

Abstract
The extracellular matrix (ECM) is a complex meshwork of cross-linked proteins that provides biophysical and biochemical cues that are major regulators of cell proliferation, survival, migration, etc. The ECM plays important roles in development and in diverse pathologies including cardio-vascular and musculo-skeletal diseases, fibrosis, and cancer. Thus, characterizing the composition of ECMs of normal and diseased tissues could lead to the identification of novel prognostic and diagnostic biomarkers and potential novel therapeutic targets. However, the very nature of ECM proteins (large in size, cross-linked and covalently bound, heavily glycosylated) has rendered biochemical analyses of ECMs challenging. To overcome this challenge, we developed a method to enrich ECMs from fresh or frozen tissues and tumors that takes advantage of the insolubility of ECM proteins. We describe here in detail the decellularization procedure that consists of sequential incubations in buffers of different pH and salt and detergent concentrations and that results in 1) the extraction of intracellular (cytosolic, nuclear, membrane and cytoskeletal) proteins and 2) the enrichment of ECM proteins. We then describe how to deglycosylate and digest ECM-enriched protein preparations into peptides for subsequent analysis by mass spectrometry.
Keywords: Biochemistry, Issue 101, Extracellular Matrix, Matrisome, Proteins, Decellularization, Sequential solubilization, Deglycosylation, Tryptic digestion, Proteomics, Mass Spectrometry
Introduction
The extracellular matrix (ECM) is a complex meshwork of cross-linked and glycosylated proteins that provides architectural support and anchorage for cells and defines, in part, tissues’ biomechanical properties1,2. ECM proteins also signal to the cells either directly through their receptors (e.g., integrins, syndecans, etc.) or by modulating growth factor signaling3. The ECM thus provides biophysical and biochemical cues that are major regulators of cellular processes such as proliferation, survival, polarization, differentiation, migration, etc.
The ECM plays key roles in physiology, development and aging4. Moreover, several pathologies, such as cardio-vascular diseases, fibrosis, musculo-skeletal diseases, cancers, are caused by, or result in, ECM alterations. Furthermore, the ECM contributes to the maintenance of stem cell niches and identifying key ECM molecules that support stemness will have direct application in tissue engineering and regenerative medicine5. However, despite its importance, the ECM has remained, until recently, underexplored6.
In silico analysis has revealed that the matrisome, defined as the ensemble of ECM and ECM-associated proteins, comprises the products of several hundred genes in both the human and mouse genomes1,7,8. However, the insolubility of ECM proteins has hindered the systematic characterization of the composition of in vivo extracellular matrices of normal and pathological specimens. We recently demonstrated that this insolubility could be turned to advantage and be used to enrich ECM proteins8–10. We and others further demonstrated that mass spectrometry was a method of choice to characterize the composition of ECMs8–10, 13.
We describe here a decellularization procedure that consists of sequential incubations in buffers of different pH and salt and detergent concentrations. This procedure results in the extraction (or depletion) of cytosolic, nuclear, membrane and cytoskeletal proteins and the enrichment of ECM proteins. We then describe how to digest ECM-enriched protein preparations into peptides for subsequent analysis by mass spectrometry.
Using the procedures detailed and illustrated here, we have successfully enriched and characterized by mass spectrometry the extracellular matrices from ten different tissues and tumor types: normal murine lung8, human and murine colon8,9, human liver9, human colorectal tumors and derived liver metastases9, melanoma xenografts8, mammary tumor xenografts10, murine pancreatic islets and murine insulinomas (Naba et al., unpublished). Comparison of the different matrisomes revealed tissue- and tumor-specific ECM signatures that could further be used as potential diagnostic or prognostic biomarkers.
We believe that this procedure can be applied to other specimens with no or relatively minor modifications.
Protocol
NOTE: The procedure can be conducted on fresh or flash frozen tissues. We recommend perfusing highly vascularized tissues with PBS at the time of dissection to eliminate red blood cells and plasma proteins. We do not recommend conducting the procedure on fixed tissues as fixation (i.e., chemical crosslinking) interferes with decellularization and can also significantly compromise subsequent mass spectrometry analysis. For the entire procedure, we recommend the use of low-retention tubes and pipette tips to maximize protein and peptide recovery.
1. Decellularization of Tissues or Tumors
NOTE: Before starting, prepare the reagents and add protease inhibitors (provided with the Compartment Protein Extraction kit) to the desired volume of each buffer. All the buffers and samples should be kept on ice for the duration of the experiment except the Buffer CS that needs to be kept at RT to prevent SDS precipitation. NOTE: The protocol uses a series of incubation in buffers of different pH and containing different amount of salts and detergents to sequentially extract intracellular proteins and enrich for insoluble ECM proteins (Figure 1, Table 1; see also Discussion for alternatives to the use of the commercial kit detailed here). The volumes of reagents given below are for 100 mg of tissues or tumors (see Table 1) and need to be adjusted appropriately.
Homogenize 100 mg of tissue in 500 μl of Buffer C containing protease inhibitors using a tissue homogenizer until the tissue is completely disrupted and a homogenous suspension is obtained. NOTE: Add deoxyribonuclease I (final concentration: 200 μg/ml, reconstituted according to the manufacturer’s instructions) and ribonuclease A (final concentration: 20 μg/ml, reconstituted according to the manufacturer’s instructions) to Buffer N.
- Sequential extraction of intracellular soluble proteins
- Extraction of cytosolic proteins. Incubate the homogenate on a tube rotator for 20 min at 4 °C. Save a small aliquot (10 μl to 20 μl) of the homogenate for subsequent western blot analysis (see Expected Results section and Figure 2).
- Centrifuge the homogenate at 16,000 x g for 20 min at 4 °C. Collect the supernatant in a clean tube, this will constitute the cytosolic (C) fraction of the western blot analysis. Flash freeze this fraction and store at -80 °C.
- To wash, resuspend the pellet in 400 μl of Buffer W containing protease inhibitors and incubate the sample on a tube rotator for 20 min at 4 °C. Centrifuge the homogenate at 16,000 x g for 20 min at 4 °C. Discard the supernatant.
- To extract, nuclear proteins, resuspend the pellet in 150 μl of Buffer N containing protease inhibitors, deoxyribonuclease I and ribonuclease A and incubate the sample on a tube rotator for 30 min at 4 °C.
- Centrifuge the sample at 16,000 x g for 30 min at 4 °C and collect the supernatant in a clean tube. Repeat this step once: after centrifuging the sample for the second time, add the supernatant to the previous supernatant: this will constitute the nuclear (N) fraction of the western blot analysis. Flash freeze this fraction and store at -80 °C. Then, perform washings as per step 1.2.3.
- To extract membrane proteins, resuspend the pellet in 100 μl of Buffer M containing protease inhibitors and incubate the sample on a tube rotator for 30 min at 4 °C. Centrifuge the sample at 16,000 x g for 30 min at 4 °C
- Collect the supernatant in a clean tube: this will constitute the membrane (M) fraction of the western blot analysis. Flash freeze this fraction and store at -80 °C.
- To extract cytoskeletal proteins, resuspend the pellet in 200 μl of Buffer CS containing protease inhibitors and incubate the sample on a tube rotator for 30 min at RT. Note that the pellet will not fully dissolve. We suggest disrupting the pellet by pipetting up and down until observing disruption of the pellet.
- Centrifuge the sample at 16,000 x g for 30 min at RT. Collect the supernatant in a clean tube. Note at this point a further marked decrease in the size of the pellet (see video).
- Resuspend the pellet in 150 μl of Buffer C containing protease inhibitors and incubate the sample on a tube rotator for 20 min at 4 °C. Centrifuge the sample at 16,000 x g for 20 min at 4 °C.
- Collect the supernatant and add it to the previous supernatant: this will constitute the cytoskeletal (CS) fraction of the western blot analysis. Flash freeze this fraction and store at -80 °C.
- Perform additional washes. Resuspend the pellet in 500 μl of PBS containing protease inhibitors and incubate the sample on a tube rotator for 5 min at 4 °C. Centrifuge the sample at 16,000 x g for 5 min at 4 °C. Discard the supernatant. Repeat this step three times. NOTE: All traces of detergents need to be removed by extensive washes prior to digestion of the proteins into peptides (see Section 3). At this point, the ECM-enriched pellet can be flash-frozen and kept at -80 °C. Note that the size of the pellet will depend on the amount of insoluble (ECM) proteins in the starting material and the efficiency of the decellularization.
2. Monitoring the Quality of the Decellularization/ECM Enrichment by SDS-PAGE and Western Blot
Mix 10–20 μl aliquot of total tissue extract and 50 μl aliquots of intermediate fractions with Laemmli Buffer containing 100 mM DTT. Resuspend the ECM-enriched, insoluble fraction in 3x Laemmli Buffer containing 100 mM DTT. Note: he elevated concentrations of DTT and SDS assist in solubilization of ECM proteins that are relatively insoluble.
Separate the proteins by SDS-PAGE and transferred onto nitrocellulose membranes.
Perform immune-blots using antibodies to monitor proteins representative of each of the cytosolic, nuclear, membrane, cytoskeletal and ECM fractions (see Representative Results section, Table 2 and Figure 2).
3. In-solution Digestion of Proteins to Peptides for Mass Spectrometry Analysis
NOTE: The pellet obtained after the decellularization procedure and removal of SDS is highly enriched in insoluble ECM proteins For further analysis by mass spectrometry these proteins need to be digested into peptides. Note that, as a consequence of ECM protein insolubility, it is not possible at this step to measure the protein concentration of the sample. We thus provide volumes of reagents to digest the ECM-enriched samples into peptides based on the size (mm) or dry weight of the ECM-enriched pellet (Table 3). The solutions of ammonium bicarbonate, urea, DTT, iodoacetamide and trifluoroacetic acid all need to be freshly prepared.
Resuspend the ECM-enriched sample by adding the appropriate volume of 8 M urea to the ECM-enriched pellet and add DTT at a final concentration of 10mM (see Table 3). Incubate with continuous agitation at 1,400 rpm for 2 hr at 37 °C. NOTE: at this point the ECM proteins will not be fully dissolved and the visibly large protein particles should not be discarded by centrifugation or filtration. The suspension will clear upon deglycosylation and digestion (see video).
- Alkylation
- Prepare the iodoacetamide solution in HPLC-grade water. Cool the sample to RT and add the iodoacetamide to a final concentration of 25 mM. For complete alkylation, the DTT:iodoacetamide ratio should be between 1:2.5 and 1:3.
- Incubate in the dark for 30 min at RT.
- Deglycosylation: Note: deglycosylation is needed to remove carbohydrate side chains that interfere with identification of peptides modified by N-linked glycosylation.
- Dilute to 2 M urea with 100 mM ammonium bicarbonate pH 8.0 and add the appropriate amount of PNGaseF (see Table 3). Incubate with continuous agitation at 1,400 rpm for 2 hr at 37 °C.
- Digestion
- Add Lys-C and incubate with continuous agitation at 1,400 rpm for 2 hr at 37 °C. Add the trypsin and incubate with continuous agitation at 1,400 rpm O/N at 37 °C. NOTE: the ECM-rich suspension that began cloudy upon initial reconstitution in 8M urea appears clear after O/N digestion (see video).
- Add a second aliquot of trypsin to the sample and incubate with continuous agitation at 1,400 rpm for an additional 2 hr at 37 °C.
- Acidification
- Upon completion of the digestion, inactivate the trypsin by acidifying the sample with freshly prepared 50% trifluoro-acetic acid (TFA). The sample should reach pH < 2. We suggest adding 1–1.5 μl of 50% TFA at a time and using 1 μl of the peptide solution to measure the pH of the solution using pH paper (see video).
- Centrifuge the acidified sample at 16,000 x g for 5 min at RT. Collect the supernatant in a clean low-retention tube. At this point, the peptide solution can be stored at -20 °C.
- Desalting Note: that this last step is usually conducted at a mass spectrometry facility according to their own preferred methods.
- Prior to proteomics analysis, desalt the samples and peptides eluted with freshly prepared 60% acetonitrile, 0.1% trifluoroacetic acid, followed by concentration in a vacuum concentrator. Resuspend the peptides in freshly prepared 3% acetonitrile, 0.1% trifluoroacetic acid8. NOTE: that after desalting, the concentration of the peptide solution can be measured by spectrophotometry (see Expected Results section).
- Now analyze the sample by mass spectrometry, again according to the optimal procedures of the mass spectrometry facility. NOTE: We encourage researchers interested in characterizing the composition of ECMs using mass spectrometry to refer to our other publications8–10 and website that provide further detailed explanation, including LC-MS/MS parameters, mass spectrometry data search for protein identification and data analysis using the in silico matrisome annotation tool we developped8.
Representative Results
Quality control of the decellularization procedure
The efficiency of the decellularization can be monitored by analyzing the protein content of each fraction by western blot. Table 2 lists proteins of diagnostic value to assess the quality of the decellularization procedure. Figure 2A shows the efficient extraction in the intermediate fractions of cytosolic (GAPDH), nuclear (histones), membrane (β1 integrin) and cytoskeletal (actin) proteins, whereas no ECM proteins (collagen I) is detected in these fractions (Figure 2). In turn, the final pellet is enriched for ECM proteins and largely depleted of intracellular proteins (Figure 2A). Figure 2B presents satisfactory intracellular protein depletion (no histone is detected in the ECM-rich fraction), although, actin can still be detected in the ECM-rich fraction and depletion of monomeric collagen I — apparent molecular weight ~110 kDa, presumptively corresponding to unassembled collagen I — can be observed in the CS fraction.
We also routinely monitor additional ECM proteins such as fibronectin and laminin, although, in some tissues these components can be partially solubilized in earlier fractions8-10. For example, fibronectin also occurs as soluble plasma fibronectin that is not incorporated into the ECM. Perfusion of the tissue prior to extraction reduces plasma fibronectin concentration but does not always eliminate it. In some tissues, laminins are found loosely associated with the cell surface or the ECM and extract in intermediate fractions. If this occurs it can be addressed by altering extraction conditions (see Discussion).
Note that the enrichment of ECM proteins and concomitant depletion of intracellular components is based on the relative solubility of proteins in the different buffers. This differs among different tissues — in some cases the histones and actin are more readily extracted than in others. Also note that, although histones are expected to be extracted in the N fraction (Figure 2B), we often observe a more complete depletion of histones in the M or CS fraction (Figure 2A and B).
Indicative peptide concentration expected
The concentration of the peptide solution obtained after digestion, acidification and desalting can be measured by spectrophotometry either by measuring the absorbance of the peptide solution using the 280 nm wavelength corresponding to tryptophan, tyrosine, or using the 205 nm wavelength corresponding to absorbance of the peptide bonds.
We measured the concentration of the peptide solutions obtained from the decellularization of three murine lungs samples (82 mg, 100 mg, and 100 mg, respectively) prepared in parallel and obtained from each: 424 ng/µl, 450 ng/µl, and 580 ng/µl of peptides respectively.
Identification of ECM peptides by mass spectrometry
Mass spectrometric analysis of the composition of ECM-enriched protein samples, prepared as described here, showed that >70% of the signal intensity corresponds to ECM and ECM-associated proteins8-10.
Table 1. Volume of reagents from Compartmental Extraction kit to decellularize 100 mg (wet weight) of tissue or tumor. This table lists the composition and the volume of each buffer used to conduct the decellularization of 100 mg of tissue or tumor. A cocktail of protease inhibitors is provided as a 50x solution and needs to be added to each buffer2.
| Reagents from Compartment Protein Extraction Kit | Volume for 100 mg of tissue | Composition (based on Millipore datasheet cat#21451) |
| Buffer C | 500 μl | HEPES (pH 7.93), MgCl2, KCl, EDT4, Sucrose, Glycerol, Sodium Orthovanadate5 |
| Buffer W | 400 μl | HEPES (pH 7.9), MgCl2, KCl, EDTA, Sucrose, Glycerol, Sodium Orthovanadate |
| Buffer N | 150 μl x 2 | HEPES (pH 7.9), MgCl2, NaCl, EDTA, Glycerol, Sodium Orthovanadate |
| Buffer W | 400 μl | HEPES (pH 7.9), MgCl2, KCl, EDTA, Sucrose, Glycerol, Sodium Orthovanadate |
| Buffer M | 100 μl | HEPES (pH 7.9), MgCl2, KCl, EDTA, Sucrose, Glycerol, Sodium deoxycholate (DOC)6, NP-406, Sodium Orthovanadate |
| Buffer CS | 200 μl | PIPES (pH 6.8), MgCl2, NaCl, EDTA, Sucrose, Sodium Dodecyl Sulfate (SDS)7, Sodium Orthovanadate |
| Buffer C | 150 μl | HEPES (pH 7.9), MgCl2, KCl, EDTA, Sucrose, Glycerol, Sodium Orthovanadate |
| 1x PBS | 500 μl/wash | - |
Notes: 1 For proprietary reasons, we were unable to obtain the precise composition of the buffers from the supplier of the kit, but we include here some notes based on our own experience using home-made buffers to conduct similar extractions. 2 Protease inhibitor: it is advisable to include a variety of inhibitors against cysteine, serine and threonine peptidases, serine esterases, divalent cation-dependent metalloproteinases etc. Many commercially available protease inhibitor cocktails exist. 3 pH above 7.0 is an effective inhibitor of lysosomal proteases. 4 EDTA (usually used at 2 mM) is an effective inhibitor of divalent cation-dependent proteases. 5 Sodium orthovanadate is a phosphatase inhibitor. A typical effective concentration would be 0.5–5 mM. 6 NP40 at 0.1%–0.5% is sufficient to solubilize most membrane lipids. The combination of NP40 and DOC — often used at equal concentrations (e.g., 0.5% of each) — is often used as a more stringent extraction that still leaves many protein-protein interactions intact. 7 SDS is a more stringent ionic detergent (CMC 0.1%). It can also be used in combination with the other two detergents SDS/NP40/DOC 0.1/0.5/0.5% as an intermediate stringency buffer.
Table 2. Diagnostic proteins to monitor the quality of the decellularization procedure. This table lists examples of proteins that are characteristics of each subcellular compartment (cytosol, nucleus, plasma membrane, cytoskeleton and ECM) that can be used to monitor the quality of the decellularization procedure and the efficiency of the ECM-enrichment.
| Intracellular Compartment | Diagnostic Proteins |
| Cytosolic proteins | Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) |
| Nuclear proteins | Histones, Lamins, Nucleoporin |
| Membrane proteins | Integrins, Transferrin Receptor |
| Cytoskeletal proteins | Actin, Tubulin, Vimentin |
| Basement membrane ECM proteins | Collagen IV, Nidogens, Laminins |
| Interstitial ECM proteins (interstitial) | Collagen I, Collagen III, Collagen VI, Fibronectin |
For recommendation on antibodies, see our publications8-10.
Table 3. Volume of reagents to digest ECM-enriched samples into peptides. This table lists the reagents used to resuspend ECM-enriched protein samples and reduce, alkylate, deglycosylate and digest proteins samples into peptides prior to mass spectrometry analysis.
| Reagents | Preparation | Final Concentration / Amount for 1 mm-thick pellet (~5–10 mg dry weight) | Volume for 1 mm-thick pellet (~5–10 mg dry weight) |
| Ammonium bicarbonate (NH4HCO3) | 100 mM solution in HPLC-grade water | - | - |
| Urea | 8 M solution in 100 mM ammonium bicarbonate | 8 M | 50 μl |
| Dithiothreitol | Reconstitute in HPLC-grade water at 500 mM | 10 mM | 1 μl |
| Iodoacetamide | Reconstitute in HPLC-grade water at 500 mM | 25 mM | 2.5 μl |
| Peptide-N-Glycosidase F (PNGaseF) | Commercial solution at 500 U/μl | 1,000 U | 2 μl |
| Endoproteinase LysC, mass spectrometry-grade | Reconstitute in HPLC-grade water at 0.5 μg/μl | 1 μg | 2 μl |
| Trypsin, mass spectrometry-grade (round 1) | Commercial solution at 0.5 μg/μl | 3 μg | 6 μl |
| Trypsin, mass spectrometry-grade (round 2) | Commercial solution at 0.5 μg/μl | 1.5 μg | 3 μl |
| Trifluoro-acetic acid (TFA) | 50% solution in HPLC-grade water | - | 2–5 μl |
| Acetonitrile (elution) | 60% solution with 0.1% TFA in HPLC-grade water | - | 500 μl |
| Acetonitrile (reconstitution) | 3% solution with 0.1% TFA in HPLC-grade water | - | 100 μl |
 Figure 1. Scheme of the experimental procedure. Schematic workflow of the protocol to decellularize tissues (Section 1), control the quality of the decellularization and evaluate the ECM enrichment (Section 2) and digest ECM-enriched protein samples into peptides prior to mass spectrometry analysis (Section 3).
Figure 1. Scheme of the experimental procedure. Schematic workflow of the protocol to decellularize tissues (Section 1), control the quality of the decellularization and evaluate the ECM enrichment (Section 2) and digest ECM-enriched protein samples into peptides prior to mass spectrometry analysis (Section 3).
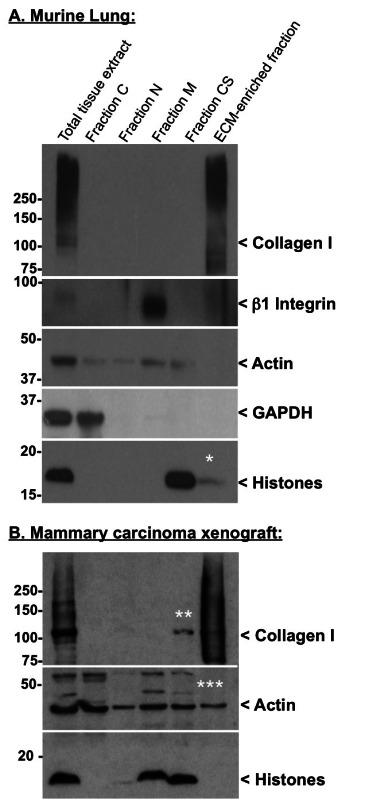 Figure 2. Quality control of the decellularization procedure by western blot. Western blots were performed on murine lung (A) and human mammary carcinoma xenograft (B) samples using the following antibodies: rabbit anti-actin (clone 14-1) and rabbit anti-β1 integrin antibodies produced in our laboratory; rabbit anti-collagen I, mouse anti-GAPDH and rabbit anti-pan-histones antibodies were from Millipore. Following primary antibody incubation, the membranes were washed and incubated in the presence of HRP-conjugated goat anti-rabbit or goat anti-mouse secondary antibody from Jackson ImmunoResearch Laboratory. Finally, the membranes were washed and incubated in Western Lightning™ Chemiluminescence Reagent (PerkinElmer LAS). * indicates minimal residual histone contamination of the ECM-rich fraction. ** indicates partial depletion of monomeric (presumptively unassembled) collagen I in the CS fraction. *** indicates residual actin contamination of the ECM-rich fraction. The smear detected with the anti-collagen antibody represents different levels of post-translational modifications (e.g., cross-linking, glycosylation).
Figure 2. Quality control of the decellularization procedure by western blot. Western blots were performed on murine lung (A) and human mammary carcinoma xenograft (B) samples using the following antibodies: rabbit anti-actin (clone 14-1) and rabbit anti-β1 integrin antibodies produced in our laboratory; rabbit anti-collagen I, mouse anti-GAPDH and rabbit anti-pan-histones antibodies were from Millipore. Following primary antibody incubation, the membranes were washed and incubated in the presence of HRP-conjugated goat anti-rabbit or goat anti-mouse secondary antibody from Jackson ImmunoResearch Laboratory. Finally, the membranes were washed and incubated in Western Lightning™ Chemiluminescence Reagent (PerkinElmer LAS). * indicates minimal residual histone contamination of the ECM-rich fraction. ** indicates partial depletion of monomeric (presumptively unassembled) collagen I in the CS fraction. *** indicates residual actin contamination of the ECM-rich fraction. The smear detected with the anti-collagen antibody represents different levels of post-translational modifications (e.g., cross-linking, glycosylation).
Discussion
Modifications
Although we employed this exact procedure to enrich the ECM from ten tissues and tumor types8–10, modifications of the protocol should be considered in the following instances:
1) Detection of ECM proteins in the intermediate fractions.
Extracellular matrices from different tissues or tumor types may differ in their extractability/insolubility, as discussed above for fibronectin and laminins. For example, it is thought that the ECM of fibrotic tissues or remodeling tissues turns over very dynamically and thus one might observe a higher proportion of ECM proteins in those tissues to be more readily extractable12. Depending on the fraction in which ECM proteins are detected, we suggest reducing the incubation time of the step causing the extraction of ECM proteins or omitting that step.
2) Detection of a significant proportion of intracellular components in the ECM-enriched pellet. In some tissues or tumor types the ratio cells:ECM is particularly high (e.g., liver, spleen, non-desmoplastic tumors). In that case, a significant contamination of the ECM-enriched fraction by intracellular proteins (in particular cytoskeletal proteins and/or histones) can be observed. To deplete intracellular proteins efficiently, we suggest repeating twice the incubation in Buffer M and/or Buffer CS (both containing detergents, this usually depletes abundant intracellular proteins). Another alternative would be to use alternative buffers with higher detergent concentrations, with the caveat that this may lead to the depletion of relatively more soluble ECM proteins as well (see next paragraph).
3) Alternative to using a commercial kit.
We were unable, for proprietary reasons, to obtain the precise composition of the buffers from the supplier of the Compartmental Protein Extraction kit. However, we have included in Table 1 notes based on our own experience using home-made buffers with defined detergent (NP-40, sodium deoxycholate and SDS) concentrations to conduct similar extractions. A recent study also highlighted the importance of the pH of decellularization buffers to retain ECM proteins13.
Limitations of the technique
The method presented here relies on the fact that ECM proteins are intrinsically more insoluble than most intracellular proteins. However, the decellularization method described here certainly extracts soluble components present within the ECM such as some growth factors or ECM-remodeling enzymes. Although ECM-associated proteins tightly bound to ECM proteins were detected by proteomics in samples prepared as described, this method may be too stringent to fully profile the composition of matrisome-associated proteins.
Significance of the technique with respect to other methods
The advantage of the method presented here over other methods is that it can be tailored to the nature of the ECM of interest: intermediate steps can be omitted or repeated to prevent the loss of ECM proteins or increase the depletion of contaminating intracellular proteins respectively. This method also only uses minimal amounts of detergents that are rinsed off to prevent their interference with subsequent peptide preparation and mass spectrometry. Finally, the method described here to digest ECM-rich protein preparations into peptides has also the advantage of not requiring proteins to be soluble and can be conducted on “crude” ECM-enriched fractions.
Alternative decellularization methods utilizing chaotropes (such as guanidine hydrochloride)to study the composition of ECMs by mass spectrometry have been reported in the literature (reviewed in 14) and have been used in combination with mass spectrometry to characterize the ECM composition of cartilage15,16, heart17, mammary gland18 and vascular19 and glomerular20 basement membranes.
Recommendations
Decellularization methods employing trypsin to digest out cells should not be used if ECMs are enriched for subsequent proteomics analyses, as trypsinization will result in partial ECM digestion and loss of ECM proteins and peptides. Similarly, if collagenase digestion were to be used to aid tissue disruption, it would need to be monitored carefully as it causes ECM digestion and loss of ECM proteins and peptides.
In-solution vs. in-gel digestion? ECM proteins are cross-linked and highly insoluble and, even when resuspended in 3x Laemmli buffer (containing 6% SDS) and 100mM DTT, separate poorly on SDS gels. Thus in-gel digestion is not a preferred method.
Future applications
It is worth noting that, while not discussed here, the composition of each of the intermediate fractions collected during the decellularization could also be analyzed by mass spectrometry. This may be particularly valuable when studying very small samples (i.e., human biopsies) or when information is desired on other cellular fractions.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors would like to thank Drs. Amanda Del Rosario and Guillaume Carmona for critical reading of the manuscript. This work was supported by grants from the Department of Defense DOD Innovator Award (W81XWH-14-1-0240 to ROH); the National Cancer Institute (U54 CA126515/CA163109); the Broad Institute of MIT and Harvard; the Howard Hughes Medical Institute, of which ROH is an Investigator; and in part by the Support Grant from the National Cancer Institute to the Koch Institute for Integrative Cancer Research at MIT (P30-CA14051). AN received postdoctoral fellowships from the Howard Hughes Medical Institute and the Ludwig Center for Cancer Research at MIT.
References
- Hynes RO, Naba A. Overview of the matrisome--an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol. 2012;4(1):a004903. doi: 10.1101/cshperspect.a004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014;15(12) doi: 10.1038/nrm3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326(5957):1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014;15(12):786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvir T, Timko BP, Kohane DS, Langer R. Nanotechnological strategies for engineering complex tissues. Nat Nanotechnol. 2011;6(1):13–22. doi: 10.1038/nnano.2010.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R. The extracellular matrix: an underexplored but important proteome. Expert Rev Proteomics. 2010;7(6):803–806. doi: 10.1586/epr.10.93. [DOI] [PubMed] [Google Scholar]
- Naba A, Hoersch S, Hynes RO. Towards definition of an ECM parts list: an advance on GO categories. Matrix Biol. 2012;31(7-8):371–372. doi: 10.1016/j.matbio.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo. characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteomics. 2012;11(4):M111.014647. doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Whittaker CA, Carr SA, Tanabe KK, Hynes RO. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer. 2014;14(1):518. doi: 10.1186/1471-2407-14-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Lamar JM, Carr SA, Hynes RO. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. eLife. 2014;3:e01308. doi: 10.7554/eLife.01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A, Ding H, Whittaker CA, Hynes RO, et al. Proteomic analysis of altered extracellular matrix turnover in bleomycin-induced pulmonary fibrosis. Mol. Cell Proteomics. 2014;13(7):1741–1752. doi: 10.1074/mcp.M113.037267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya T, Balestrini JL, Mendez JJ, Calle EA, Zhao L, Niklason L. Influence of pH on extracellular matrix preservation during lung decellularization. Tissue Eng Part C Methods. 2014;20(12):1028–1036. doi: 10.1089/ten.tec.2013.0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byron A, Humphries JD, Humphries MJ. Defining the extracellular matrix using proteomics. Int J Exp Pathol. 2013;94(2):75–92. doi: 10.1111/iep.12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Diseberg AF, et al. Comprehensive profiling of cartilage extracellular matrix formation and maturation using sequential extraction and label-free quantitative proteomics. Mol. Cell Proteomics. 2010;9(6):1296–1313. doi: 10.1074/mcp.M000014-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluoccio D, Wilson R, Thornton DJ, Wallis TP, Gorman JJ, Bateman JF. Proteomic analysis of mouse growth plate cartilage. Proteomics. 2006;6(24):6549–6553. doi: 10.1002/pmic.200600191. [DOI] [PubMed] [Google Scholar]
- Barallobre-Barreiro J, Didangelos A, Yin X, Doménech N, Mayr M. A sequential extraction methodology for cardiac extracellular matrix prior to proteomics analysis. In. Heart Proteomics: Methods and Protocols. 2013;1005:215–223. doi: 10.1007/978-1-62703-386-2_17. [DOI] [PubMed] [Google Scholar]
- Hansen KC, Kiemele L, et al. An in-solution ultrasonication-assisted digestion method for improved extracellular matrix proteome coverage. Mol. Cell Proteomics. 2009;8(7):1648–1657. doi: 10.1074/mcp.M900039-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didangelos A, Yin X, Mandal K, Baumert M, Jahangiri M, Mayr M. Proteomics characterization of extracellular space components in the human aorta. Mol. Cell Proteomics. 2010;9(9):2048–2062. doi: 10.1074/mcp.M110.001693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon R, Byron A, et al. Global analysis reveals the complexity of the human glomerular extracellular matrix. J. Am. Soc. Nephrol. 2014;25(5):939–951. doi: 10.1681/ASN.2013030233. [DOI] [PMC free article] [PubMed] [Google Scholar]