

Abstract
Accidental bony injuries are common in children. Children may also present with bony injuries following non-accidental injuries. Pathological fractures, though extremely rare, are an important entity and constitute fractures that occur in abnormal bones, usually after minor trauma. Pycnodysostosis is a rare skeletal dysplasia characterised by a clinical phenotype that includes short stature, skull deformities, osteosclerosis, acroosteolysis and bone fragility. Often the disease is diagnosed at an early age as a result of the investigation of short stature. However, the diagnosis is sometimes delayed and must be considered in any child with a history of recurrent or multiple bone fractures and dysmorphic features. The purpose of this report is to describe the clinical, radiological and genetic issues of a 9-year-old girl with a long history of multiple bone fractures. She had been subjected to safeguarding investigations previously and was identified to have dysmorphic features diagnosed as pycnodysostosis associated with craniosynostosis.
Background
Pycnodysostosis (PYCD, OMIM:265 800) is an autossomal recessive skeletal dysplasia, first described in 1962 by Maroteaux and Lamy (O'Neill M personal communication).1 It is caused by the absence of active cathepsin K (CTSK) due to a mutation in the CTSK gene, located on chromosome 1q21 (O'Neill M personal communication).2–8 CTSK, abundantly expressed in osteoclasts, is a lysossomal cysteine protease that plays a role in degrading the organic matrix of bones, acting in bone resorption and remodeling (O'Neill M personal communication).2–10 As a result there is a decrease of bone turnover and deterioration of bone structure, which lead to bone fragility and increased tendency for pathological fractures. Radiological examination shows osteosclerosis, delayed closure of cranial sutures with wormian bones, open fontanels, hypoplasia of facial bones and of paranasal sinuses. An obtuse mandible angle and dental irregularities are present on skull films. Acroosteolysis of the distal phalanges is usually present and with diffuse osteosclerosis flagging the disorder.1 11–15 17 The diagnosis is based on characteristic clinical and radiological features confirmed by genetic studies.2 3 5
Craniosynostosis is defined as premature fusion or growth arrest at one or more of the cranial sutures. Commonly, it occurs sporadically as an isolated defect. The presence of craniosynostosis in patients with PYCD is of uncertain clinical relevance and rarely described.18–20 For that reason, the authors report a case of a child with clinical, radiological and genetic features of PYCD diagnosed in the setting of multiple fractures and dysmorphic features in whom the presence of metopic and coronal craniosynostosis was also found.
Case presentation
The authors present the case of a 9- year-old Caucasian girl, the eldest of two siblings of non-consanguineous parents, with no family history of genetic, skeletal or metabolic disorder. The prenatal course was uneventful and she was born at a 39-week postconception age. The birth weight was 3010 g (25th percentile). The postnatal period was also uneventful, with normal developmental milestones.
She was investigated for unexplained history of failure to thrive since the first month of life. At 12 and 16 months of age she was admitted to hospital after a minor fall with head trauma. An uncomplicated linear skull fracture was diagnosed. At the age of 3 years she suffered a second degree facial burn with hot water. The father had a history of alcohol and substance abuse. Non-accidental injury was considered and the suspicion of neglected care was thoroughly investigated with the assistance of child protection services, without evidence neglect or non-accidental cause identified. At 9 years of age, after another minor fall, she was diagnosed to have sustained fracture of the right tibia, which was treated conservatively (figure 1).
Figure 1.
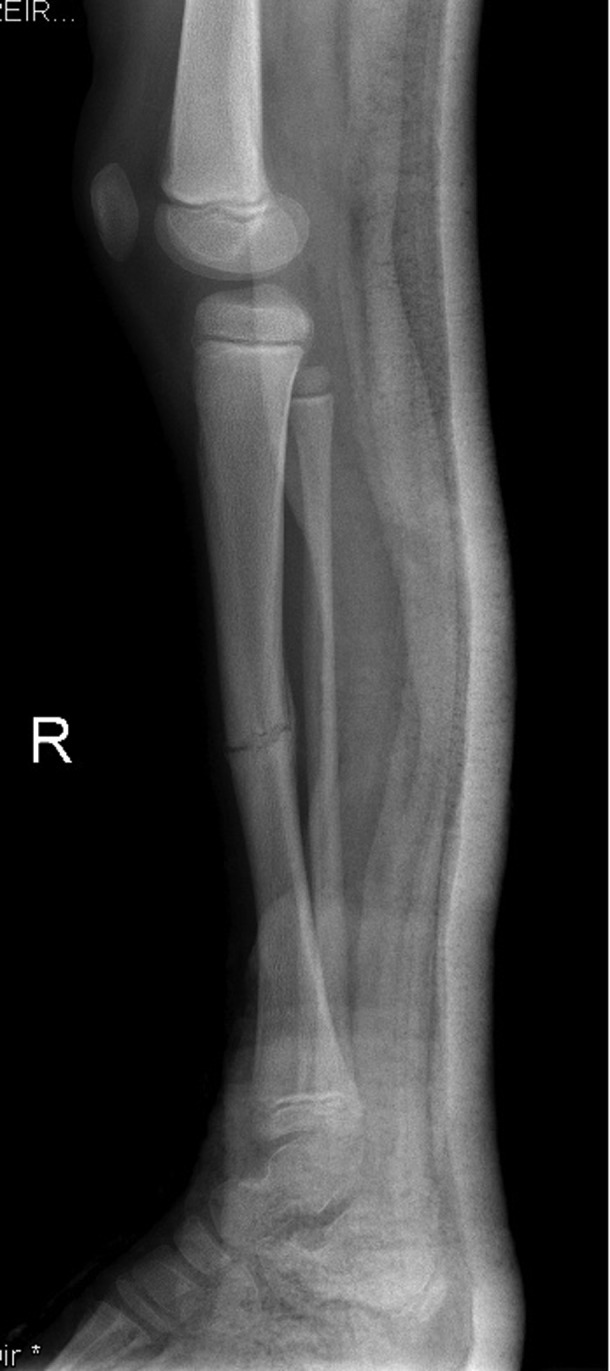
Right lower limb x-ray showing increased bone density of right tibia with transverse fracture though the middle part of the shaft, aged 8 years.
On admission, the patient showed the following growth parameters: weight of 17.4 kg (<3rd percentile), height of 114 cm (<3rd percentile). Several dysmorphic features were perceptible: facial dysmorphisms with bilateral frontal bossing, proptosis, mid-facial hypoplasia and micrognathia (figure 2). The examination of the mouth revealed a narrow arched groove palate, impacted and malposicioned teeth with persistence of decidual dentition (figure 3). Also evident was brachydactyly, large distal phalanges with flattened and watch-glass nails (figure 4). There was no pallor, hepatomegaly or lymphadenopathy. The physical examination was otherwise unremarkable. The clinical diagnosis of dysostosis was considered.
Figure 2.
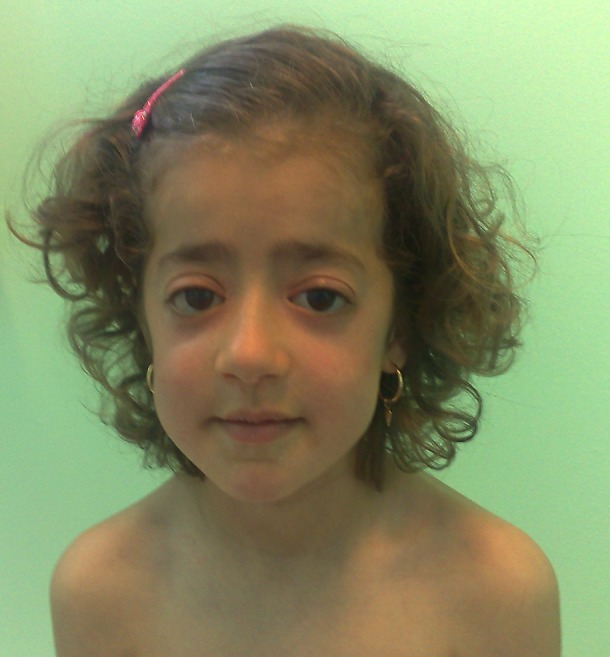
Frontal close-up photographic image of the child: frontal bossing, proptosis, mid-facial hypoplasia and micrognathia.
Figure 3.
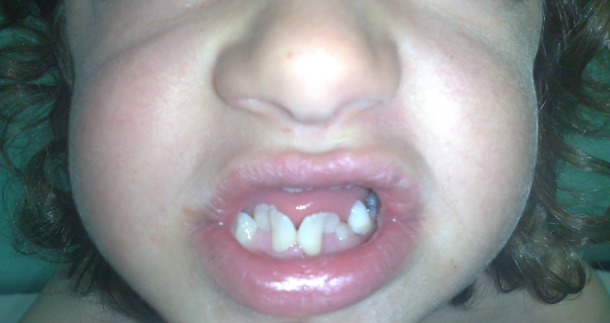
Frontal close-up photographic image of the mouth: dental crowding and over-retained deciduous teeth.
Figure 4.
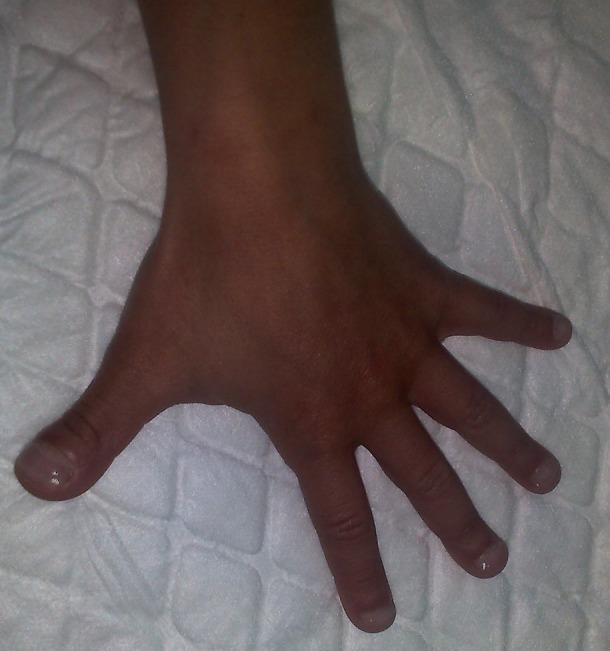
Detail of the left hand: brachydactyly, large distal phalanges and flattened, watch-glass nails.
Investigations
Blood assays, including complete blood count, electrolytes, renal and liver function tests, urinalysis, serum thyroid stimulating hormone, T4, growth hormone (GH), insulin-like growth factor 1 (IGF-1) and cortisol gave results within normal limits. Radiological investigation showed brachycephaly with basal sclerosis and separated cranial sutures (figure 5). The angle of mandible was obtuse and maxillary sinuses were non-pneumatised (figure 5). There was diffuse skeletal hyperostosis (figure 6 figure 7) sparing most of the medullary cavity and acroosteolysis (figure 6).
Figure 5.
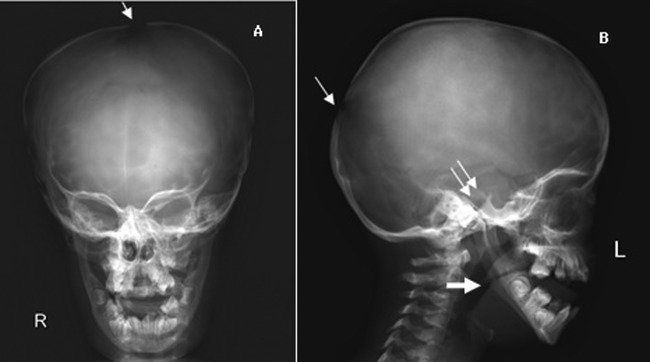
Skull x-ray showing a brachycephalic skull with basal sclerosis (double arrow), open sutures (narrow arrow), hypoplasy of maxilla, non-pneumatised sinuses and obtuse mandibular angle (large arrow).
Figure 6.
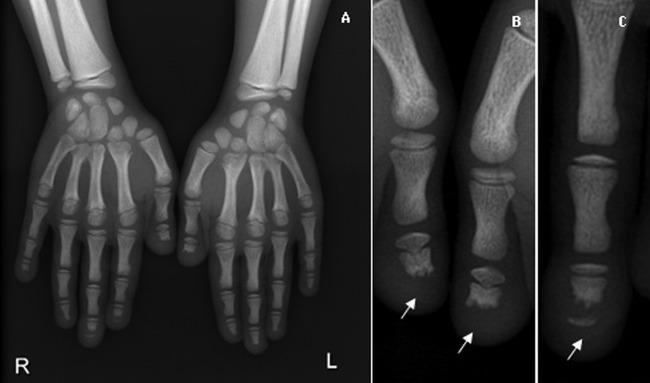
Hand x-ray showing dense aspect of metacarpals and distal portion of radius and ulna (A). Phalanges with partial bone resorption of distal phalanges (arrow)—acroosteolysis (B and C).
Figure 7.
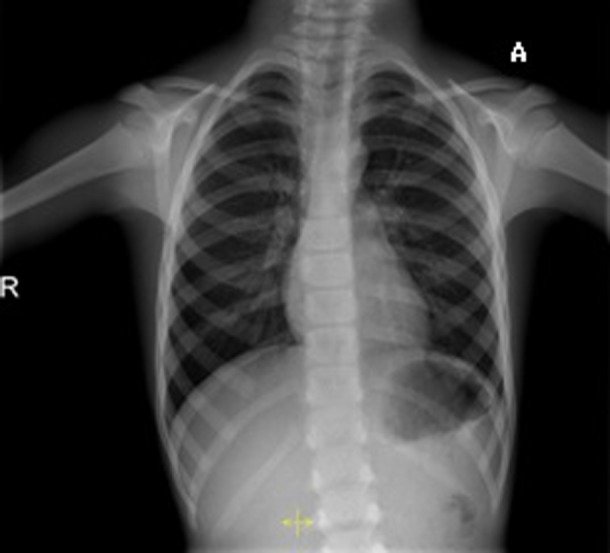
Chest x-ray showing dense aspect of the rib cage and spine.
Three-dimensional cranial CT scans confirmed the x-ray findings: brachycephaly, separated sagittal and lambdoid sutures with presence of wormian bones. An incidental finding was noted: the presence of bilateral coronal and metopic craniosynostosis, a rarely reported condition associated with PYCD (figure 8).
Figure 8.

Three-dimensional cranial CT scans: brachycephaly. Extinction of coronal and metopic sutures compatible with craniosynostosis (arrow). Wide lambdoidal with wormian bones and open sagittal sutures compatible with pycnodysostosis (double arrows).
Differential diagnosis
Non-accidental injuries in the paediatric population can be easily missed unless healthcare professionals are attuned to subtle clues in both the history and physical examination. Fractures are the second most common manifestation of child abuse after soft tissue injuries. In our case, the history of multiples fractures, soft tissue injury, social background of substance-abusing parent alerted to this issue.
Various bone diseases should also be considered, particularly cleidocranial dysostosis, idiopathic acroosteolysis, osteogenesis imperfecta and osteopetrosis. In cleidocranial dysostosis, the pattern of inheritance is autosomal dominance. Although open fontanels and cranial sutures are also observed the clavicle is severely involved, a bone less affected in PYD. Frequent fractures and bone fragility also may suggest the possibility of osteogenesis imperfecta. Nevertheless, fractures in this condition are much more severe than PYCD with other associated features. In idiopathic acro-osteolysis, patients usually present phalange acro-osteolysis and the absence of other cranial or skeletal dysplasia. In osteopetrosis, haematopoietic alterations as well signs of compression of cranial nerves, such as facial paralysis or deafness, are frequent.9 15
Outcome and follow-up
The final diagnosis contributed not only to providing specific preventive measures and genetic counselling but also to reinforce the adequate care provided by the mother who, in the meantime, had suffered long-term depression.
Discussion
PYCD is a rare genetic disease with less than 200 patients been described worldwide.7 19 The cardinal clinical features are increased bone density, osteosclerosis, bone fragility with tendency towards pathological fractures, short stature, cranial dysplasia, obtuse angle of mandible and total or partial dysplasia of terminal phalanges. The exfoliation of deciduous teeth is usually altered as well the eruption of permanent dentition.1 11–16 Short broad hands and feet with dystrophic nails and trunk deformities such as kyphosis, scoliosis, increased lumbar lordosis, recurrent chest infections, snoring and narrow chest had also been highlighted.15 Visual and auditory disorders have also been described.15 The diagnosis usually takes place at an early age, on investigation of short stature.12 14 21 The association of dysmorphic features and history of multiple unexplained bone fractures should flag the diagnosis, as in the presented case.
Laboratory investigations usually give results within normal limits as verified in the case presented. However, several previous reports addressed that patients with PYCD may present defective GH and low IGF-1 concentrations.12 21 In those cases, GH therapy increased IGF-1 secretion and improved linear growth. MRI studies of the brain including the hypothalamic-pituitary area have been recommended in these children because of the high incidence of pituitary hypoplasia.
The diagnosis of PYCD is primarily based on the clinical features and radiological studies. There is limited published material describing CT findings in patients with PYCD.22 Comparing CT with reconstructions made with radiographs provides greater anatomical detail and may identify incidental associations, as in the case presented.
The CTSK gene mutation analysis is the confirmatory test. Thirty-three different CTSK mutations have been found in 59 unrelated PYCD families.7 The candidate gene for PYCD was identified as coding for CTSK, in 1996.2 3 The genetic mutation identified in our case (p.Arg241x and p.Gly146Arg) has already been found in patients with PYCD of different ethnicity (Moroccan, Tunisian, Algerian, Spanish, Italian and Brazilian).3 5 18 19
Craniosynostosis has been reported in condensing osteopathies but the association with PYCD has only been reported in three previous reports.18–20 Craniosynostosis in patients with PYCD was not associated with a specific genetic alteration. As previously proposed,18–20 further studies are important to corroborate this specific cranial finding in order to evaluate whether any modification in the current follow-up of these patients will be necessary.
The treatment for this disorder is exclusively supportive.11 15 18 Since bone fractures are the main threat to these patients, it is crucial that all care must be taken to prevent or minimise the tendency for fractures to occur. Dental anomalies require regular dental check-ups. Fracture of the mandible and osteomyelitis of the jaw bones have been reported following dental extraction, thereby requiring close monitoring.11 Osteomyelitis of the maxilla can even extend to involve the base of the skull resulting in serious complications. Active eye and hearing examinations have also been proposed.15 Life expectancy is considered normal.15
Learning points.
Pycnodysostosis (PYCD) is a rare osteosclerotic skeletal disease.
The combination of dysmorphic features and history of bone fractures should provide an alert for this disorder.
The diagnosis is clinically and radiologically confirmed by genetic assessment of cathepsin K (CTSK) gene mutation.
Look also for craniosynostosis in patients with clinical features of pycnodysostosis.
Since there is no specific treatment available for PYCD to date, measures remain supportive.
Footnotes
Competing interests: None.
Patient consent: Obtained.
References
- 1.Maroteaux P, Lamy M. Pycnodysostosis. Presse Med 1962;25:999–1002. [PubMed] [Google Scholar]
- 2.Gelb BD, Edelson JG, Desnick RJ. Linkage of pycnodysostosis to chromosome 1q21 by homozygosity mapping. Nat Genet 1995;10:235–7. [DOI] [PubMed] [Google Scholar]
- 3.Gelb BD, Shi G-P, Chapman HA, et al. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 1996;273:1236–8. [DOI] [PubMed] [Google Scholar]
- 4.Ho N, Punturieri A, Wilkin D, et al. Mutations of CTSK result in pycnodysostosis via a reduction in cathepsin K protein. J Bone Miner Res 1999;14:1649–53. [DOI] [PubMed] [Google Scholar]
- 5.Donnarumma M, Regis S, Tappino B, et al. Molecular analysis and characterization of nine novel CTSK mutations in twelve patients affected by pycnodysostosis. Hum Mutat 2007;28:524. [DOI] [PubMed] [Google Scholar]
- 6.Naeem M, Sheikh S, Ahmad W. A mutation in CTSK gene in an autosomal recessive pycnodysostosis family of Pakistani origin. BMC Med Genet 2009;10:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xue Y, Cai T, Shi S, et al. Clinical and animal research findings in pycnodysostosis and gene mutations of cathepsin K from 1996 to 2011. Orphanet J Rare Dis 2011;6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toral–López J, Gonzalez–Huerta LM, Sosa B, et al. Familial pycnodysostosis: identification of a novel mutation in the CTSK gene (cathepsin K). J Investig Med 2011;59:277–80. [DOI] [PubMed] [Google Scholar]
- 9.Fratzl–Zelman N, Valenta A, Roschger P, et al. Decreased bone turnover and deterioration of bone structure in two cases of pycnodysostosis. J Clin Endocrinol Metab 2004;89:1538–47. [DOI] [PubMed] [Google Scholar]
- 10.Schilling AF, Mülhausen C, Lehmann W, et al. High bone mineral density in pycnodysostotic patients with a novel mutation in the propeptide of cathepsin K. Osteoporos Int 2007;18:659–69. [DOI] [PubMed] [Google Scholar]
- 11.Bathi RJ, Naik Masur V. Pyknodysostosis—a report of two cases with a brief review of the literature. Int J Oral Maxillofac Surg 2000;29:439–42. [PubMed] [Google Scholar]
- 12.Soliman AT, Ramadan MAF, Sherif A, et al. Pycnodysostosis: clinical, radiologic, and endocrine evaluation and linear growth after growth hormone therapy. Metabolism 2001;50:905–11. [DOI] [PubMed] [Google Scholar]
- 13.Quezado R, Montenegro RM, Jr, Araripe FFA, et al. Picnodisostose: relato de dois casos. Arq Bras Endocrinol Metabol 2003;47:95–101. [Google Scholar]
- 14.Alves Pereira D, Berini AL, Gay EC. Pycnodysostosis. A report of 3 clinical cases. Med Oral Patol Oral Cir Bucal 2008;13:633–5. [PubMed] [Google Scholar]
- 15.Mujawar Q, Naganoor R, Patil H, et al. Pycnodysostosis with unusual findings: a case report. Cases J 2009;2:6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fonteles CSR, Chaves CM, Da Silveira A, et al. Cephalometric characteristics and dentofacial abnormalities of pycnodysostosis: report of four cases from Brazil. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007;104:e83–90. [DOI] [PubMed] [Google Scholar]
- 17.Della Marca G, Scarano E, Leoni C, et al. Pycnodysostosis with extreme sleep apnea: a possible alternative to tracheotomy. Sleep Breath 2012;16:5–10. [DOI] [PubMed] [Google Scholar]
- 18.Bertola D, Amaral C, Kim C, et al. Craniosynostosis in pycnodysostosis: broadening the spectrum of the cranial flat bone abnormalities. Am J Med Genet A 2010;152A:2599–603. [DOI] [PubMed] [Google Scholar]
- 19.Osimani S, Husson I, Passemard S, et al. Craniosynostosis: a rare complication of pycnodysostosis. Eur J Med Genet 2010;53:89–92. [DOI] [PubMed] [Google Scholar]
- 20.Caracas HP, Figueiredo PS, Mestrinho HD, et al. Pycnodysostosis with craniosynostosis: case report of the craniofacial and oral features. Clin Dysmorphol 2012;21:19–21. [DOI] [PubMed] [Google Scholar]
- 21.Soliman AT, Rajab A, AlSalmi I, et al. Defective growth hormone secretion in children with pycnodysostosis and improved linear growth after growth hormone treatment. Arch Dis Child 1996;75:242–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fleming KW, Barest G, Sakai O. Dental and facial bone abnormalities in pyknodysostosis: CT findings. Am J Neuroradiol 2007;28:132–4. [PMC free article] [PubMed] [Google Scholar]