

Abstract
Parenteral and oral routes have been the traditional methods of administering cytotoxic agents to cancer patients. Unfortunately, the maximum potential effect of these cytotoxic agents has been limited because of systemic toxicity and poor tumor perfusion. In an attempt to improve the efficacy of cytotoxic agents while mitigating their side effects, we have developed modalities for the localized iontophoretic delivery of cytotoxic agents. These iontophoretic devices were designed to be implanted proximal to the tumor with external control of power and drug flow. Three distinct orthotopic mouse models of cancer and a canine model were evaluated for device efficacy and toxicity. Orthotopic patient-derived pancreatic cancer xenografts treated biweekly with gemcitabine via the device for 7 weeks experienced a mean log2 fold change in tumor volume of −0.8 compared to a mean log2 fold change in tumor volume of 1.1 for intravenous (IV) gemcitabine, 3.0 for IV saline, and 2.6 for device saline groups. The weekly coadministration of systemic cisplatin therapy and transdermal device cisplatin therapy significantly increased tumor growth inhibition and doubled the survival in two aggressive orthotopic models of breast cancer. The addition of radiotherapy to this treatment further extended survival. Device delivery of gemcitabine in dogs resulted in more than 7-fold difference in local drug concentrations and 25-fold lower systemic drug levels than the IV treatment. Overall, these devices have potential paradigm shifting implications for the treatment of pancreatic, breast, and other solid tumors.
INTRODUCTION
Chemotherapy has had an immeasurable impact on the field of oncology since its inception in the 1940s (1). Cytotoxic and molecularly targeted agents have become a mainstay of cancer therapy and play a large role in curative resection and high-risk operations of solid tumors. Tumors at high risk of recurrence or that may involve close margins at the time of operation such as head and neck, rectal, gastroesophageal, advanced gastric, and pancreatic cancers, and soft tissue sarcomas benefit from neoadjuvant chemotherapy and/or radiation (2–7). However, dense stromal environments and poor vascularization impede drug diffusion, reducing exposure to the primary tumor (8–11). This impaired drug delivery has contributed to the recurrence in and overall dreadful prognosis of certain solid tumors (9, 11). To improve local control at the primary tumor, new drug delivery strategies are necessary to effectively concentrate the active drug in the tumor site.
Local drug delivery technologies offer a promising adjunct to systemic delivery. They exist in a variety of form factors designed to facilitate the delivery of drug directly to the site of disease in a controlled manner. Many of these are biodegradable polymeric depots designed to maintain therapeutic concentrations of drug at the tumor site for a prolonged period. However, only a small number of these technologies have demonstrated potentially curative preclinical results for cancer applications, and far fewer have progressed toward clinical practice. A key challenge of many of these local drug delivery systems, particularly polymeric drug–eluting technologies like the Gliadel wafer, has been diffusion limitations (12, 13). The lack of spatial distribution of drugs and elevated interstitial fluid pressures in solid tumors have relegated the use of many local drug delivery systems to postsurgical therapy (12).
A subset of local drug delivery devices involves the use of electric fields to drive drugs into tissues, using a technique known as iontophoresis. Iontophoretic devices are capable of overcoming diffusion barriers by electromigratory and electroosmotic forces (14, 15). Advances in ophthalmologic and urologic devices have enabled the effective iontophoretic and electroosmotic delivery of mitomycin C and dexamethasone to tissues while reducing the systemic effects of these drugs (16, 17). Here, we developed and investigated a new iontophoretic device platform for the local delivery of cytotoxic therapies to solid tumors. These iontophoretic devices were designed to be implanted proximal to the tumor with external user control of power and drug flow.
To evaluate the broad application of iontophoretic devices as potential anticancer therapies, we elected to test the devices in a diverse set of orthotopic mouse models of cancer, including pancreatic and breast cancer models, and a canine model for pharmacokinetic (PK) studies (18–21). These cancer types were chosen as models because of their major local control issues—nearly 40% of patients with locally advanced, nonmetastatic pancreatic cancer do not have the opportunity to undergo surgery because of tumor invasion into adjacent vessels, and inflammatory breast cancers have significant chest wall involvement, where extensive surgery may be needed (22, 23). We describe an in-depth preclinical characterization of iontophoretic delivery of cytotoxic agents. We report that these devices deliver high levels of cytotoxic drugs to the tumor, reduce systemic exposure of the drugs, and potently prevent tumor growth. Iontophoretic devices offer entirely new modalities for the treatment of cancer by improving local tumor control and organ preservation.
RESULTS
Design and fabrication of the devices
Our devices were designed to adapt conventional iontophoretic drug delivery techniques (Fig. 1, A and B). The devices could be either implanted, such as in the case of pancreatic tumors, or used transdermally, for breast tumors, and primarily consisted of an electrode in direct contact with the drug solution, a polyurethane or polydimethylsiloxane (PDMS) reservoir surrounding the electrode, and an inlet and outlet for drug flow-through where the solution in contact with the biological tissue was continuously replaced and replenished (Fig. 1C). This continuous flow design maintained a constant drug concentration around the electrode and pressure within the device. Under an applied electric potential, the drug was transported in the direction normal to the electrode and into the tissue. Initially, we fabricated our devices using silver because it is the most efficient material for drug transport into tissue surrogates (15). However, the electrochemical reaction at the silver anode interface prohibited its long-term use owing to the buildup of silver chloride and depletion of silver. Platinum was chosen for long-term implanted device studies, whereas silver was used for nonimplanted device studies.
Fig. 1. Iontophoretic devices used for the delivery of cytotoxic agents to solid tumors.

(A) Front and side images of the implantable and transdermal devices. (B) Device components and assemblies. (C) Device treatment setups in the pancreatic (implanted device) and breast (transdermal device) cancer models where the drug is supplied to the device, using a syringe pump and electrical current via a DC power supply. Positive and negative leads connect to the device and counter electrode, respectively. (D) Device parameters of drug concentration (at constant current and time) and applied current (at constant concentration and time) were evaluated in mice with patient-derived pancreatic cancer xenografts. Data are means ± SD (n = 5). P values were determined by one-way ANOVA with unpaired t test. (E) Role of current on drug transport in ex vivo tumor and human skin tissue. Gemcitabine transport through PDX tumor tissue was evaluated by applying a current of 2 or 0 mA for 10 min and comparing drug transport into tumor. Data are means ± SD (n = 6). Cisplatin transport into human skin was evaluated by applying a current of 1 or 0 mA for 25 min and comparing drug transport into and through the skin. Data are means ± SD (n = 5). P values were determined by unpaired t test. NS, not significant.
Iontophoretic drug delivery optimization in tumors and in human skin
Several device parameters were evaluated to determine the optimal drug transport conditions, including drug concentration (Fig. 1D) and applied current (Fig. 1E). To test these parameters, we implanted the device into an orthotopic patient-derived xenograft (PDX) model of pancreatic cancer. The mice were allowed to recover for 1 week after device implantation, to allow wound healing and encapsulation. A single treatment of gemcitabine was administered, and tumors and plasma were harvested directly thereafter. Drug transport into PDX tumors with a constant current of 2 mA for 10 min was highest for 40 versus 20 and 10 mg/ml gemcitabine (Fig. 1D and fig. S1). There was a significantly higher concentration of gemcitabine in plasma as well (Fig. 1D). Furthermore, the application of 2 mA of constant current for 10 min using 40 mg/ml gemcitabine resulted in 1.3-fold greater drug transport compared to 1 mA. Plasma levels of gemcitabine were significantly greater for 2 mA compared to 1 mA (Fig. 1E). The most beneficial drug concentration is one that has highest uptake in the tumor with low plasma levels, thus 20 mg/ml gemcitabine at 2 mA was used in PK and efficacy studies.
Ex vivo drug transport studies were conducted using pancreatic cancer PDX tumors and human skin. To test the transport of gemcitabine in the ex vivo PDX tumors, we sutured the devices onto the tumors and placed the counter electrode on the contralateral side of the tumor. A current of 2 or 0 mA was applied for 10 min, and the tumors were subsequently analyzed. The application of 2 mA constant current resulted in an 8.9-fold increase in drug transport compared to the passive diffusion control (0 mA) into the tumor (Fig. 1E). The pH of the gemcitabine solution was measured before and after device treatment, and the treatment reduced the pH of the solution by 1.3 units, indicating a more acidic environment that may have assisted in gemcitabine transport.
The transport of cisplatin into ~1-mm-thick human skin ex vivo was evaluated. A current of 1 or 0 mA was applied for 25 min, and the skin and solution were snap-frozen, processed, and analyzed by inductively coupled plasma mass spectrometry (ICP-MS). The application of 1 mA constant current resulted in an 11.4-fold increase in platinum transported into the human skin compared to passive diffusion (Fig. 1E). Murine skin was evaluated using the same method, revealing similar drug transport into the skin with current but larger transport through the skin (fig. S2). There was a minor drop in pH (~0.2 pH units) for the cisplatin solution after device treatment.
PK and biodistribution of cytotoxic drugs in mice
Iontophoretic delivery of gemcitabine was further characterized with respect to PK in an orthotopic PDX model of pancreatic cancer (18, 19). Recent reports in lung, pancreatic, breast, and colon cancers, as well as glioblastoma, suggest that patient tumors directly implanted in immunocompromised mice exhibit response rates to cytotoxic or targeted therapies more similar to patient responses (24–27). In the PDX model, devices were surgically implanted when the tumor reached a median size of 260 mm3. One week after device implantation, a single treatment was administered, and tumors were harvested at designated time points. We performed a PK study comparing device delivery of gemcitabine to intravenous (IV) delivery (80 mg/kg) (Fig. 2). Gemcitabine IV dosing at 80 mg/kg was chosen because in mice it more appropriately matches the gemcitabine PK profile in humans than the maximum tolerated dose of 120 mg/kg (28–30).
Fig. 2. PK of gemcitabine and cisplatin delivered by iontophoretic devices in mouse models of human pancreatic and breast cancers.

Iontophoretic device delivery was compared with IV delivery. PK of gemcitabine (20 mg/ml) delivered by device compared to IV was evaluated in an orthotopic PDX model of pancreatic cancer. Mice were administered a single treatment of gemcitabine through the device. Organs were collected from each animal at various times, and total gemcitabine concentrations were analyzed. Data are means ± SD (n = 3 to 5 animals per group). The limit of gemcitabine quantitation was 1 μg/ml. P values were determined by unpaired t test. PK of cisplatin delivered by device compared to IV and device + IV was evaluated in SUM149 orthotopic xenografts of breast cancer. Mice were administered a single treatment of cisplatin. Organs were collected from each animal at various times, and total platinum concentrations were analyzed. Data are means ± SD (n = 5 animals per group). The limit of platinum quantitation was 5 ng/ml. P values were determined by one-way ANOVA with unpaired t test comparing device cisplatin and device + IV cisplatin.
Gemcitabine plasma exposure as measured by the area under the curve (AUC) for IV delivery was 52.0 hour*μg/ml, with no detectable gemcitabine in the plasma of the animals receiving gemcitabine via iontophoretic device. Gemcitabine tumor AUC for iontophoretic delivery was an order of magnitude greater than IV delivery (344.4 versus 30.8 hour*μg/g, respectively) (Fig. 2). The average penetration distances that gemcitabine was detected in the tumors in the direction away from the devices were 4.7 mm at 0 hours and 3 mm at 3 and 6 hours; for IV gemcitabine–treated mice, gemcitabine was detected throughout the entire tumor, but at lower drug concentrations. The pancreas had a better blood supply than the tumor, which resulted in greater gemcitabine exposure after IV compared to device delivery. Drug accumulation in the pancreatic tumor and distance of drug transport were dependent on the inflow gemcitabine concentration, which correlated with the in vitro drug transport results (fig. S3).
Device delivery of cisplatin in an orthotopic SUM149 xenograft model of human breast cancer was compared with IV delivery of cisplatin (Fig. 2). Concurrent device and IV delivery of cisplatin was added as an arm of the study based on the low systemic exposure of cisplatin from device treatments (plasma exposure was 2.0 hour*μg/ml). A single treatment was administered after adhesion of the device to the skin above the tumor. Platinum plasma exposures as measured by area under the concentration versus time curve for device delivery, IV (5 mg/kg), and device + IV (5 mg/kg) delivery were comparable at 2.0, 9.9, and 10.7 hour*μg/ml, respectively. However, tumor AUC for device + IV delivery was nearly twofold greater than that for IV delivery alone (83.4 versus 42.4 hour*μg/g, respectively) (Fig. 2).
In PK studies in mice, device delivery resulted in lower kidney, left inguinal lymph node, and right inguinal mammary exposure to cisplatin (1 mg/ml) but more skin exposure compared with IV delivery (fig. S4). The combination of device + IV delivery resulted in the largest cisplatin exposure to all organs.
Device delivery of gemcitabine reduces tumor volume and cancer cell proliferation compared with IV delivery
To evaluate the antitumor activity of gemcitabine and cisplatin delivered by the iontophoretic devices, we performed efficacy studies in orthotopic pancreatic and breast cancer models. In the pancreatic cancer model, devices were surgically implanted onto orthotopic human pancreatic tumors when their size reached a median size of 260 mm3. Mice were treated twice per week for up to 7 weeks with device gemcitabine (20 mg/ml), device saline (0.9% NaCl), IV gemcitabine (80 mg/kg), or IV saline. Implanted devices impaired ultrasound imaging, and thus, tumor volumes were measured only after completion of the treatment.
Device gemcitabine resulted in significant tumor regression in seven of seven mice, outperforming IV gemcitabine and the control arms of IV and device saline over the 7-week period of the study (Fig. 3A). Mice treated with device gemcitabine had a mean log2 fold change in tumor volume of −0.8 compared to a mean log2 fold change in tumor volume of 1.1 for IV gemcitabine, 3.0 for IV saline, and 2.6 for device saline groups. Mice treated with device saline had tumor volumes that were not statistically different from those of mice treated with IV saline. Device gemcitabine was better tolerated on the basis of greater body weight gain compared to IV gemcitabine (fig. S5), but no statistical difference in alanine transaminase, aspartate transaminase, blood urea nitrogen (BUN), and lipase was noted for the different delivery routes (fig. S6). Histological samples from the tumors after 7 weeks of treatment revealed a significant decrease in Ki-67, a marker of cell proliferation, in mice treated with gemcitabine from the iontophoretic device compared with tumors from mice that received IV gemcitabine (Fig. 3B).
Fig. 3. Therapeutic effect of gemcitabine delivered iontophoretically in a pancreatic cancer PDX model.
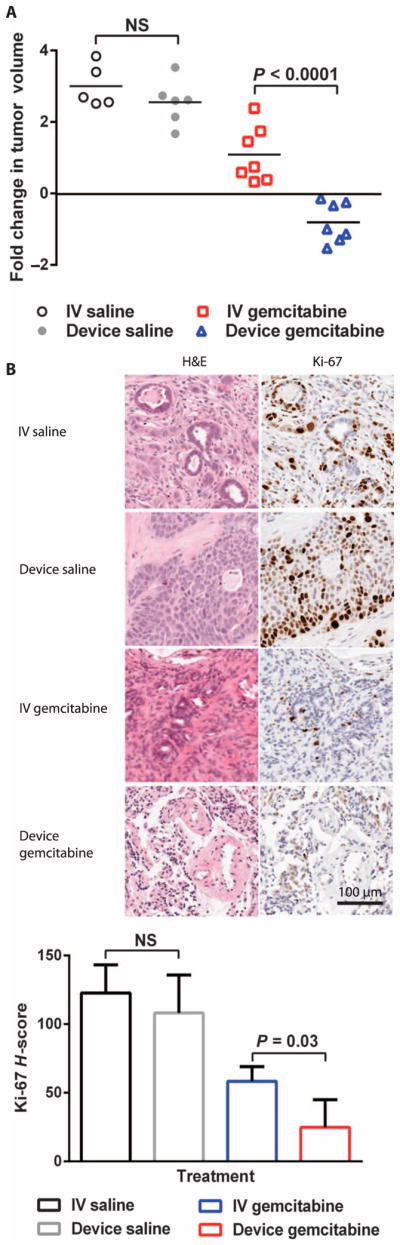
(A) Efficacy of device gemcitabine, IV gemcitabine, device saline, and IV saline in PDX mice treated twice per week for 7 weeks. Data are fold change in tumor volume (log2) (n = 7 for IV and device gemcitabine, n = 5 to 6 for IV and device saline). (B) Histological staining of representative tumors in (A) for Ki-67. Ki-67 staining was quantified according to H-score. P values were determined by one-way ANOVA with unpaired t test.
Device delivery of cisplatin combined with radiotherapy prolongs survival of mice with human breast cancer
This iontophoretic delivery technology was further evaluated in orthotopic breast cancer models: SUM149 human xenograft and T11 syngeneic. In both models, we compared the efficacy of device cisplatin, IV cisplatin (5 mg/kg), device + IV cisplatin (5 mg/kg), device saline, and IV saline (Fig. 4A). Once the human xenograft breast tumors reached ~50 mm3, the mice were treated every week for a total of four doses. The skin of mice after four device-based treatments showed no scarring or deformation (Fig. 4B). Mice bearing T11 tumors received two doses of the same test arms 1 week apart beginning 5 days after inoculation (~20 mm3). The number of treatments varied across different tumor models owing to differences in tolerability of treatments.
Fig. 4. Therapeutic effect of cisplatin delivered iontophoretically in mouse tumor xenograft and syngeneic models of breast cancer.

(A) Treatment schedule according to mouse model. (B) Efficacy and survival of animals treated with device cisplatin, IV cisplatin (5 mg/kg), device + IV cisplatin (5 mg/kg), device saline, and IV saline. SUM149 tumor xenografts were treated once per week for a total of four doses (n = 8 to 9 per treatment group). T11 syngeneic tumors were treated once per week for a total of two doses (n = 9 per treatment group). The study endpoint was time to tumor progression to 2.0 cm in one dimension. Volume data are means ± SD. (C) Representative images of murine skin before and after 4 weeks of transdermal device treatment. (D) γH2AX staining of tumors harvested from SUM149 xenografts 24 hours after a single treatment. γH2AX staining was quantified according to H-score. P values were determined by one-way ANOVA with unpaired t test. (E) Efficacy and survival of animals with T11 syngeneic tumors after a single treatment of radiation (dose), device cisplatin, device cisplatin + radiation, IV cisplatin (dose), IV cisplatin (dose) + radiation, device + IV cisplatin, or device + IV cisplatin + radiation. Data are mean tumor volumes ± SEM (n = 8 per treatment group). P values for tumor growth inhibition were determined by one-way ANOVA with unpaired t test; P values for survival determined by log-rank test.
Both device cisplatin and IV cisplatin resulted in significant tumor growth inhibition compared to controls in the SUM149 and T11 models (Fig. 4C). Device + IV cisplatin, however, outperformed device cisplatin and IV cisplatin in both tumor models. Device cisplatin, IV cisplatin, and device + IV cisplatin extended the life span from a median of 49 days without treatment to 60, 68, and >100 days, respectively, in the SUM149 model. Device cisplatin, IV cisplatin, and device + IV cisplatin extended the life span from a median of 10 days without treatment to 20, 22, and 32 days, respectively, in the T11 model (Fig. 4C).
Device cisplatin was better tolerated compared to IV cisplatin and device + IV cisplatin as shown by the staining of kidneys for a molecular marker of DNA damage and repair, γH2AX, and platinum-DNA adducts (fig. S7). In addition, histological samples from tumors revealed almost equivalent γH2AX staining after treatment with device cisplatin and IV cisplatin and significantly greater γH2AX staining in device + IV cisplatin–treated mice (Fig. 4D).
We next determined whether the addition of radiotherapy to the device delivery of cisplatin could improve the chemotherapeutic effect of device + IV cisplatin. Mice bearing orthotopic T11 tumors received a single dose of radiation (10 Gy); device cisplatin; device cisplatin + radiation (10 Gy); IV cisplatin (5 mg/kg); IV cisplatin (5 mg/kg) + radiation (10 Gy); device + IV cisplatin (5 mg/kg); or device cisplatin + IV cisplatin (5 mg/kg) + radiation (10 Gy) 5 days after tumor inoculation (~20 mm3). There were three major cohorts of response (Fig. 4E). Mice treated with a single dose of radiation, device cisplatin, or IV cisplatin showed similar tumor growth rates and survival (~17 days), which were significantly improved compared with control animals that did not receive treatment. Mice treated with a single dose of combination therapy—device cisplatin + radiation, IV cisplatin + radiation, or device + IV cisplatin—also showed similar tumor growth rates and survival (~23 days).
Device + IV cisplatin + radiation outperformed all other treatment groups in tumor growth inhibition and survival (~26 days) (P < 0.0002, log-rank test). Tumor growth inhibition in the device + IV + radiation group was not significant compared to the other groups [P = 0.084, analysis of variance (ANOVA)]. The discrepancy between the mice treated with radiation and without radiation is a result of the treatment schedule. In the nonradiation T11 study, we treated the mice twice to establish a difference in effect. For the radiation T11 study, we treated the mice only once because of the radiation dose administered. Overall, the addition of radiation significantly improved survival in the T11 breast tumor mouse model compared with the other device cisplatin groups (P < 0.0001, log-rank test).
PK of iontophoretic delivery in dogs
To further evaluate the implantable iontophoretic device, we used a non–tumor-bearing dog model. A laparotomy was performed to expose the pancreas, and a device (Fig. 5A) was sutured directly onto the pancreas. A constant current of 10 mA was applied for 60 min using either 40 or 10 mg/ml gemcitabine. For the IV treatment arm, the human dose of 1 g/m2 was administered for 30 min.
Fig. 5. Evaluation of single device treatments in dogs.
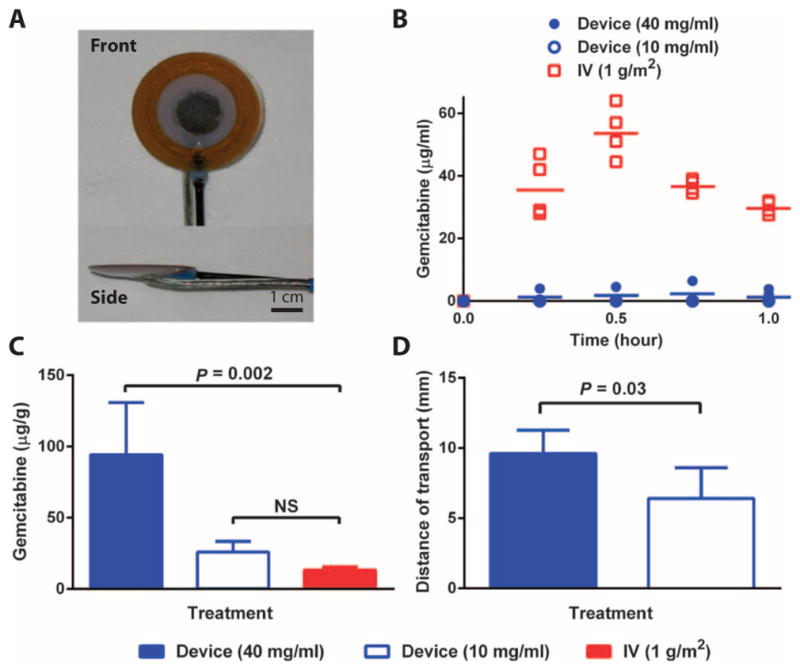
(A) Device to be implanted directly onto the canine pancreas. (B) Plasma PK of gemcitabine during the single device (10 or 40 mg/ml) or IV (1 g/m2) treatment. (C) Organs were removed 1 hour after the initiation of treatment, and gemcitabine content was quantified in the pancreas of dogs after the administration of a single treatment. (D) Distance of gemcitabine transport away from the device and into the pancreatic tissue. Data are means ± SD (n = 5). P values were determined by Wilcoxon rank sum tests.
There was no drug detected in the plasma of the animals when a low gemcitabine concentration (10 mg/ml) was used in the device treatment (Fig. 5B). In the plasma of dogs treated with the high gemcitabine concentration (40 mg/ml) delivered by the device, two of five dogs had detectable levels of gemcitabine, but the levels of gemcitabine detected were more than 25-fold lower than the IV treatment. There was a higher concentration of gemcitabine in the pancreas after device delivery of 40 mg/ml gemcitabine versus IV and 10 mg/ml gemcitabine (Fig. 5C). The distances of gemcitabine transport after device treatment using high and low gemcitabine concentrations were statistically different at 9.6 and 6.3 mm away from the electrode, respectively (Fig. 5D).
DISCUSSION
Local delivery of chemotherapies could have a revolutionary impact on the treatment of cancer by maximizing the effect at the target site and sparing off-target tissue toxicities (11). Yet, the translation of the local drug delivery technologies from basic research into everyday cancer treatments has remained elusive. We decided to take an iontophoretic approach to improve local delivery and drug penetration into the tumor. Iontophoresis is capable of overcoming considerable flow and pressure gradients, particularly those found in solid tumors (16). In addition, a large number of small molecule drugs are capable of being delivered by iontophoresis (31–34).
Here, we show that iontophoretic devices can deliver substantial amounts of drugs to the tumor site of interest with limited systemic exposure in mice and in dogs. Our preclinical results suggest that device delivery of gemcitabine and cisplatin may potentiate the current treatment of solid tumors by enhancing the therapeutic index of the drugs. In addition, the coadministration of systemic therapy, device therapy, and radiotherapy proved feasible and significantly improved tumor growth inhibition and survival in breast cancer models. These studies showcase the versatility of iontophoresis, building on the work of Di Stasi et al. (16).
Surgery remains the optimal therapy for most solid tumors, yet only 10 to 15% of pancreatic cancer patients, 20% of hepatocellular carcinoma patients, 28% of non–small cell lung cancer patients, 45% of gastric cancer patients, and 15 to 20% of esophageal cancer patients undergo curative resection, because the tumor has either invaded critical vessels or metastasized to more distant organs (35–40). In addition, tumor resection—even when successful—can negatively affect quality of life owing to disfiguration or high-risk operations. Radiation therapy is used in combination with surgery; however, it can also have major side effects, including skin reactions, fatigue, poor surgical wound healing, and secondary malignancies (41). Our device has the potential to be an adjunct to surgery, which is supported by the significant tumor regression in the PDX model of pancreatic cancer achieved by device gemcitabine treatments. The device’s ability to increase intratumoral drug concentrations well above current methods of drug administration while maintaining low systemic exposure may increase rates of tumor regression and margin-negative surgical resection and thus improve long-term patient outcomes.
For patients with metastatic disease and debilitating local symptoms such as pain due to nerve involvement or obstruction, an iontophoretic, local delivery device could provide a modality for palliative care. Moreover, this therapy could be used as an adjunct treatment to systemically administered therapy to better treat the primary tumors while systemic therapies concomitantly treat metastatic disease; our data in mice revealed a reduction in tumor growth when the device treatment was combined with IV treatments. One tremendous advantage would be to leverage our device for the delivery of agents that are limited by systemic toxicity. For example, FOLFIRINOX is a promising cytotoxic combination but with limited utility in many patients because of its high level of systemic toxicity (42). The combination iontophoretic delivery of two drugs (5-fluorouracil and leucovorin) from the FOLFIRINOX regimen in healthy pig buccal tissue has shown potential for treatment of head and neck cancer (34).
Although our iontophoretic devices improved local drug transport compared with IV routes, their efficacy may be further improved by potentially altering the dosing schedule, changing the electrode material, optimizing drug formulations, and configuring the device according to anatomy. Because of the high drug concentrations in the tumor and low level of systemic exposure after a single treatment, the number of device treatments could be increased. Because Ag/AgCl electrodes are more efficient in drug delivery and minimize pH changes in the drug solution, these electrodes should be investigated for human studies. Drug formulation plays an important role in the iontophoretic delivery of drugs through competing ions and electroosmotic flow, and the evaluation of formulation excipients could improve total drug delivered and distance of drug transport (15). Configuring these devices to fit the shape of the tumor and surrounding anatomy could improve efficacy by creating better contact between the device and tumor tissue and, in the case of pancreatic cancer, strategically delivering the cytotoxic agents to the area that would enable tumor resection. For the device treatment of breast cancer, the skin acts as a barrier reducing drug transport; thus, placing the device subcutaneously would increase the amount of drug delivered.
Some key parameters that need to be further understood for translation are safe and tolerated current densities at the device-tumor interface in humans and iontophoretic drug transport in human solid tumors. These data will build on the large amount of research about drug transport through the layers of the skin, using microneedle patches and transdermal iontophoretic devices (14). In a similar fashion to the scale-up of devices from our mouse to dog studies, the treatments will need to be scaled to address larger human tumors. Evaluation of toxicity in large animals, such as dogs, is a necessary step in the progression of the device toward clinical trials. There are several devices guiding our path forward with the U.S. Food and Drug Administration, including an iontophoretic Foley catheter by Physion EMDA, with indications for bladder cancer and Peyronie’s disease, and the InfusAid Pump for intrahepatic artery injection of chemotherapeutic agents, among others (43, 44).
Our iontophoretic device technology may be co-opted for several clinical applications, including as an adjunct to surgery or to systemic therapy, similar to the current practice of using radiation therapy in the setting of metastatic cancer. Looking beyond pancreatic and breast cancers, this iontophoretic approach can be applied to a variety of other cancers, including head and neck cancer, sarcomas, and colorectal and gastric cancers. The device could be extended to the delivery of a multiplicity of drugs to improve primary tumor resection, preserve organ function, and improve quality of life for patients. Overall, our devices could potentially offer entirely new modalities for the treatment of cancer under the emerging field of interventional oncology.
MATERIALS AND METHODS
Study design
The aim of this study was to develop iontophoretic devices for the local treatment of solid tumors. It was hypothesized that iontophoretic devices could be used to deliver chemotherapies directly to tumors, overcoming major barriers of drug efficacy. In vitro work was first performed to optimize and test the iontophoretic delivery of gemcitabine and cisplatin into tissues. For PK and efficacy studies, mice were randomized into the experimental and control groups. Tumor measurements for the efficacy studies were performed by blinded animal handlers from the University of North Carolina (UNC) Animal Studies Core Facility. The study endpoint for efficacy in the mouse model of pancreatic cancer was an interval of 7 weeks; the study endpoint for efficacy in the mouse model of breast cancer was time to tumor progression to 2.0 cm in one dimension. For histological analysis, tumors were annotated, and the number of positively stained cells according to stain intensity was quantified. Finally, we tested these devices in randomized dogs to highlight the translatability of the technology; for these studies, power analysis involving Wilcoxon rank sum tests was used to obtain the sample sizes needed. No outlying data points were excluded in the studies.
Device fabrication
Iterative device design was performed using a three-dimensional computer-aided design software (SolidWorks). For the long-term implanted murine devices, a 5-mm platinum (Strem Chemicals) disc was soldered to a stainless steel cable wire and embedded in a polyurethane (Hapco Steralloy 2056) reservoir before cross-linking. A steel ring was also embedded in the polyurethane reservoir to act as an anchor for suturing the device onto the tumor. The steel wire was then threaded through the multiluminal tubing, and the tubing-reservoir interface was encased in heat-shrink tubing. A semipermeable 14K cellulose membrane (Fisher Scientific) was adhered to the reservoir for enclosure. For the short-term murine transdermal devices, an 8.6-mm silver disc was soldered to a 36-gauge copper wire and embedded in a PDMS (Sylgard 184) reservoir before cross-linking. The copper wire was threaded through a multiluminal tube, and the tube was inserted into the reservoir for inflow of drug. Another multiluminal tube was inserted on the contralateral side of the reservoir for drug efflux. For the short-term implanted canine devices, a 1-cm silver (Fisher Scientific) circular disc was soldered to an insulated copper conducting wire and embedded in a polyethylene reservoir covered by a 14K semi-permeable cellulose membrane. A dual lumen tube was inserted into the reservoir for drug flow into and out of the reservoir.
Animal studies
All procedures were approved by the appropriate Institutional Animal Care and Use Committee before initiation. All animals used in PK, biodistribution, and efficacy studies were allowed to acclimate for at least 1 week to the animal facilities before experimentation. Animals were exposed to a 12-hour light/dark cycle and received food and water ad libitum through the studies. Each tissue required a different current density for functionality and safety of the device treatment. For quantitation of gemcitabine and cisplatin plasma concentrations, blood samples were collected into heparinized tubes, and plasma was generated and frozen with liquid nitrogen; tissue and device were extracted and snap-frozen with liquid nitrogen and stored at −80°C. Total gemcitabine was extracted from frozen plasma and organs with a protein precipitation method and analyzed by ultraviolet–high-performance liquid chromatography (UV-HPLC) (45). Total platinum was extracted using a nitric acid degradation method and analyzed by ICP-MS (46).
Ex vivo studies
Pancreatic cancer PDXs
De-identified tumors of pancreatic cancer patients were grafted and passaged in athymic nude mice as described previously (18, 19). Upon reaching ~200 mm3, the tumors were directly removed and placed into petri dishes with 2 ml of saline. Devices were sutured directly onto the tumor using 6-0 nylon, followed by either no current (control) or 2 mA of current (current density, 4 mA/cm2) for 10 min with a drug flow of 50 μl/min into the device. After treatment, the tumors were snap-frozen and sectioned using a cryostat microtome. The sections were analyzed by UV-HPLC.
Human skin
De-identified human skin was provided by the cooperative human tissue network. The frozen skin was thawed and sectioned into 2 cm × 2 cm squares before drug transport studies. The transport of cisplatin in skin was evaluated using a modified Franz diffusion cell with the device directly above the skin. The receptor chamber was filled with 5 ml of saline. No current or 1 mA of current (current density, 0.5 mA/cm2) was applied for 25 min, and the skin and solution were snap-frozen, processed, and analyzed by ICP-MS.
PK studies
PKs were evaluated in pancreatic cancer PDXs in mice, SUM149 orthotopic xenografts in mice, and in healthy dogs, as described in Supplementary Methods.
Efficacy studies
Pancreatic cancer PDXs
The technical protocols for surgical implantation of the device and device treatments were identical to the PK studies. Mice were treated biweekly for 7 weeks using device gemcitabine (20 mg/ml), device saline (0.9% NaCl), IV saline, or IV gemcitabine (80 mg/kg).
SUM149 orthotopic xenograft and T11 orthotopic syngeneic models
The technical protocol for device treatments was identical to the PK studies. Mice were treated two or four times per week using device cisplatin, IV cisplatin (5 mg/kg), device + IV (5 mg/kg) cisplatin, device saline (0.9% NaCl), or IV saline. Tumors derived from BALB/c TP53−/−orthotopic mammary gland transplant line (T11) were passage in BALB/c wild-type mice by subcutaneous injection of 500,000 cells re-suspended in Matrigel (50:50) into the left inguinal mammary gland. The day before the scheduled treatment, the mice were shorn with clippers, and the residual hair was removed using Nair. For the radiation studies, at 2 hours after injection, the tumors were subjected to radiation of 10 Gy with XRAD 320. Mice were shielded with a lead shield to reduce irradiation of other organs.
PK and statistical analyses
Data are expressed as means ± SD. Graphs were created with GraphPad Prism software. All analyses were done using SAS v9.3. PK parameters were assessed with Phoenix WinNonLin (version 6.0). ANOVA methods were used for comparisons of continuous values between groups. Unpaired t tests were used when an overall difference was detected. Unadjusted P values were reported for pairwise comparisons when an overall difference was detected. For the breast cancer efficacy studies, comparisons were made between groups at two time points: (i) when all mice were still alive (days 28, 9, and 9) and (ii) when all mice on treatment were still alive (days 42, 16, and 15). For survival data, the Kaplan-Meier method and log-rank tests were used for comparisons between groups. To compare the distribution in absolute mass of gemcitabine per mass of tissue and distance of transport between different pairs of methods, Wilcoxon rank sum tests were used and exact nominal P values were reported.
Supplementary Material
Acknowledgments
We dedicate this paper to the memory of G. W. Roberts, Professor Emeritus of Chemical and Biomolecular Engineering at North Carolina State University, an esteemed collaborator and dear friend who succumbed to pancreatic cancer during these research efforts. We would like to thank X. Wang, C. Santos, S. G. Herrera-Loeza, UNC Animal Studies Core, PDX Program, the Tissue Procurement Facility, the Translational Pathology Laboratory, and the Center for Gastrointestinal Biology Disease Histology Core for their contributions to this work.
Funding: The University Cancer Research Fund, Synecor LLC, NIH Pioneer Award, UNC Lineberger Comprehensive Cancer Center Postdoctoral Training Grant T32-CA009156, and NIH R01CA178748–01. J.D.B. was supported by a National Defense Science and Engineering Graduate Fellowship, UNC Medical Scientists Training Program NIGMS-2-T32-GM008719, and PhRMA Foundation Fellowship.
Footnotes
www.sciencetranslationalmedicine.org/cgi/content/full/7/273/273ra14/DC1
Materials and Methods
Fig. S1. Gemcitabine transport into PDX tumors after a single device treatment.
Fig. S2. Drug transport through freshly excised murine skin.
Fig. S3. Role of gemcitabine concentration in drug transport in vitro.
Fig. S4. PK of cisplatin delivered transdermally by iontophoretic devices.
Fig. S5. Body weight changes in response to chemotherapy.
Fig. S6. Change in laboratory values after the device gemcitabine treatment schedule.
Fig. S7. Short-term renal toxicity after a single cisplatin treatment in mice.
Author contributions: J.D.B., A.T.O., L.R.B., M.N., and C.R.B. designed and fabricated the devices. J.D.B., M.R.N.J., A.T.O., L.R.B., and A.W.K. performed the in vitro studies. J.D.B., M.R.N.J., A.T.O., R.E.L., A.W.K., and W.L. completed the PK and device efficacy studies in mice and analytics. N.H. and K.W. assisted with the radiation treatments. J.D.B., A.T.O., and C.S. performed the large animal studies. J.D.B., M.R.N.J., and A.D. completed the statistical analysis of the data. R.A.M., J.C.L., M.E.N., D.D., C.K.A., R.S., J.E.T., A.Z.W., W.C.Z., J.J.Y., and J.M.D. consulted, analyzed, and interpreted the data. J.D.B., M.R.N.J., A.T.O., L.R.B., J.J.Y., and J.M.D. wrote the manuscript.
Competing interests: J.D.B., M.E.N., J.J.Y., and J.M.D. have filed one patent related to the device PCT/US2010/025416. All other authors declare that they have no competing interests or patents to disclose.
Data and materials availability: Materials are available and can be provided under the material transfer policies of UNC.
REFERENCES AND NOTES
- 1.Goodman LS, Wintrobe MM, Dameshek W, Goodman MJ, Gilman MA, McLennan MT. Nitrogen mustard therapy; use of methyl-bis (β-chloroethyl) amine hydrochloride and tris (β-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J Am Med Assoc. 1946;132:126–132. doi: 10.1001/jama.1946.02870380008004. [DOI] [PubMed] [Google Scholar]
- 2.Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, Beger H, Fernandez-Cruz L, Dervenis C, Lacaine F, Falconi M, Pederzoli P, Pap A, Spooner D, Kerr DJ, Büchler; MW European Study Group for Pancreatic Cancer. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350:1200–1210. doi: 10.1056/NEJMoa032295. [DOI] [PubMed] [Google Scholar]
- 3.Sauer R, Becker H, Hohenberger W, Rödel C, Wittekind C, Fietkau R, Martus P, Tschmelitsch J, Hager E, Hess CF, Karstens JH, Liersch T, Schmidberger H, Raab; R German Rectal Cancer Study Group. Preoperative versus postoperative chemoradiotherapy for rectal cancer. N Engl J Med. 2004;351:1731–1740. doi: 10.1056/NEJMoa040694. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham D, Allum WH, Stenning SP, Thompson JN, Van de Velde CJ, Nicolson M, Scarffe JH, Lofts FJ, Falk SJ, Iveson TJ, Smith DB, Langley RE, Verma M, Weeden S, Chua; YJ MAGIC Trial Participants. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med. 2006;355:11–20. doi: 10.1056/NEJMoa055531. [DOI] [PubMed] [Google Scholar]
- 5.Cunningham D, Starling N, Rao S, Iveson T, Nicolson M, Coxon F, Middleton G, Daniel F, Oates J, Norman; AR Upper Gastrointestinal Clinical Studies Group of the National Cancer Research Institute of the United Kingdom. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med. 2008;358:36–46. doi: 10.1056/NEJMoa073149. [DOI] [PubMed] [Google Scholar]
- 6.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 7.Clark MA, Fisher C, Judson I, Thomas JM. Soft-tissue sarcomas in adults. N Engl J Med. 2005;353:701–711. doi: 10.1056/NEJMra041866. [DOI] [PubMed] [Google Scholar]
- 8.Tunggal JK, Cowan DSM, Shaikh H, Tannock IF. Penetration of anticancer drugs through solid tissue: A factor that limits the effectiveness of chemotherapy for solid tumors. Clin Cancer Res. 1999;5:1583–1586. [PubMed] [Google Scholar]
- 9.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Rückert F, Grützmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minchinton AI, Tannock IF. Drug penetration in solid tumors. Nat Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 11.Tannock IF, Lee CM, Tunggal JK, Cowan DS, Egorin MJ. Limited penetration of anticancer drugs through tumor tissue: A potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res. 2002;8:878–884. [PubMed] [Google Scholar]
- 12.Wolinsky JB, Colson YL, Grinstaff MW. Local drug delivery strategies for cancer treatment: Gels, nanoparticles, polymeric films, rods, and wafer. J Control Release. 2012;159:14–26. doi: 10.1016/j.jconrel.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fung LK, Ewend MG, Sills A, Sipos EP, Thompson R, Watts M, Colvin OM, Brem H, Saltzman WM. Pharmacokinetics of interstitial delivery of carmustine, 4-hydroperoxycyclophosphamide and paclitaxel from a biodegradable polymer implant in the monkey brain. Cancer Res. 1998;58:672–684. [PubMed] [Google Scholar]
- 14.Kalia YN, Naik A, Garrison J, Guy RH. Iontophoretic drug delivery. Adv Drug Deliv Rev. 2004;56:619–658. doi: 10.1016/j.addr.2003.10.026. [DOI] [PubMed] [Google Scholar]
- 15.Merino V, López A, Kalia YN, Guy RH. Electrorepulsion versus electroosmosis: Effect of pH on the iontophoretic flux of 5-fluorouracil. Pharm Res. 1999;16:758–761. doi: 10.1023/a:1018841111922. [DOI] [PubMed] [Google Scholar]
- 16.Di Stasi SM, Valenti M, Verri C, Liberati E, Giurioli A, Leprini G, Masedu F, Ricci AR, Micali F, Vespasiani G. Electromotive instillation of mitomycin immediately before transurethral resection for patients with primary urothelial non-muscle invasive bladder cancer: A randomised controlled trial. Lancet Oncol. 2011;12:871–879. doi: 10.1016/S1470-2045(11)70190-5. [DOI] [PubMed] [Google Scholar]
- 17.Cohen AE, Assang C, Patane MA, From S, Korenfeld M Avion Study Investigators. Evaluation of dexamethasone phosphate delivered by ocular iontophoresis for treating non-infectious anterior uveitis. Ophthalmology. 2012;119:66–73. doi: 10.1016/j.ophtha.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 18.Torphy RJ, Tignanelli CJ, Kamande JW, Moffitt RA, Herrera Loeza SG, Soper SA, Yeh JJ. Circulating tumor cells as a biomarker of response to treatment in patient-derived xenograft mouse models of pancreatic adenocarcinoma. PLOS One. 2014;9:e89474. doi: 10.1371/journal.pone.0089474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neel NF, Stratford JK, Shinde V, Ecsedy JA, Martin TD, Der CJ, Yeh JJ. Response to MLN8237 in pancreatic cancer is not dependent on RalA phosphorylation. Mol Cancer Ther. 2014;13:122–133. doi: 10.1158/1535-7163.MCT-12-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Usary J, Zhao W, Darr D, Roberts PJ, Liu M, Balletta L, Karginova O, Jordan J, Combest A, Bridges A, Prat A, Cheang MCU, Herschkowitz JI, Rosen JM, Zamboni W, Sharpless NE, Perou CM. Predicting drug responsiveness in human cancers using genetically engineered mice. Clin Cancer Res. 2013;19:4889–4899. doi: 10.1158/1078-0432.CCR-13-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang D, LaFortune TA, Krishnamurthy S, Esteva FJ, Cristofanilli M, Liu P, Lucci A, Singh B, Hung MC, Hortobagyi GN, Ueno NT. Epidermal growth factor receptor tyrosine kinase inhibitor reverses mesenchymal to epithelial phenotype and inhibits metastasis in inflammatory breast cancer. Clin Cancer Res. 2009;15:6639–6648. doi: 10.1158/1078-0432.CCR-09-0951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 23.Smoot RL, Koch CA, Degnim AC, Sterioff S, Donohue JH, Grant CS, Barnes SA, Gullerud RE, Hobday TJ, Farley DR. A single-center experience with inflammatory breast cancer, 1985–2003. Arch Surg. 2006;141:567–572. doi: 10.1001/archsurg.141.6.567. [DOI] [PubMed] [Google Scholar]
- 24.Fichtner I, Rolff J, Soong R, Hoffmann J, Hammer S, Sommer A, Becker M, Merk J. Establishment of patient-derived non–small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin Cancer Res. 2008;14:6456–6468. doi: 10.1158/1078-0432.CCR-08-0138. [DOI] [PubMed] [Google Scholar]
- 25.Kimmelman AC, Hezel AF, Aguirre AJ, Zheng H, Paik JH, Ying H, Chu GC, Zhang JX, Sahin E, Yeo G, Ponugoti A, Nabioullin R, Deroo S, Yang S, Wang X, McGrath JP, Protopopova M, Ivanova E, Zhang J, Feng B, Tsao MS, Redston M, Protopopov A, Xiao Y, Futreal PA, Hahn WC, Klimstra DS, Chin L, DePinho RA. Genomic alterations link Rho family of GTPases to the highly invasive phenotype of pancreas cancer. Proc Natl Acad Sci USA. 2008;105:19372–19377. doi: 10.1073/pnas.0809966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, Shi C, Danenberg K, Danenberg PV, Kuramochi H, Tanaka K, Singh S, Salimi-Moosavi H, Bouraoud N, Amador ML, Altiok S, Kulesza P, Yeo C, Messersmith W, Eshleman J, Hruban RH, Maitra A, Hidalgo M. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res. 2006;12:4652–4661. doi: 10.1158/1078-0432.CCR-06-0113. [DOI] [PubMed] [Google Scholar]
- 27.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Patient-derived tumor xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh M, Lima A, Molina R, Hamilton P, Clermont AC, Devasthali V, Thompson JD, Cheng JH, Bou Reslan H, Ho CC, Cao TC, Lee CV, Nannini MA, Fuh G, Carano RA, Koeppen H, Yu RX, Forrest WF, Plowman GD, Johnson L. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol. 2010;28:585–593. doi: 10.1038/nbt.1640. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Li M, Rinehart JJ, Zhang R. Pretreatment with dexamethasone increases anti-tumor activity of carboplatin and gemcitabine in mice bearing human cancer xenografts: In vivo activity, pharmacokinetics, and clinical implications for cancer chemotherapy. Clin Cancer Res. 2004;10:1633–1644. doi: 10.1158/1078-0432.ccr-0829-3. [DOI] [PubMed] [Google Scholar]
- 30.Morfouace M, Shelat A, Jacus M, Freeman BB, III, Turner D, Robinson S, Zindy F, Wang YD, Finkelstein D, Ayrault O, Bihannic L, Puget S, Li XN, Olson JM, Robinson GW, Guy RK, Stewart CF, Gajjar A, Roussel MF. Pemetrexed and gemcitabine as combination therapy for the treatment of group3 medulloblastoma. Cancer Cell. 2014;25:516–529. doi: 10.1016/j.ccr.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komuro M, Suzuki K, Kanebako M, Kawahara T, Otoi T, Kitazato K, Inagi T, Makino K, Toi M, Terada H. Novel iontophoretic administration method for local therapy of breast cancer. J Control Release. 2013;168:298–306. doi: 10.1016/j.jconrel.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 32.Chang BK, Guthrie TH, Jr, Hayakawa K, Gangarosa LP., Sr A pilot study of iontophoretic cisplatin chemotherapy of basal and squamous cell carcinomas of the skin. Arch Dermatol. 1993;129:425–427. [PubMed] [Google Scholar]
- 33.Welch ML, Grabski WJ, McCollough ML, Skelton HG, Smith KJ, Menon PA, Anderson LL. 5-Fluorouracil iontophoretic therapy for Bowen’s disease. J Am Acad Dermatol. 1997;36:956–958. doi: 10.1016/s0190-9622(97)80280-0. [DOI] [PubMed] [Google Scholar]
- 34.Gratieri T, Kalia YN. Targeted local simultaneous iontophoresis of chemotherapies for topical therapy of head and neck cancers. Int J Pharm. 2014;460:24–27. doi: 10.1016/j.ijpharm.2013.10.053. [DOI] [PubMed] [Google Scholar]
- 35.Ferrone CR, Brennan MF, Gonen M, Coit DG, Fong Y, Chung S, Tang L, Klimstra D, Allen PJ. Pancreatic adenocarcinoma: The actual 5-year survivors. J Gastrointest Surg. 2008;12:701–706. doi: 10.1007/s11605-007-0384-8. [DOI] [PubMed] [Google Scholar]
- 36.Gillen S, Schuster T, Meyer Zum Büschenfelde C, Friess H, Kleeff J. Preoperative/neoadjuvant therapy in pancreatic cancer: A systematic review and meta-analysis of response and resection percentages. PLOS Med. 2010;7:e1000267. doi: 10.1371/journal.pmed.1000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berry MF. Esophageal cancer: Staging system and guidelines for staging and treatment. J Thorac Dis. 2014;6:S289–S297. doi: 10.3978/j.issn.2072-1439.2014.03.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin S, Hoffmann K, Schemmer P. Treatment of hepatocellular carcinoma: A systematic review. Liver Cancer. 2012;1:144–158. doi: 10.1159/000343828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laroche C, Wells F, Coulden R, Stewart S, Goddard M, Lowry E, Price A, Gilligan D. Improving surgical resection rate in lung cancer. Thorax. 1998;53:445–449. doi: 10.1136/thx.53.6.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Msika S, Tazi MA, Benhamiche AM, Couillault C, Harb M, Faivre J. Population-based study of diagnosis treatment and prognosis of gastric cancer. Br J Surg. 1997;84:1474–1478. [PubMed] [Google Scholar]
- 41.Suit H, Goldberg S, Niemierko A, Ancukiewicz M, Hall E, Goitein M, Wong W, Paganetti H. Secondary carcinogenesis in patients treated with radiation: A review of data on radiation-induced cancers in human, non-human primate, canine and rodent subjects. Radiat Res. 2007;167:12–42. doi: 10.1667/RR0527.1. [DOI] [PubMed] [Google Scholar]
- 42.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, Bennouna J, Bachet JB, Khemissa-Akouz F, Péré-Vergé D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M Groupe Tumeurs Digestives of Unicancer; PRODIGE Intergroup, FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 43.Rose AE, Azevedo KJ, Payne CK. Office bladder distention with Electromotive Drug Administration (EMDA) is equivalent to distention under General Anesthesia (GA) BMC Urol. 2005;5:14. doi: 10.1186/1471-2490-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kemeny N, Daly J, Reichman B, Geller N, Botet J, Oderman P. Intrahepatic or systemic infusion of fluorodeoxyuridine in patients with liver metastases from colorectal carcinoma. A randomized trial. Ann Intern Med. 1987;107:459–465. doi: 10.7326/0003-4819-107-4-459. [DOI] [PubMed] [Google Scholar]
- 45.Kirstein MN, Hassan I, Guire DE, Weller DR, Dagit JW, Fisher JE, Remmel RP. High performance liquid chromatographic method for the determination of gemcitabine and 2′,2′-difluorodeoxyuridine in plasma and tissue culture media. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;835:136–142. doi: 10.1016/j.jchromb.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 46.Caron WP, Lay JC, Fong AM, La-Beck NM, Kumar P, Newman SE, Zhou H, Monaco JH, Clarke-Pearson DL, Brewster WR, Van Le L, Bae-Jump VL, Gehrig PA, Zamboni WC. Translational studies of phenotypic probes for the mononuclear phagocyte system and liposomal pharmacology. J Pharmacol Exp Ther. 2013;347:599–606. doi: 10.1124/jpet.113.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.