

Abstract
Somatic cells can be reprogrammed into induced pluripotent stem cells (iPSCs) by forcing the expression of four transcription factors (Oct-4, Sox-2, Klf-4, and c-Myc), typically expressed by human embryonic stem cells (hESCs). Due to their similarity with hESCs, iPSCs have become an important tool for potential patient-specific regenerative medicine, avoiding ethical issues associated with hESCs. In order to obtain cells suitable for clinical application, transgene-free iPSCs need to be generated to avoid transgene reactivation, altered gene expression and misguided differentiation. Moreover, a highly efficient and inexpensive reprogramming method is necessary to derive sufficient iPSCs for therapeutic purposes. Given this need, an efficient non-integrating episomal plasmid approach is the preferable choice for iPSC derivation. Currently the most common cell type used for reprogramming purposes are fibroblasts, the isolation of which requires tissue biopsy, an invasive surgical procedure for the patient. Therefore, human peripheral blood represents the most accessible and least invasive tissue for iPSC generation.
In this study, a cost-effective and viral-free protocol using non-integrating episomal plasmids is reported for the generation of iPSCs from human peripheral blood mononuclear cells (PBMNCs) obtained from frozen buffy coats after whole blood centrifugation and without density gradient separation.
Keywords: Developmental Biology, Issue 100, Stem cell biology, cellular biology, molecular biology, induced pluripotent stem cells, peripheral blood mononuclear cells, reprogramming, episomal plasmids.
Introduction
In 2006, the group of Shinya Yamanaka1 demonstrated for the first time that somatic cells from adult mice and humans can be converted into a pluripotent state by ectopic expression of four reprogramming factors (Oct-4, Sox-2, Klf-4, and c-Myc), generating the so-called Induced Pluripotent Stem Cells (iPSCs)2. Patient-specific iPSCs closely resemble human embryonic stem cells (hESCs) in terms of morphology, proliferation and ability to differentiate into the three-germ cell types (mesoderm, endoderm and ectoderm) while lacking the ethical concerns associated to the use of hESCs and bypassing possible immune rejection3. Thus, iPSCs appear as one of the most important sources of patient-specific cells for basic research, drug screening, disease modeling, evaluation of toxicity, and regenerative medicine purposes4.
Several approaches have been used for iPSC generation: viral integrating vectors (retrovirus5, lentivirus6), viral non-integrating vectors (adenovirus7), Sendai-virus8, BAC transposons9, episomal vectors10, proteins11 or RNA delivery12. Although the use of virus-mediated methods can lead to high efficiency reprogramming, viral vectors integrate into the genome of host cells and therefore potential random insertional mutagenesis, permanent alteration of gene expression, and reactivation of silenced transgenes during differentiation cannot be excluded13.
To make iPSCs safer for regenerative medicine, efforts have been made to derive iPSCs without the integration of exogenous DNA into cellular genomes. Although excisable viral vectors and transposons have been developed, it is still unclear whether short vector sequences, which inevitably remain in the transduced cells after excision, and transposase expression, could induce alteration in cellular function13. Despite its high reprogramming efficiency, Sendai virus represents an expensive approach and reach-through licensing concerns with the company that developed this system have the potential to limit its application in translational studies. Furthermore, the need for direct introduction of proteins and RNA requires multiple delivery of reprogramming molecules with the inherent technical limitations this introduces, and overall reprogramming efficiency is very low14. Of note, cost-effective viral-free and non-integrating methods based on the use of episomal plasmids have been successfully reported for the reprogramming of skin fibroblasts15. Specifically, in the present work we decided to use commercial available integration-free episomal plasmids, as previously reported10,15.
To date, skin fibroblasts represent the most popular donor cell type5. However, other cell sources have been successfully reprogrammed into iPSCs including keratinocytes16, bone marrow mesenchymal stem cells17, adipose stromal cells18, hair follicles19, and dental pulp cells20. The isolation of these cells requires surgical procedures, and several weeks are needed for in vitro cell expansion in order to establish a primary cell culture.
In this light, the selection of starting cell type is critical and it is equally important to be able to produce iPSCs from easily accessible and less invasive tissues such as blood. Both cord blood mononuclear cells (CBMNCs)21,22 and peripheral blood mononuclear cells (PBMNCs)14,22-24 represent suitable sources of cells for the derivation of iPSCs.
Although the efficiency of adult PBMNC reprogramming is 20–50 times lower than that of CBMNCs22, they remain the most convenient cell type for sampling purpose. In fact, PBMNC sampling has the advantage of being minimally invasive, and in addition, these cells do not require extensive expansion in vitro before reprogramming experiments. To date, different protocols have reported that PBMNCs after density gradient separation can be frozen and thawed days to several months after freezing and expanded for few days before reprogramming into iPSCs22,23. Nevertheless, as far as we are aware no reports have described reprogramming of PBMNCs from frozen buffy coats. Importantly, frozen buffy coats collected without density gradient separation represent the most common blood samples stored in large scale biobanks from population studies, thus representing an easily accessible pool of material for iPSC production that avoids further sample collection.
Herein we report for the first time the generation of viral-free iPSCs from human frozen buffy coats, based on a previously described protocol22. In addition, iPSCs were generated from frozen PBMNCs obtained after density gradient separation, as a control protocol for the non-density gradient purified PBMNC results.
Protocol
Peripheral blood mononuclear cells (PBMNCs) were isolated from human peripheral blood samples of healthy donors after signed informed consent and approval of the Ethical Committee of the Province of South Tyrol. Experiments were conducted in accordance with the principles expressed in the Declaration of Helsinki. All data were collected and analyzed anonymously.
1. Isolation of Peripheral Blood Mononuclear Cells (PBMNCs)
- PBMNCs obtained from buffy coats after whole blood centrifugation, without density gradient separation.
- Collect 8 mL of venous peripheral blood into a sodium citrate buffered plastic tube, and store at RT (25 °C). Process blood within 12 hr after collection.
- Centrifuge the tube at 2,000 x g for 15 min at 4 °C in a swinging bucket rotor, switching-off the centrifuge brakes.
- Collect the cloudy buffy coat (the layer between the upper plasma phase and the lower phase containing most of the erythrocytes), containing the enriched PBMNC fraction (500 µl) into a cryovial tube.
- Resuspend 500 µl of buffy coat with 500 µl of 2x freezing medium containing 90% FBS and 20% DMSO, in order to obtain a total volume of 1 ml with 10% DMSO concentration.
- Freeze the vial in a controlled-rate freezing container at -80 °C. Store cells in liquid nitrogen for longer periods.
- PBMNCs obtained after density gradient separation
- Collect 12 ml of venous peripheral blood into a sodium citrate buffered plastic tube, and store at RT. Process blood within 12 hr after collection.
- Dilute the blood with sterile PBS to a final volume of 35 ml.
- Carefully and slowly, layer 35 ml of diluted blood on 15 ml of polysucrose solution (used for density gradient separation) (p = 1.077) in a 50 ml conical tube (ratio 3:1).
- Centrifuge the tube at 800 x g for 30 min at RT in a swinging bucket rotor, switching-off the centrifuge brakes.
- Aspirate the upper, yellow phase containing plasma and discard it. Carefully, transfer the opaque white interphase layer containing PBMNCs to a new 50 ml conical tube.
- Bring total volume to 30 ml with sterile PBS and centrifuge at 300 x g for 10 min at RT.
- Discard the supernatant and resuspend the cells with 30 ml of PBS. Count the number of viable cells, based on trypan blue exclusion25.
- Centrifuge PBMNCs at 300 x g for 10 min at RT, aspirate the supernatant and discard it.
- Resuspend the cell pellet in an appropriate amount of freezing medium containing 90% FBS and 10% DMSO, in order to obtain a concentration of 5 x 106 cells/ml.
- Aliquot 1 ml per vial (about 5 x 106 cells/ml) and freeze the vial in a controlled-rate freezing container at – 80 °C. Store cells in liquid nitrogen for longer periods.
2. Thawing and Plating of PBMNCs (DAY 0)
- PBMNCs obtained from buffy coats after whole blood centrifugation, without density gradient separation.
- To start the protocol using PBMNCs from frozen buffy coats, thaw 1 vial of cells in a water bath at 37 °C and dilute the buffy coat with sterile PBS to a final volume of 2 ml.
- Transfer the diluted buffy coat into a 50 ml conical tube, add 10 ml of red cell lysis buffer and incubate for 10 min at RT.
- To prepare 500 ml of red cell lysis buffer, add 0.5 g of potassium bicarbonate, 4.145 g of ammonium chloride, 100 µl of 0.5 M EDTA solution to 500 ml of ultrapure water, pH 7.2-7.4.
- Bring total volume to 50 ml with sterile PBS and centrifuge at 300 x g for 10 min at RT.
- Discard the supernatant and wash the pellet with 50 ml of sterile PBS, centrifuge the buffy coat at 300 x g for 10 min at RT.
- Resuspend the cells in 1 ml of PBMNC medium, as previously described22, composed of: IMDM and Ham’s F-12 (ratio 1:1), 1% of Insulin-Transferrin-Selenium-Ethanolamine (ITS-X), 1% of Chemically Defined Lipid Concentrate (CDL), 1% Penicillin/Streptomycin, 0.005% of L-ascorbic acid, 0.5% of Bovine Serum Albumin (BSA), 1-Thioglycerol (final concentration 200 µM), Stem Cell Factor SCF (100 ng/ml), Interleukin-3 IL-3 (10 ng/ml), Erythropoietin EPO (2 U/ml), Insulin-like Growth Factor IGF-1 (40 ng/ml), Dexamethasone (1 µM) and holo-transferrin (100 µg/ml).
- Transfer them into one well of a 12-well plate with standard tissue culture treated surface, without coating, at a density of 2 x 106 cells/ml. Incubate the cells at 37 °C, 5% CO2 in a humidified incubator for 48 hr, until the changing medium step (see Section 3).
- PBMNCs obtained after density gradient separation
- To start the protocol using frozen PBMNCs after density gradient separation, thaw 1 vial of cells in a water bath at 37 °C, transfer the cells into 5 ml of PBMNC medium. Centrifuge at 300 x g for 10 min at RT.
- Resuspend the cells in 1 ml of PBMNC medium and transfer them into one well of a 12-well plate, at a density of 2 x 106 cells/ml. Incubate the cells at 37 °C, 5% CO2 in a humidified incubator for 48 hr, until the changing medium step (see Section 3).
3. Culture and Expansion of PBMNCs (DAY 2-13)
Collect PBMNCs in suspension culture, using a pipette with a 1 ml tip, in a 15 ml conical tube and centrifuge at 300 x g for 10 min at RT.
Discard the supernatant and resuspend the cells in 1 ml of fresh PBMNC medium.
Transfer the cells into 1 well of 12-well plate with standard tissue culture treated surface, without coating, and incubate them at 37 °C, 5% CO2 in a humidified incubator.
Repeat these steps every two days, and split the cells at a ratio 1:2 when they reach the appropriate confluence (around 80%).
4. Transfection of PBMNCs with Episomal Plasmids (DAY 14)
Note: Perform the isolation of the four commercial intergation-free episomal plasmids, carrying the pluripotency genes using a commercial kit for plasmid purification and assess the quality of the purified plasmids using an agarose gel analysis, according to the manufacturer’s instructions.
Collect PBMNCs in 15 ml conical tube and count the number of viable cells (on the basis of trypan blue exclusion)25.
Centrifuge 2 x 106 cells at 300 x g for 10 min at RT.
Resuspend 2 x 106 cells in transfection mix containing 100 µl of Resuspension Buffer T and 1 µg of each plasmid DNA (one plasmid carrying OCT3/4 and shRNA against p53, one plasmid carrying SOX2 and KLF4, one plasmid carrying L-MYC and LIN28, and one plasmid carrying EGFP, to check the transfection efficiency).
Aspirate the transfection mix containing the cells into a 100 µl tip and insert the pipette with the sample vertically into the tube, filled with 3 ml of Electrolytic Buffer E2, placed in the pipette station of electroporation machine, according to manufacturer’s instructions.
Efficiently electroporate cells using the following program: 1,650 V, 10 msec, 3 pulses.
Resuspend the electroporated cells (2 x 106 cells) into 2 ml of pre-warmed PBMNC medium, adding 0.25 mM Sodium Butyrate (NaB) and count the number of viable cells after electroporation protocol (on the basis of trypan blue exclusion)25.
Transfer the cells into one well of a 6-well plate without coating, gently rock the plate and incubate the cells at 37 °C, 5% CO2 in a humidified incubator.
The day after electroporation, count the number of GFP-positive cells under fluorescence microscopy from 8-10 random fields, in order to assess the transfection efficiency.
Maintain the transfected cells without splitting for 3 days, until plating them on MEFs (see Section 6).
Replace 2 ml of fresh PBMNCs medium, plus 0.25 mM NaB every two days.
5. Prepare Dishes with Feeder-cells for the Co-culture (DAY 16)
Coat the wells of a 6-well plate with 1 ml of basement membrane matrix for 15 min at 37 °C and plate Mouse Embryonic Fibroblasts (MEFs) at a density of 2 x 105 cells/well with 2 ml of 10% FBS contained DMEM (MEF medium) one day before passaging the electroporated PBMNCs onto MEF feeder-cells.
6. Plating Transfected PBMNCs onto MEFs (DAY 17-19)
Collect PBMNCs in suspension culture, using a pipette with 1 ml tip, in 15 ml conical tube and centrifuge transfected PBMNCs at 300 x g for 10 min at RT.
Discard the supernatant and resuspend the pellet in 2 ml of fresh PBMNC medium, plus 0.25 mM NaB.
Plate the cells onto MEF-coated 6 well-plate and incubate the cells at 37 °C, 5% CO2 in a humidified incubator.
After two days, aspirate the medium and replace it with 2 ml of iPSC medium composed of knockout DMEM (KO-DMEM), 20% KO-Serum Replacement (KOSR), 1 mM NEAAs, 1% Penicillin/Streptomycin, 20 mM L-Glutamine, 0.1 mM β-mercaptoethanol, 10 ng/ml FGF (basic bFGF) and 0.25 mM NaB.
Replace 2 ml of fresh iPSC medium every day and check the cells daily. Note: At about day 22-25, the first colonies of iPSCs should be visible.
7. Picking and Expansion of Induced Pluripotent Stem Cells (iPSCs) Clones (DAY 30-35)
Note: At about 30-35 days after transfection, iPSC colonies should be ready for the picking and replating. The reprogramming efficiency should be estimated as the percentage of number of iPSC colonies/total number of electroporated cells, as previously reported26.
The day before passaging iPSC colonies, prepare the MEF feeder in a 12-well plate (1.25 x 105 cells/well).
Aspirate the MEF medium and change with 1 mL of iPSC medium with 10 ng/ml bFGF and 10 µM Y-27632. Manually dissect with a needle one colony at each time under the dissecting microscope in the picking-hood in order to obtain a grid, collect smaller clumps by pipetting and transfer them individually each into one well of 12-well plate coated with MEFs.
Passage and expand each 12-well plate to a 6-well plate in a ratio 1:2, then each 6-well plate in a ratio 1:4 for further propagation. Dissect and expand iPSCs manually for the first 2-3 passages (as thoroughly described in step 7.2), then use an enzyme dissociation procedure (start from step 7.4).
For an enzyme dissociation protocol, clean iPSC colonies by aspirating differentiated portions, under the dissecting microscope in the picking-hood.
Incubate iPSCs with 1 ml of collagenase IV (1 mg/ml) for 10 min at 37 °C.
Aspirate the enzyme solution, rinse the cells with 1 ml of KO-DMEM and collect the colonies in a 15 ml conical tube. Centrifuge at 100 x g for 2 min at RT.
Aspirate the supernatant, resuspend the colonies in 2 ml of iPSC medium with 10 ng/ml bFGF and 10 µM Y-27632 and plate them on MEF-coated 6-well plate (ratio 1:4).
8. Characterization of iPSCs (Between 5-15 Passages)
- Alkaline Phosphatase Staining
- Culture the iPSC colonies for 3-5 days in iPSC medium with 10 ng/ml bFGF.
- For starting an alkaline phosphatase staining protocol, rinse iPSCs once with PBS and then fix them with 1 ml of 4% PFA for 20 min at RT.
- Wash the cells three times with PBS, to remove residual PFA.
- Stain fixed cells using a commercial alkaline phosphatase staining kit according to the manufacturer’s instructions.
- Immunofluorescence
- Culture the iPSC colonies for 3-5 days in iPSC medium with 10 ng/ml bFGF.
- To start an immunofluorescence assay, wash iPSCs once with PBS and then fix them with 1 ml of 4% PFA for 20 min at RT.
- Wash the cells three times with PBS, to remove residual PFA.
- Treat fixed iPSCs with 1 ml of permeabilization/blocking solution containing 4% goat serum, 0.1% BSA, 0.1% Triton X-100 and 0.05% sodium azide (NaN3) for 1 hr at RT.
- Aspirate the blocking solution and incubate the fixed cells with primary antibodies in 3% BSA and 0.05% NaN3 solution O/N at 4 °C (check the material table for primary antibody list and suggested antibody dilution factor).
- The next day, wash iPSCs three times with PBS, then incubate the cells with Alexa Fluor 488 goat anti-mouse and Alexa Fluor 555 goat anti-rabbit antibodies, diluted 1:1,000 in 3% BSA and 0.05% NaN3 solution, for 1 hr at 37 °C.
- Stain the nuclei with DAPI (1µg/ml) for 10 min at RT in dark, followed by three time washing with PBS.
- Differentiation of iPSCs in Embryoid Bodies (EBs)
- Culture the iPSC colonies for 5-6 days in iPSC medium with 10 ng/ml bFGF.
- Remove differentiated portions from iPSC colonies by aspirating, under the dissecting microscope in the picking-hood.
- Incubate iPSCs with 1 ml of collagenase IV (1 mg/ml) for 10 min at 37 °C.
- Aspirate the enzyme solution, rinse the cells with 1 ml of KO-DMEM and collect the colonies in a 15 ml conical tube. Centrifuge at 100 x g for 2 min at RT.
- Aspirate the supernatant and resuspend the colonies in 2 ml of EB medium composed of KO-DMEM, 20% defined fetal bovine serum (FBS), 1 mM NEAAs, 1% Penicillin/Streptomycin, 20 mM L-Glutamine, 0.1 mM β-mercaptoethanol.
- Plate iPSC colonies in suspension for 6 days onto ultra-low attachment 6-well plates, in order to allow the formation of spheroidal three-dimensional embryoid bodies (EBs). Change 2 ml of fresh EB medium every two days.
- On day 7, collect EBs and plate them onto 0.1% gelatin coated 6-well plates in 2 ml of EB medium and maintain EBs in differentiation for 20 days.
- Change 2 ml of fresh EB medium every two days.
- qRT-PCR for Gene Expression Analysis
- Culture the iPSC colonies for 5-6 days in iPSC medium with 10 ng/ml bFGF.
- Extract total RNA from PBMNCs, iPSCs or EBs using 1 ml of commercial reagent in accordance with manufacturer’s instructions.
- Evaluate RNA concentration and purity by suitable methods such as use of a spectrophotometer (high quality RNA with A260/A280 and A260/A230 ratios > 1.8)27.
- Check RNA integrity using a commercial kit according to manufacturer’s protocol (high quality RNA RQI >9.5)28.
- Perform a reverse transcription reaction on 1 µg of total RNA using a commercial reverse transcriptase kit according to manufacturer’s instructions.
- Prepare the qPCR mixtures containing 5 ng of cDNA template, 7.5 µl SYBR GREEN Supermix, 2 µl of 5 µM primers (forward: 1 µl and reverse: 1 µl) and 6 µl of RNase/DNase-free water for each reaction29.
- Perform the qRT-PCR using the following program: an initial denaturation step at 95 °C for 10 min, followed by 40 cycles divided into a denaturation step at 95 °C for 15 sec and primer annealing and extension step at 60°C for 45 sec29.
- Normalize the expression of other genes using GAPDH as housekeeping gene28 (see the primers listed in Table 1).
- qPCR for Transgene Exclusion
- Extract DNA from PBMNCs or iPSCs using 1 ml of lysis buffer containing 50 mM Tris, 100 mM EDTA, 100 mM NaCl, 1% Sodium Dodecyl Sulphate (SDS) and 20 µg/ml Proteinase K.
- Add an equal volume (1 ml) of 100% isopropanol, mix by inversion three times in order to allow DNA precipitation and centrifuge at 10,000 x g for 5 min at RT.
- Discard the supernatant, wash the DNA pellet with 1 ml of 70% ethanol and centrifuge at 10,000 x g for 5 min at RT. Aspirate and discard the supernatant and resuspend it in RNase/DNase-free water.
- Evaluate DNA concentration and purity by suitable methods such as use of a spectrophotometer (high quality RNA with A260/A280 and A260/A230 ratios >1.8)27.
- Prepare the qPCR mixtures containing 5 ng of DNA template, 7.5 µl SYBR GREEN Supermix, 2 µl of 5 µM primers (forward: 1 µl and reverse: 1 µl) and 6 µl of RNase/DNase-free water for each reaction29.
- Normalize the expression of specific episomal plasmid EBNA-1 gene using FBX-15 as housekeeping gene29(see the primers listed in Table 1).
- Karyotype Analysis
- Culture the iPSC colonies for 5-6 days onto feeder-free basement membrane matrix coated plate with a commercial defined, feeder-free maintenance medium for iPSCs, following manufacturer’s instructions.
- To start the karyotype analysis protocol, detach iPSC colonies with 1 ml of cell detachment solution for 5 min at 37 °C, until obtaining single-cell dissociation.
- Collect cells and centrifuge at 400 x g for 5 min at RT.
- Resuspend iPSCs in 2 ml of a medium specifically developed for culturing cells for prenatal diagnostic techniques. Plate single-cell iPSCs onto basement membrane matrix coated glass dishes and incubate them in a 37 °C, 5% CO2 incubator.
- After 2-4 days, add 10 µl/ml colchicine directly to the cultures and incubate for 3 hr at 37 °C, 5% CO2 in a humidified incubator.
- Aspirate the medium and incubate iPSCs in 1 ml of a hypotonic solution (0.6% sodium citrate and 0.13% potassium chloride) for 10 min at RT.
- Rinse cells with 1 ml of 5% acetic acid solution and fix iPSCs with methanol/acetic acid solution (ratio 3:1) in a machine with controlled temperature and humidity (28 °C, 42% rH).
- Immerse and stain fixed cells with Quinacrine solution for 15-20 min at RT in dark (to obtain Q-banding)30.
- Acquire cell metaphases under a fluorescence microscope at 100X magnification29.
Representative Results
In this study a simple and effective protocol for the generation of viral-free iPSCs by reprogramming of PBMNCs isolated from frozen buffy coats after whole blood centrifugation and comparison with reprogramming of PBMNCs obtained after density gradient separation is reported. Figure 1A shows a schematic representation of the detailed protocol. After thawing, the isolated PBMNCs, showing a typical rounded shape, are expanded in specific blood culture medium for 14 days (Figure 1B) and then transfected with episomal plasmids. After 10-15 days post-transfection, small rounded bright colonies with defined margins and embryonic stem cell-like morphology start to appear (Figure 1C). Approximately 20-30 days after transfection, iPSC colonies become highly compact, with sharp edges, increase their size and are ready for picking and expansion (Figure 1D). After the first passaging steps (2-3 passages), iPSC colonies maintain their undifferentiated state and show a typical human embryonic-stem cell morphology. During the first passages, manual picking is a more efficient expansion way compared to enzyme dissociation in order to select fully undifferentiated, compact and tightly packed colonies (Figures 2A). Larger magnification of iPSC colonies shows individual cells within the colonies and the high nucleus to cytoplasm ratio is more easily observed (Figure 2B). After initial mechanical picking, selected iPSC clones are expanded with enzyme dissociation using Collagenase IV. After 10-15 passages of in vitro expansion, cytogenetic analysis is conducted on iPSC colonies. About 20 metaphases from at least two independent cultures, at approximately 300–400 band level, are analyzed and all samples show a normal karyogram (Figure 2C). This observation suggests that iPSCs are genetically stable.
After in vitro expansion (5-10 passages), iPSCs are characterized for the expression of stemness markers and their pluripotency potential. Figures 3A, B show that iPSCs are positive for alkaline phosphatase staining. Using qRT-PCR analysis, we have verified that iPSCs express endogenous pluripotent genes (SOX-2, OCT3/4, NANOG) at significantly higher levels than starting PBMNCs, while expression levels of endogenous C-MYC and KLF-4 are similar before and after iPSC reprogramming (Figure 3C). After only 5 passages in culture, expression of exogenous episomal transgenes cannot be detected in iPSCs, thus indicating the successful activation of endogenous pluripotent genes. PBMNCs 5 days after transfection, with strong transgene expression level, evaluated by episomal specific primers for EBNA-1, are used as positive control. PBMNCs that are never exposed to plasmids carrying the 4 exogenous pluripotency genes, are used as negative control(Figure 3D). Finally, immunofluorescence analysis reveals that iPSCs express pluripotency markers, such as OCT3/4, SOX-2, SSEA-4, TRA-1-60, and TRA-1-81 (Figure 4).
iPSCs are differentiated into embryoid bodies (EBs) using a spontaneous differentiation protocol, as shown in Figure 5A, in order to evaluate their in vitro pluripotency potential. qRT-PCR experiments reveal that iPSCs are able to differentiate into cells of the three germ layers; indeed, EBs obtained after 20 days of differentiation display significant up-regulation of lineage markers representative of the endoderm (SOX-7, AFP), mesoderm (CD31, ACTA-2, CDH-5, RUNX-1) and ectoderm (KTR-14, TH, GABRR-2) (Figure 5B). Confocal microscopy analysis shows that single-cell dissociated beating clusters express typical cardiomyocyte markers such as Troponin I (Tn-I) and α-Actinin, organized in a clear sarcomeric pattern (Figure 6A). In addition, immunofluorescence analysis of EBs shows a positive staining for the neuron-specific marker class III β-Tubulin (TUJ1) as well for the dopaminergic neuronal marker tyrosine hydroxylase (TH) (Figure 6B).
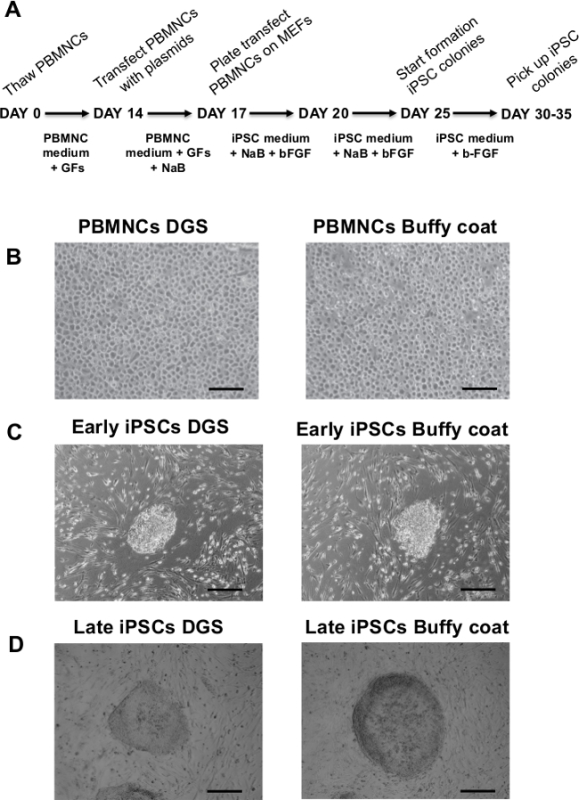 Figure 1. Protocol for the generation of iPSCs from human peripheral blood and cell morphological changes during the protocol. (A) Diagram of the protocol used to generate iPSCs obtained from frozen PBMNCs after DGS and PBMNCs isolated from frozen buffy coats, using a single transfection of non-integrating episomal plasmids. (B) Representative images of proliferating PBMNCs obtained from DGS and buffy coats after thawing and expansion in culture for 14 days, before being transfected. Scale bar 100 µm. (C) Examples of iPSC colonies one week after the cells are plated on MEFs. Scale bar 500 µm. (D) Representative images of iPSC colonies at 30-35 days after re-plating on a MEF feeder-layer. Scale bar 500 µm. iPSCs = induced pluripotent stem cells; DGS = density gradient separation; PBMNCs = peripheral blood mononuclear cells; MEFs = mouse embryonic fibroblasts; GFs = growth factors; bFGF = basic fibroblast growth factor; NaB = sodium butyrate. Please click here to view a larger version of this figure.
Figure 1. Protocol for the generation of iPSCs from human peripheral blood and cell morphological changes during the protocol. (A) Diagram of the protocol used to generate iPSCs obtained from frozen PBMNCs after DGS and PBMNCs isolated from frozen buffy coats, using a single transfection of non-integrating episomal plasmids. (B) Representative images of proliferating PBMNCs obtained from DGS and buffy coats after thawing and expansion in culture for 14 days, before being transfected. Scale bar 100 µm. (C) Examples of iPSC colonies one week after the cells are plated on MEFs. Scale bar 500 µm. (D) Representative images of iPSC colonies at 30-35 days after re-plating on a MEF feeder-layer. Scale bar 500 µm. iPSCs = induced pluripotent stem cells; DGS = density gradient separation; PBMNCs = peripheral blood mononuclear cells; MEFs = mouse embryonic fibroblasts; GFs = growth factors; bFGF = basic fibroblast growth factor; NaB = sodium butyrate. Please click here to view a larger version of this figure.
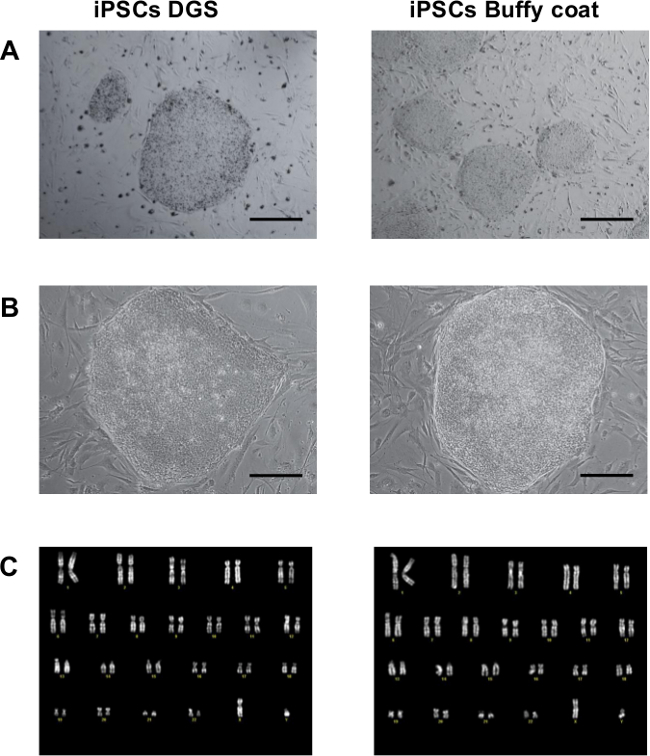 Figure 2. Morphology and karyotype analysis. (A) Representative images of iPSC colonies after 2-3 manual passaging steps. Scale bar 500 µm. (B) Larger magnification of iPSC colonies. Scale bar 200 µm. (C) Representative normal karyograms from iPSCs after 10-15 passages of in vitro expansion. Please click here to view a larger version of this figure.
Figure 2. Morphology and karyotype analysis. (A) Representative images of iPSC colonies after 2-3 manual passaging steps. Scale bar 500 µm. (B) Larger magnification of iPSC colonies. Scale bar 200 µm. (C) Representative normal karyograms from iPSCs after 10-15 passages of in vitro expansion. Please click here to view a larger version of this figure.
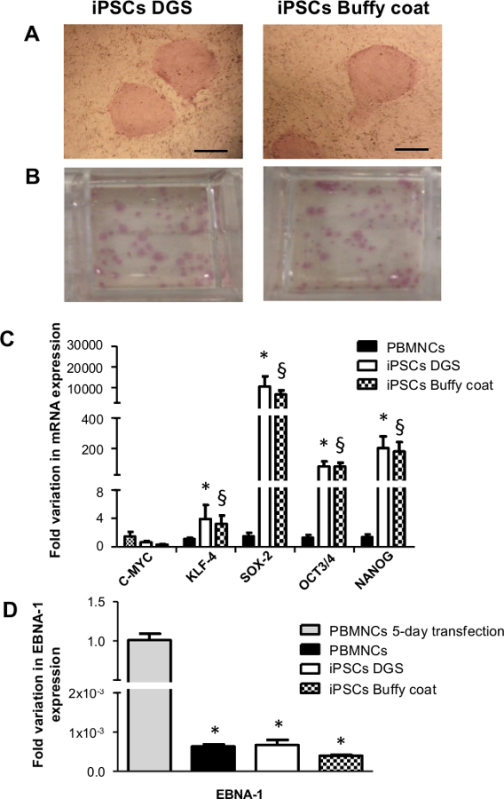 Figure 3. iPSC characterization: alkaline phosphatase activity, re-expression of endogenous pluripotency genes of iPSCs and evaluation of transgene silencing. (A) and (B) Representative images showing the positive staining for alkaline phosphatase. Scale bar 500 µm. (C) The bar graph showing the re-expression of endogenous pluripotency genes (C-MYC, KLF-4, SOX-2, OCT3/4 and NANOG) in both iPSCs obtained from DGS and buffy coats, using qRT-PCR analysis (n=4, Student t-test : * p <0.05, § p <0.005 vs corresponding PBMNCs). (D) qRT-PCR with episomal-specific primers (EBNA-1) (relative to control FBOX15 primers) shows no genome-integration of episomal vectors. (n = 4, Student t-test: * p <0.001 vs corresponding PBMNCs 5-day transfection). Please click here to view a larger version of this figure.
Figure 3. iPSC characterization: alkaline phosphatase activity, re-expression of endogenous pluripotency genes of iPSCs and evaluation of transgene silencing. (A) and (B) Representative images showing the positive staining for alkaline phosphatase. Scale bar 500 µm. (C) The bar graph showing the re-expression of endogenous pluripotency genes (C-MYC, KLF-4, SOX-2, OCT3/4 and NANOG) in both iPSCs obtained from DGS and buffy coats, using qRT-PCR analysis (n=4, Student t-test : * p <0.05, § p <0.005 vs corresponding PBMNCs). (D) qRT-PCR with episomal-specific primers (EBNA-1) (relative to control FBOX15 primers) shows no genome-integration of episomal vectors. (n = 4, Student t-test: * p <0.001 vs corresponding PBMNCs 5-day transfection). Please click here to view a larger version of this figure.
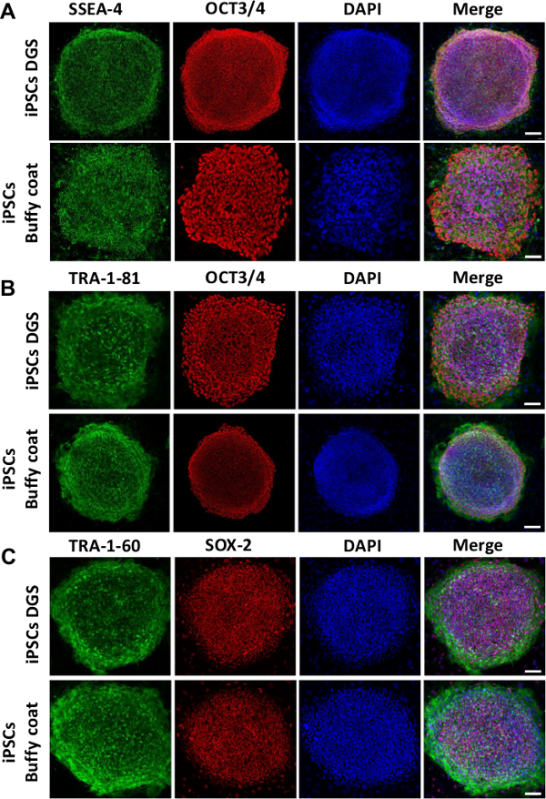 Figure 4. Immunofluorescence analysis of embryonic stem cells pluripotency markers. Representative immunofluorescence staining showing significant expression of pluripotency proteins SSEA-4, TRA-1-60, TRA-1-81, OCT3/4 and SOX-2. Nuclei are counterstained with DAPI. Scale bar 200 µm for iPSC DGS (A) and iPSC Buffy coat (B). Scale bar 100 µm for iPSC Buffy coat (A), iPSC DGS (B) and (C). Please click here to view a larger version of this figure.
Figure 4. Immunofluorescence analysis of embryonic stem cells pluripotency markers. Representative immunofluorescence staining showing significant expression of pluripotency proteins SSEA-4, TRA-1-60, TRA-1-81, OCT3/4 and SOX-2. Nuclei are counterstained with DAPI. Scale bar 200 µm for iPSC DGS (A) and iPSC Buffy coat (B). Scale bar 100 µm for iPSC Buffy coat (A), iPSC DGS (B) and (C). Please click here to view a larger version of this figure.
 Figure 5. Protocol for the differentiation of iPSCs and expression of three germ layer genes in EBs obtained from iPSCs. (A) Schematic representation of the protocol inducing spontaneous differentiation of iPSCs from PBMNCs after DGS and buffy coats in EBs. (B) qRT-PCR experiments showing the expression of all three germ layer genes: endoderm (SOX-7, AFP), mesoderm (CD31, ACTA-2, CDH-5, RUNX-1), and ectoderm (KRT-14, TH, GABRR-2) in EBs after 20 days of differentiation (n = 4, Student t-test: * p <0.05 vs corresponding iPSCs counterpart). EBs = embryoid bodies. Please click here to view a larger version of this figure.
Figure 5. Protocol for the differentiation of iPSCs and expression of three germ layer genes in EBs obtained from iPSCs. (A) Schematic representation of the protocol inducing spontaneous differentiation of iPSCs from PBMNCs after DGS and buffy coats in EBs. (B) qRT-PCR experiments showing the expression of all three germ layer genes: endoderm (SOX-7, AFP), mesoderm (CD31, ACTA-2, CDH-5, RUNX-1), and ectoderm (KRT-14, TH, GABRR-2) in EBs after 20 days of differentiation (n = 4, Student t-test: * p <0.05 vs corresponding iPSCs counterpart). EBs = embryoid bodies. Please click here to view a larger version of this figure.
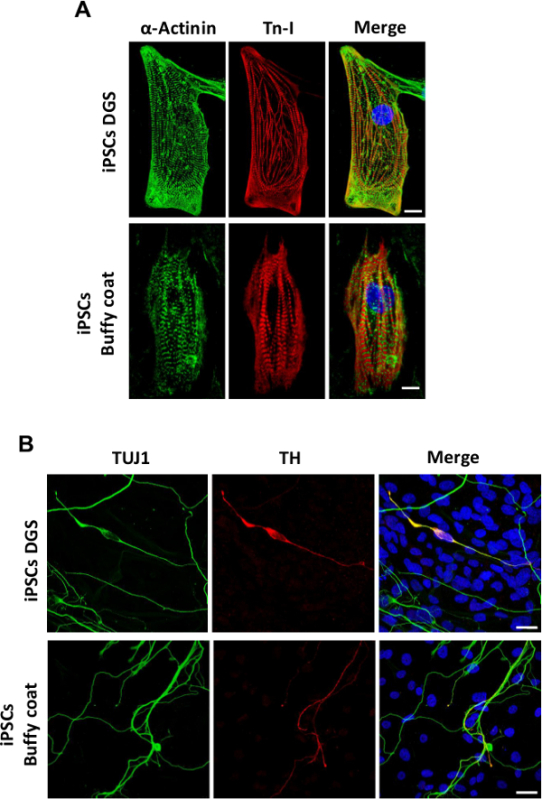 Figure 6. Confocal microscopy analysis of cardiac and neuronal proteins in EBs at day 20 of differentiation obtained from PBMNCs after DGS and buffy coats-derived iPSCs. (A) Representative images (maximum projection of confocal image stack) for cardiac sarcomeric proteins α-Actinin and Troponin I (Tn-I) in single-cell dissociated beating areas (scale bar 10 µm) and (B) for neuron-specific marker class III β-Tubulin (TUJ1) and the dopaminergic neuronal marker tyrosine hydroxylase (TH) (scale bar 20 µm). Nuclei are counterstained with DAPI. Please click here to view a larger version of this figure.
Figure 6. Confocal microscopy analysis of cardiac and neuronal proteins in EBs at day 20 of differentiation obtained from PBMNCs after DGS and buffy coats-derived iPSCs. (A) Representative images (maximum projection of confocal image stack) for cardiac sarcomeric proteins α-Actinin and Troponin I (Tn-I) in single-cell dissociated beating areas (scale bar 10 µm) and (B) for neuron-specific marker class III β-Tubulin (TUJ1) and the dopaminergic neuronal marker tyrosine hydroxylase (TH) (scale bar 20 µm). Nuclei are counterstained with DAPI. Please click here to view a larger version of this figure.
| Primer | Sequence |
| C-MYC fw | 5’-AGAAATGTCCTGAGCAATCACC-3’ |
| C-MYC rev | 5’-AAGGTTGTGAGGTTGCATTTGA-3’ |
| KLF-4 fw | 5′-ATAGCCTAAATGATGGTGCTTGG-3’ |
| KLF-4 rev | 5′- AACTTTGGCTTCCTTGTTTGG-3’ |
| SOX-2 fw | 5′- GGGAAATGGGAGGGGTGCAAAAGAGG-3’ |
| SOX-2 rev | 5′- TTGCGTGAGTGTGGATGGGATTGGTG-3 |
| OCT3/4 fw | 5′- GACAGGGGGAGGGGAGGAGCTAGG-3’ |
| OCT3/4 rev | 5′- CTTCCCTCCAACCAGTTGCCCCAAAC-3’ |
| NANOG fw | 5′-TGCAAGAACTCTCCAACATCCT-3’ |
| NANOG rev | 5′-ATTGCTATTCTTCGGCCAGTT-3’ |
| SOX-7 fw | 5′-TGAACGCCTTCATGGTTTG-3’ |
| SOX-7 rev | 5′-AGCGCCTTCCACGACTTT-3’ |
| AFP fw | 5′-GTGCCAAGCTCAGGGTGTAG -3’ |
| AFP rev | 5′-CAGCCTCAAGTTGTTCCTCTG-3’ |
| CD31 fw | 5′-ATGCCGTGGAAAGCAGATAC-3’ |
| CD31 rev | 5′-CTGTTCTTCTCGGAACATGGA-3’ |
| ACTA-2 fw | 5′-GTGATCACCATCGGAAATGAA-3’ |
| ACTA-2 rev | 5′-TCATGATGCTGTTGTAGGTGGT-3’ |
| CDH-5 fw | 5′-GAGCATCCAGGCAGTGGTAG-3’ |
| CDH-5 rev | 5′-CAGGAAGATGAGCAGGGTGA-3’ |
| RUNX-1 fw | CCCTAGGGGATGTTCCAGAT-3' |
| RUNX-1 rev | TGAAGCTTTTCCCTCTTCCA-3' |
| KRT-14 fw | 5′-CACCTCTCCTCCTCCCAGTT-3’ |
| KRT-14 rev | 5′-ATGACCTTGGTGCGGATTT-3’ |
| TH fw | 5′-TGTACTGGTTCACGGTGGAGT-3’ |
| TH rev | 5′-TCTCAGGCTCCTCAGACAGG-3’ |
| GABBR-2 fw | 5′-CTGTGCCTGCCAGAGTTTCA-3’ |
| GABBR-2 rev | 5′-ACGGCCTTGACGTAGGAGA-3’ |
| EBNA-1 fw | 5′-ATCAGGGCCAAGACATAGAGATG-3’ |
| EBNA-1 rev | 5′-GCCAATGCAACTTGGACGTT-3’ |
| FBX-15 fw | 5′-GCCAGGAGGTCTTCGCTGTA-3’ |
| FBX-15 fw | 5′-AATGCACGGCTAGGGTCAAA-3’ |
| GAPDH fw | 5′-CCACCCATGGCAAATTCC-3’ |
| GAPDH rev | 5’-TCGCTCCTGGAAGATGGTG-3’ |
Table 1: List of qRT-PCR primers. Primer sequences used for evaluation of endogenous pluripotency gene expression, transgene silencing and expression of three germ layer markers.
Discussion
In the past, the only way to obtain human pluripotent stem cells carrying a particular genetic mutation was to recruit parents undergoing pre-implantation genetic diagnosis and generate embryonic stem cells from their discarded blastocysts31,32. Using a reprogramming approach, researchers can now generate iPSCs from patients carrying virtually any genotype. The possibility to start from a patient-specific cell line and an easily accessible source is very important since it allows an investigation of the pathophysiology of disorders and subsequent identification of novel therapeutic approaches.
In the present study, we combined a plasmid approach15 with a method already applied to produce iPSCs from PBMNCs previously frozen after density gradient separation22 and successfully generated virus-free iPSCs from frozen buffy coat PBMNCs. The use of buffy coats instead of density gradient separated PBMNCs allows us to reduce costs, working time and manual steps for operators during sample processing. The set-up of a low-cost protocol using PBMNCs from frozen buffy coats is essential for studies with elevated number of participants and for sample collections in multi-center studies. Importantly, the ability to produce iPSCs from frozen buffy coats allows access to numerous frozen biosamples already stored in blood banks, using a simple, cost-effective and not so time consuming method for sample collection.
This induction method may be useful for the derivation of patient-specific integration-free iPSCs, where colonies are characterized by the absence of episomal transgene after only few passages (≥5 passages), in both starting materials. Remarkably, we observed that all the selected iPSC colonies obtained from both types of starting material display comparable pluripotency features and the preservation of cellular and molecular similarities to hESCs, indicating a successful iPSC derivation and a consistent reproducibility of the protocol.
Moreover, we have successfully generated iPSCs from more than 10 human skin fibroblast and 3 cardiac mesenchymal stromal cell lines (both from healthy donors and patients with cardiac and skeletal muscle diseases) using this method (data not shown), thus suggesting that the described protocol can be used for the successful reprogramming of different cell types. However, the choice of blood as starting material is advantageous compared to fibroblasts, especially when considering that extensive expansion of fibroblasts in vitro may affect the efficiency of reprogramming, based on fibroblast passage number and proliferation rate33,34. Our protocol also allows the generation of transgene-free iPSCs from frozen buffy coats after a minimal culture time, thus reducing by about 3-4 weeks the time needed for derivation of iPSCs compared to other sources, such as fibroblasts. Furthermore, recent studies revealed that blood cell-derived iPSCs display an epigenetic signature that more closely resembles hESCs than those obtained from mesenchymal stem cells (MSC)/fibroblasts14,35.
Despite of the successful generation of iPSCs from frozen buffy coats, a few limitations need to be considered in this protocol. Before reprogramming induction, the amplification of PBMNCs from frozen buffy coats could represent a critical step of the protocol. The quality of starting material can strongly influence the selection and isolation of PBMNCs, in order to achieve sufficient cell numbers for reprogramming procedure (at least 2 x 106 cells for electroporation). This is mostly due to intrinsic biological variability of the samples, but also to technical issues. Indeed, the amplification of PBMNCs from frozen buffy coats is less efficient compared to PBMNCs obtained from density gradient separation, due to a strong dependence on manual operator variability. In order to enhance reproducibility and efficiency of PBMNC amplification from frozen buffy coats without density gradient separation, the use of automated systems is strongly recommended to collect buffy coat fractions. Although it is more difficult to expand a sufficient number of PBMNCs from buffy coats than PBMNCs after density gradient separation, the efficiency of reprogramming (about 0.001-0.0005%) of the two starting materials is comparable, according to blood reprogramming efficiency previously reported14,23. Since the reprogramming efficiency is low for regenerative medicine purposes, the use of improved EBNA1/OriP (from Epstein-Barr virus, EBV) episomal vectors may increase the number of iPSC colonies for future cell therapy development22,36. Additionally, a MEF feeder layer for iPSC generation and maintenance is used in our protocol. In order to obtain clinical-grade iPSCs, a feeder-free culture could be an alternative method for further studies.
In conclusion, this protocol represents a valid method to generate iPSCs from an easily accessible patient cell source with the great advantage of using a non-integrative system that can potentially be used for the derivation of clinical-grade iPSCs, not only for disease modelling but also for a future personalized medicine.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The study was supported by the Ministry of Health and Department of Educational Assistance, University and Research of the Autonomous Province of Bolzano and the South Tyrolean Sparkasse Foundation.
References
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Okita K, Nakagawa M, Yamanaka S. Induction of pluripotent stem cells from fibroblast cultures. Nat. Protoc. 2007;2(12):3081–3089. doi: 10.1038/nprot.2007.418. [DOI] [PubMed] [Google Scholar]
- Drews K, Jozefczuk J, Prigione A, Adjaye J. Human induced pluripotent stem cells--from mechanisms to clinical applications. J. Mol. Med. (Berl) 2012;90(7):735–745. doi: 10.1007/s00109-012-0913-0. [DOI] [PubMed] [Google Scholar]
- Bayart E, Cohen-Haguenauer O. Technological overview of iPS induction from human adult somatic cells) Curr. Gene Ther. 2013;13(2):73–92. doi: 10.2174/1566523211313020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Sommer AG, et al. Generation of human induced pluripotent stem cells from peripheral blood using the STEMCCA lentiviral vector. J. Vis. Exp. 2012. p. e4327. [DOI] [PMC free article] [PubMed]
- Zhou W, Freed CR. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells. 2009;27(11):2667–2674. doi: 10.1002/stem.201. [DOI] [PubMed] [Google Scholar]
- Lieu PT, Fontes A, Vemuri MC, Macarthur CC. Generation of induced pluripotent stem cells with CytoTune, a non-integrating Sendai virus. Methods Mol. Biol. 2013;997:45–56. doi: 10.1007/978-1-62703-348-0_5. [DOI] [PubMed] [Google Scholar]
- Woltjen K, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458(7239):766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods. 2011;8(5):409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- Kim D, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell. Stem Cell. 2009;4(6):472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren L, Ni Y, Wang J, Guo X. Feeder-free derivation of human induced pluripotent stem cells with messenger RNA. Sci. Rep. 2012;2:657. doi: 10.1038/srep00657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Hochedlinger K. Induced pluripotency: history, mechanisms, and applications. Genes Dev. 2010;24(20):2239–2263. doi: 10.1101/gad.1963910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou BK, et al. Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res. 2011;21(3):518–529. doi: 10.1038/cr.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494(7435):105–110. doi: 10.1038/nature11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aasen T, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008;26(11):1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- Streckfuss-Bomeke K, et al. Comparative study of human-induced pluripotent stem cells derived from bone marrow cells, hair keratinocytes, and skin fibroblasts. Eur. Heart J. 2013;34(33):2618–2629. doi: 10.1093/eurheartj/ehs203. [DOI] [PubMed] [Google Scholar]
- Miyoshi N, et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell. Stem Cell. 2011;8(6):633–638. doi: 10.1016/j.stem.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Wang Y, et al. Induced pluripotent stem cells from human hair follicle mesenchymal stem cells. Stem Cell. Rev. 2013;9(4):451–460. doi: 10.1007/s12015-012-9420-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao-Braga PC, et al. Feeder-free derivation of induced pluripotent stem cells from human immature dental pulp stem cells. Cell Transplant. 2011;20(11-12):1707–1719. doi: 10.3727/096368911X566235. [DOI] [PubMed] [Google Scholar]
- Haase A, et al. Generation of induced pluripotent stem cells from human cord blood. Cell. Stem Cell. 2009;5(4):434–441. doi: 10.1016/j.stem.2009.08.021. [DOI] [PubMed] [Google Scholar]
- Dowey SN, Huang X, Chou BK, Ye Z, Cheng L. Generation of integration-free human induced pluripotent stem cells from postnatal blood mononuclear cells by plasmid vector expression. Nat. Protoc. 2012;7(11):2013–2021. doi: 10.1038/nprot.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staerk J, et al. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell. Stem Cell. 2010;7(1):20–24. doi: 10.1016/j.stem.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geti I, et al. A practical and efficient cellular substrate for the generation of induced pluripotent stem cells from adults: blood-derived endothelial progenitor cells. Stem Cells Transl. Med. 2012;1(12):855–865. doi: 10.5966/sctm.2012-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober W. Trypan blue exclusion test of cell viability. Curr Protoc Immunol. 2001. pp. Appendix 3–Appendix 3B. [DOI] [PubMed]
- Hasegawa K, et al. Comparison of reprogramming efficiency between transduction of reprogramming factors, cell-cell fusion, and cytoplast fusion. Stem Cells. 2010;28(8):1338–1348. doi: 10.1002/stem.466. [DOI] [PubMed] [Google Scholar]
- Desjardins P, Conklin D. NanoDrop microvolume quantitation of nucleic acids. J Vis Exp. 2010. p. 2565. [DOI] [PMC free article] [PubMed]
- Fleige S, Pfaffl MW. RNA integrity and the effect on the real-time qRT-PCR performance. Mol Aspects Med. 2006;27(2-3):126–129. doi: 10.1016/j.mam.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Meraviglia V, et al. Human chorionic villus mesenchymal stromal cells reveal strong endothelial conversion properties. Differentiation. 2012;83(5):260–270. doi: 10.1016/j.diff.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Caspersson T, Zech L, Johansson C. Analysis of human metaphase chromosome set by aid of DNA-binding fluorescent agents. Exp Cell Res. 1999;253(2):302–304. doi: 10.1006/excr.1999.4745. [DOI] [PubMed] [Google Scholar]
- Alikani M, Munne S. Nonviable human pre-implantation embryos as a source of stem cells for research and potential therapy. Stem Cell. Rev. 2005;1(4):337–343. doi: 10.1385/SCR:1:4:337. [DOI] [PubMed] [Google Scholar]
- Tropel P, et al. High-efficiency derivation of human embryonic stem cell lines following pre-implantation genetic diagnosis. In Vitro Cell. Dev. Biol. Anim. 2010;46(3-4):376–385. doi: 10.1007/s11626-010-9300-8. [DOI] [PubMed] [Google Scholar]
- Hanna J, et al. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature. 2009;462(72-73):595–601. doi: 10.1038/nature08592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460(7259):1136–1139. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467(7313):285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner SE, Sugden B. The plasmid replicon of Epstein-Barr virus: mechanistic insights into efficient, licensed, extrachromosomal replication in human cells. Plasmid. 2007;58(1):1–12. doi: 10.1016/j.plasmid.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]