dependent.
The mRNA levels for nine genes (Hk2, Smox, Mt1a, Lpcat1, Acsl3, Best2, Fkbp5, RGD1560691 and Usp2) were found to be differential in a ZN NOLT X VEH NOLT screen (Table 3, column A). The zinc-mediated effects for eight of these genes were in turn repressed or reversed by subsequent light exposure. The nine ZN NOLT X VEH NOLT genes with differential expression profiles define proteins with a broad spectrum of cellular functions (cell stress response, metal ion regulation, fatty acid metabolism, ion channels and cell death). Pathway and network analysis of these genes resulted in the generation of a biological network that incorporates seven of them (Lpcat 1, Hk2, Acsl3, Fkbp5, Usp2, Smox and metallothionein 1A; data not shown). Genes in this network feed into hubs defined by Tnf, Pten, Il6, Tp53, Erbb2 and Ifng, genes central to apoptosis and / or cell growth and / or cell cycle processes. Moreover, zinc is known to interact with Tnf, Pten, Il6, Tp53 and metallothionein 1A. Although there are a few obvious functional interactions among these genes additional studies will be required to understand the extent of their interactions more clearly.
Potential mechanisms of action
Zinc is known to enhance catalysis for numerous metalloenzymes by directly binding within the active center (14). Likewise, by its site specific binding to DNA zinc can activate several hundred nuclear regulatory binding elements (23,45). This can lead to the induction of metallothionein synthesis, with an associated decrease in peroxide levels (24). Another potential mechanism for zinc’s antioxidant action is its ability to reduce superoxide formation by indirectly inhibiting NADPH oxidase (26). Superoxide production by NADPH oxidase results in the simultaneous extrusion of protons from subcellular organelles. Zinc inhibits their reuptake by altering voltage-gated membrane proton channels (25). As proton availability is linked to NADPH oxidase activity, a lack of protons would lead to enzyme inhibition and decreased superoxide production (26). Thus, unlike traditional antioxidants, which can interact directly with reactive oxygen, zinc’s antioxidative and neuroprotective effect may depend on an indirect mechanism. The ability of zinc to inhibit oxidation also appears to be concentration dependent as high-zinc levels have been reported to be toxic in retina (14) and RPE (27,28). In retinal homogenates Ugarte and Osborne (14) actually found an increase in the formation of reactive oxygen species upon incubation at high-zinc levels. The reasons for zinc’s toxicity are not currently known; however, its deleterious effects were found to be diminished by divalent metal ion chelators and antioxidants (14,27).
AREDS and implications for the use of zinc
It is now clear that zinc has multiple effects in cells and that it has the potential for synergistic effects beneficial for metabolism. It is also clear that high levels of cellular zinc can be toxic. For example, micromolar zinc enhances CFH-mediated inhibition of lysis in intact host cells, through the alternative complement pathway (29,30), whereas higher zinc levels are detrimental to RPE viability (27,28). Drusen in RPE from AMD patients may contain higher levels of oxidative lipid– protein adducts, including CEP, than from unaffected individuals (8,9) and macular drusen appear to contain an abnormally high level of protein-bound zinc (46). Although this suggests a possible role for zinc in the etiology of disease, its toxic effects can be over ridden by antioxidants (27) and by restoration of cellular NAD levels (47). CFH is also a major serum binding protein for MDA, a common lipid oxidation product (31), which would be expected to further reduce oxidative stress. Additional study will be required to determine whether zinc can enhance the ability of CFH to bind oxidative metabolites in vivo. In AREDS I, high levels of circulating antioxidants were found to correlate with a reduced risk of advanced AMD (10).
In addition, zinc and antioxidants, alone or in combination, were found to be beneficial (11). This confirms earlier work showing a beneficial effect of zinc supplementation, although the antioxidant index of patients was not determined (12). Fortunately, the AREDS formulation contains high levels of vitamins C and E, which may help to reduce any potential oxidative burden from accumulating zinc. In this study, we used green light to induce rhodopsin-mediated retinal degeneration (5) and to test for zinc’s protective effect. We also tested zinc oxide in rats exposed to broad spectrum white light, which mimics the wave lengths of light normally encountered in the environment, and more narrowly focused blue light. In each case, rhodopsin and retinal DNA recoveries were better in the zinc-treated rats (data not shown). Thus, whether white, blue or green light is used to induce retinal damage zinc exhibits a strong protective effect, although its mechanism of action remains to be determined. Similarly, how the uptake of zinc by the RPE affects retinal metabolism and or protection against light damage is an open question. We have made a start by determining genetic changes in the retinas of zinc-treated rats. Sorting through the multitude of possibilities and teasing out the actual mechanisms will require additional work and perhaps additional studies with experimental models of retinal degeneration.
Acknowledgments
This study was funded by support from Alcon Research Ltd. and the Ohio Lions Research Foundation (DTO), a NEI Core Grant for Vision Research (P30 EY 006360) and an unrestricted departmental grant from Research to Prevent Blindness (RPB) Department of Ophthalmology, Emory School of Medicine, Atlanta, GA, USA (PW). Special thanks to Dr. Larry Donoso and Dr. John Crabb for their generous gifts of anti-S-ag (arrestin) and anti- CEP antibodies.
Footnotes
This invited paper is part of the Symposium-in-Print “Retinal Photodamage.”
References
- 1.Shahinfar S, Edward DP, Tso MO. A pathological study of photoreceptor cell death in retinal photic injury. Curr Eye Res. 1991;10:47–59. doi: 10.3109/02713689109007610. [DOI] [PubMed] [Google Scholar]
- 2.Reme C, Weller M, Szczyensy P, Munz P, Hafezi K, Reinboth J-J, Clausen M. Light-induced apoptosis in the rat retina in vivo. In: Anderson RE, LaVail MM, Hollyfield JG, editors. Degenerative Diseases of the Retina. Plenum Press; New York: 1995. pp. 19–25. [Google Scholar]
- 3.Gordon WC, Casey DM, Lukiw WJ, Bazan NG. DNA damage and repair in light-induced photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2002;43:3511–3521. [PubMed] [Google Scholar]
- 4.Organisciak DT, Vaughan DK. Retinal light damage: mechanisms and protection. Prog Retin Eye Res. 2010;29:113–134. doi: 10.1016/j.preteyeres.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noell WK, V, Walker S, Kang BS, Berman S. Retinal damage by light in rats. Invest Ophthalmol. 1966;5:450–473. [PubMed] [Google Scholar]
- 6.Marc RE, Jones BW, Watt CB, Vazquez-Chona F, Vaughan DK, Organisciak DT. Extreme retinal remodeling triggered by light damage: implications for age related macular degeneration. Mol Vis. 2008;14:782–806. [PMC free article] [PubMed] [Google Scholar]
- 7.Rozanowska M, Rosanowski B, Boulton M. Light induced damage to the retina. Photobiology of the Retina. Smith KC, editor. Photobiol Sci. 2009 Available at: http://www.photobiology.info/
- 8.Crabb JW, Miyagi M, Gu X, Shadrach K, West K, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Solomon RG, Hollyfield JG. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakata K, Crabb JW, Hollyfield JG. Crystallin distribution in Bruch’s membrane-choroid complex from AMD and age-matched donor eyes. Exp Eye Res. 2005;80:821–826. doi: 10.1016/j.exer.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 10.Eye Disease Case-Control Study Group. Risk factors for neovascular age-related macular degeneration. Arch Ophthalmol. 1992;110:1701–1708. doi: 10.1001/archopht.1992.01080240041025. [DOI] [PubMed] [Google Scholar]
- 11.Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene and zinc for agerelated macular degeneration and vision loss. AREDS Report 8. Arch Ophthalmol. 2001;119:1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newsome DA, Swartz M, Leone NC, Elston RC, Miller E. Oral zinc in macular degeneration. Arch Ophthalmol. 1988;106:192–198. doi: 10.1001/archopht.1988.01060130202026. [DOI] [PubMed] [Google Scholar]
- 13.Grahm BH, Paterson PG, Gottschall-Pass KT, Zhang Z. Zinc and the eye. J Am Coll Nutr. 2001;20:106–118. doi: 10.1080/07315724.2001.10719022. [DOI] [PubMed] [Google Scholar]
- 14.Ugarte M, Osborne NN. Zinc in the retina. Prog Neurobiol. 2001;64:219–249. doi: 10.1016/s0301-0082(00)00057-5. [DOI] [PubMed] [Google Scholar]
- 15.Shuster TA, Nagy AK, Conly DC, Farber DB. Direct zinc binding to purified rhodopsin and disc membranes. Biochem J. 1992;282:123–128. doi: 10.1042/bj2820123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tam SW, Wilber KE, Wagner FW. Light sensitive zinc content of protein fractions from bovine rod outer segments. Biochem Biophys Res Commun. 1976;72:302–309. doi: 10.1016/0006-291x(76)90994-3. [DOI] [PubMed] [Google Scholar]
- 17.Ugarte M, Osborne NN. The localization of free zinc changes in rat photoreceptors during light and dark adaptation. Exp Eye Res. 1999;69:459–461. doi: 10.1006/exer.1999.0727. [DOI] [PubMed] [Google Scholar]
- 18.Wu SM, Qiao X, Noebels JL, Yang XL. Localization and modulatory actions of zinc in the vertebrate retina. Vision Res. 1993;33:2611–2616. doi: 10.1016/0042-6989(93)90219-m. [DOI] [PubMed] [Google Scholar]
- 19.Ou CZ, Ebadi M. Pineal and retinal protein kinase C isoenzymes: cooperative activation by calcium and zinc metallothionein. J Pineal Res. 1992;12:17–26. doi: 10.1111/j.1600-079x.1992.tb00021.x. [DOI] [PubMed] [Google Scholar]
- 20.Tate DJ, Miceli MV, Newsome DA, Alcock NW, Oliver PD. Influences of zinc on selected cellular functions of cultured human retinal pigment epithelium. Curr Eye Res. 1995;14:897–903. doi: 10.3109/02713689508995129. [DOI] [PubMed] [Google Scholar]
- 21.Springman E, Angleton E, Birkedal-Hansen H, Van Wart H. Multiple modes of action of latent human fibroblast collagenase: evidence for the roles of a Cys73 active-site zinc complex in latency and cysteine switch mechanism for activation. Proc Natl Acad Sci USA. 1990;87:364–368. doi: 10.1073/pnas.87.1.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oliver PD, Tate DJ, Newsome DA. Metallothionein in human retinal pigment epithelial cells; expression, induction and zinc uptake. Curr Eye Res. 1992;11:183–188. doi: 10.3109/02713689209000069. [DOI] [PubMed] [Google Scholar]
- 23.Ha KN, Chen Y, Cai J, Sternberg P. Increased glutathione synthesis through an ARE-Nrf2-dependent pathway by zinc in the RPE: implication for protection against oxidative stress. Invest Ophthalmol Vis Sci. 2006;47:2709–2715. doi: 10.1167/iovs.05-1322. [DOI] [PubMed] [Google Scholar]
- 24.Miceli MV, Tate DJ, Alcock NW, Newsome DA. Zinc deficiency and oxidative stress in the retina of pigmented rats. Invest Ophthalmol Vis Sci. 1999;40:1238–1244. [PubMed] [Google Scholar]
- 25.Cherny VV, DeCoursey TE. pH-dependent inhibition of voltage-gated H+ currents in rat alveolar epithelial cells by Zn2+ and other divalent cations. J Gen Physiol. 1999;114:819–838. doi: 10.1085/jgp.114.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henderson LM, Chappell JB, Jones OTC. Superoxide generation by the electrogenic NADPH oxidase of human neutrophils is limited by the movement of a compensating charge. Biochem J. 1988;255:285–290. [PMC free article] [PubMed] [Google Scholar]
- 27.Wood JPM, Osborne NN. Zinc and energy requirements in induction of oxidative stress to retinal pigmented epithelial cells. Neurochem Res. 2003;28:1525–1533. doi: 10.1023/a:1025622425501. [DOI] [PubMed] [Google Scholar]
- 28.Leung KW, Liu M, Xu X, Seiler MJ, Barnstable CJ, Tobran-Tink J. Expression of ZnT and ZIP zinc transporters in the human RPE and their regulation by neurotrophic factors. Invest Ophthalmol Vis Sci. 2008;49:1221–1231. doi: 10.1167/iovs.07-0781. [DOI] [PubMed] [Google Scholar]
- 29.Blom AM, Kask L, Ramesh B, Hillarp A. Effects of zinc on factor I cofactor activity of C4b-binding protein and factor H. Arch Biochem Biophys. 2003;418:108–118. doi: 10.1016/j.abb.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 30.Zarbin M, Sunness JS. Dry age-related macular degeneration and age-related degeneration pathogenesis. In: Levin LA, Albert DM, editors. Ocular Disease: Mechanisms and Management. Elsevier; London: 2010. pp. 527–535. [Google Scholar]
- 31.Weismann D, Hartvigen K, Lauer N, Bennet KL, Scholl HPN, Charbel Issa P, Cano M, Brandstatter H, Tsimikas S, Skerka C, Superti-Fuga G, Handa JT, Zipfel PF, Witztum JL, Binder CJ. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478:76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Organisciak DT, Barsalou LS, Darrow RM. Light-induced retinal degeneration. In: Levin LA, DiPolo A, editors. Ocular Neuroprotection. Marcel Dekker; New York: 2003. pp. 85–107. [Google Scholar]
- 33.Organisciak DT, Darrow RM, Barsalou L, Kutty RK, Wiggert B. Circadian-dependent retinal light damage in rats. Invest Ophthalmol Vis Sci. 2000;41:3694–3701. [PubMed] [Google Scholar]
- 34.Delmelle M, Noell WK, Organisciak DT. Hereditary retinal dystrophy in the rat: rhodopsin, retinol, vitamin A deficiency. Exp Eye Res. 1975;21:369–380. doi: 10.1016/0014-4835(75)90047-0. [DOI] [PubMed] [Google Scholar]
- 35.Organisciak DT, Darrow RM, Jiang YL, Blanks JC. Retinal light damage in rats with altered levels of rod outer segment docosahexaenoate. Invest Ophthalmol Vis Sci. 1996;37:2243– 2257. [PubMed] [Google Scholar]
- 36.Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 37.Organisciak DT, Darrow RA, Barsalou L, Darrow RM, Lininger LA. Light-induced damage to the retina: differential effects of dimethylthiourea on photoreceptor survival, apoptosis and DNA oxidation. Photochem Photobiol. 1999;70:261– 268. [PubMed] [Google Scholar]
- 38.Du P, Kibbe WA, Lin SM. Lumi: a pipeline for processing Illumina microarrays. Bioinformatics. 2008;24:1547– 1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 39.Lin SM, Du P, Huber W, Kibbe WA. Model-based variance-stabilizing transformation for Illumina microarray data. Nucleic Acids Res. 2008;36:e11. doi: 10.1093/nar/gkm1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and BioConductor. Springer; New York: 2005. pp. 379–420. [Google Scholar]
- 41.Sugawara T, Sieving PA, Bush RA. Quantitative relationship of the scotopic and photopic ERG to photoreceptor cell loss in light damaged rats. Exp Eye Res. 2000;70:693–705. doi: 10.1006/exer.2000.0842. [DOI] [PubMed] [Google Scholar]
- 42.Leure-duPree AE, McClain CJ. The effect of zinc deficiency on the morphology of the rat retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1982;23:425–434. [PubMed] [Google Scholar]
- 43.Kutty RK, Kutty G, Wiggert B, Chader GJ, Darrow RM, Organisciak DT. Induction of heme oxygenase-1 in the retina by intense light: suppression by the antioxidant dimethylthiourea. Proc Natl Acad Sci USA. 1995;92:1177–1181. doi: 10.1073/pnas.92.4.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carmody RJ, McGowan AJ, Cotter TG. Reactive oxygen species as mediators of photoreceptor apoptosis in vitro. Exp Cell Res. 1999;248:520–530. doi: 10.1006/excr.1998.4421. [DOI] [PubMed] [Google Scholar]
- 45.Beyersmann D, Haase H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals. 2001;14:331–341. doi: 10.1023/a:1012905406548. [DOI] [PubMed] [Google Scholar]
- 46.Lengyel I, Flinn JM, Peto T, Linkous DH, Cano K, Bird AC, Lanzirotti A, Frederickson CJ, van Kuijk FJGM. High concentration of zinc in sub-retinal pigment epithelial deposits. Exp Eye Res. 2007;84:772–780. doi: 10.1016/j.exer.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 47.Sheline CT, Zhou Y, Bai S. Light-induced photoreceptor and RPE degeneration involve zinc toxicity and are attenuated by pyruvate, nicotinamide, or cyclic light. Mol Vis. 2010;16:2639–2652. [PMC free article] [PubMed] [Google Scholar]