

Abstract
With the recent advances in the development of novel protein based therapeutics, controlled delivery of these biologics is an important area of research. Herein, we report the synthesis of microparticles from bovine serum albumin (BSA) as a model protein using Particle Replication in Non-wetting Templates (PRINT) with specific size and shape. These particles were functionalized at room temperature using multifunctional chlorosilane that cross-link the particles to render them to slowly-dissolving in aqueous media. Mass spectrometric study of the reaction products of diisopropyldichlorosilane with individual components of the particles revealed that they are capable of reacting and forming cross-links. Energy dispersive spectroscopy (EDS) and X-ray photoelectron spectroscopy (XPS) were also used to confirm the functionalization of the particles. Cross sectional analysis using focused ion beam (FIB) and EDS proved that the functionalization occurs throughout the bulk of the particles and is not just limited to the surface. Circular dichroism data confirmed that the fraction of BSA molecules released from the particles retains its secondary structure thereby indicating that the system can be used for delivering protein based formulations while controlling the dissolution kinetics.
Keywords: silylation, protein delivery, controlled release, functionalization, bovine serum albumin, diisopropyldichlorosilane
INTRODUCTION
Controlled delivery of protein based formulations to the site of interest is of high practical importance, due to the development of novel biomolecules with therapeutic potential.1 With the recent advances in biotechnology, monoclonal antibodies (mAb) are also being used as protein based therapeutic agents for treating different diseases.2 However, low bioavailability of these molecules makes their systemic delivery through the oral route quite challenging. This can be attributed to large molecular weight and hydrophilicity of these macromolecules that makes their absorption through biological membranes difficult.3,4 The ability to protect the active compound from the harsh conditions of the gastrointestinal tract and enzymatic barrier are other challenges in oral delivery of proteins, since the delicate structure of these molecules is prone to different modes of instabilities.4,5 Due to less than 1% oral bioavailability of the initial dose, proteins are normally administered by either injection or infusion in aqueous solutions.4,6–8 Local delivery to a specific site using intratumoral,9,10 intravitreal,11 epidural,12 and intra-articular13 routes is desirable as it significantly reduces the therapeutic drug dose and adverse side effects associated with the undesirable distribution in various tissues and organs.14 Other noninvasive alternative routes suffer from the fact that, in the absence of a membrane permeation enhancer, the absorption is much less than that of parenteral administration while toxicity is a major drawback of using these enhancers in the formulation.14,15
According to Almeida and Souto,1 the established method of protein delivery consists of attaching the drugs to a proper particulate carrier, where the fate of the drug molecule is determined by the properties of the carrier rather than those of the protein. Therefore, use of an appropriate delivery vehicle that can protect proteins from enzymatic degradation and control the release rate is a promising approach for prolonged retention while saving the biological activity of the drug.16
There are several studies that address different systems for protein/antibody delivery. Lee and Yuk16 review both polymeric hydrogel carriers and liposomal systems for delivery of proteins. One of the obstacles in the development of a suitable polymeric carrier for protein drugs is low drug loading.10 A possible solution to this problem is preparation of particles that are made from a protein based carrier instead of a polymer. Several researchers have tried to prepare particulate systems that do not rely on a carrier polymer for delivering the protein. Yang et al.17 have developed methods to produce crystalline suspensions of approved therapeutic mAbs. Miller et al.18 studied highly concentrated suspensions of lysozyme powder in a nonaqueous mixture of safflower oil and benzyl benzoate. α-Amylase crystals have been suspended in sucrose acetate isobutyrate plasticized with ethanol as a potential in situ formable protein depot.6
Preparation of cross-linked albumin nano/microparticles has been reported in the literature using suspension technology,19,20 layer-by-layer assembly,21,22 spray-drying,23 and desolvation/coacervation.24,25 These methods rely on heat treatment for self-cross-linking through formation of interchain amide links or chemical treatment with a stabilizer such as glutaraldehyde (GA) and butanedione to confer dimensional stability. GA reacts with amino groups of the protein while butanedione reacts with the guanidinyl moiety of arginine residue to form uncleavable chemical cross-links. In addition to the irreversible cross-linking reaction of these stabilizers, the shapes of the fabricated particles are limited to spheres, capsules, and rods. Bogdansky26 gives a detailed insight into the use of albumin based particles for therapeutic applications. The advantage of the albumin based particulate system is the lack of toxicity and degradation into natural products. Albumin based particles containing proteins have been examined as potential drug delivery systems by other researchers. Urokinase has been immobilized in GA cross-linked bovine serum albumin (BSA) particles by Bhargava and Ando,27 and GA cross-linked insulin-BSA microbeads were prepared by Goosen et al.28 Human serum albumin (HSA) nanocapsules were prepared by Rollett et al.29 for targeted drug delivery to activated macrophages. Application of albumin microspheres (3–4 μm) to pro-inflammatory cytokine inhibition was reviewed by Oettinger and D’Souza,30 and versatility of albumin as a drug carrier was evaluated by Kratz.31 Doshi and Mitragotri32 demonstrated that both size and shape of polymeric particles critically control their elimination by macrophages. Therefore, precise control over size and shape is desirable for controlling the clearance time of the particles upon local delivery.
According to Lee et al.,33 injectable beads prepared by cross-linking BSA under mild conditions using GA should yield a nonimmunogenic and biodegradable device for drug delivery and have been used for sustained release of progesterone. Furthermore, Ratcliffe et al.34 tested the in vivo biocompatibility of poly(lactic acid), poly(butyl cyanoacrylate), gelatin, and GA cross-linked homologous albumin microspheres (1.9 ± 1.5 μm) with rabbit’s synovial tissues upon intra-articular injection. They concluded that albumin microspheres are the most acceptable polymer among the ones studied. The slight hyperplasia observed by the authors in the synovial membrane was attributed to denaturation of albumin with GA.
Numerous pH cleavable linkers such as acetals, ketals, ortho esters,35,36 bis-pyridoxal polyphosphates,37 and the derivative of 2-methylmaleic anhydride38 have been developed for cross-linking proteins. Silyl ethers have also been successfully used for reversibly cross-linking precision particles made from hydroxyethyl acrylate and controlling the release kinetics based on the pH of the environment and size of the substituents on the silicon atom.39
The objective of this work was to fabricate suspensions of protein particles with controlled size and shape capable of slowly releasing the protein based therapeutic in aqueous media exploiting the pH cleavability of the silyl ether bond in cross-linked particles. This carrier system can be used for local delivery of the biomolecules to the site of interest while maintaining protein concentration within the therapeutic window. The outcome of this work demonstrates the ability to effectively increase the retention time of the protein based therapeutics deliverable through the parenteral route by fine-tuning the silylation reaction and can potentially maximize the efficacy of the drug.
EXPERIMENTAL SECTION
Materials
BSA, diisopropyldichlorosilane (DIDCS), and formic acid (FA) were obtained from Sigma-Aldrich. Extra-dry acetonitrile (ACN), trifluoroacetic acid (TFA), α-D-lactose monohydrate (lactose), glycerol, and extra-dry isopropanol (IPA) were all ACROS products obtained through Fisher Scientific. Standard buffers of pH 7.4 and 10.0, as well as HPLC grade water and acetonitrile, were all obtained through Fisher Scientific. The Alexa Fluor 555 conjugated BSA was purchased from Life Technologies.
Particle Fabrication
Particle Replication in Non-wetting Templates (PRINT) platform has been previously used to fabricate monodisperse, shape-specific albumin particles.40 In this work, PRINT was used to make particles from a mixture of BSA, lactose, and glycerol (40/40/20 wt %) in deionized water (18.2 MΩ·cm). The mixture was rolled and dried into the form of a solid flat sheet on the surface of a poly(ethylene terephthalate) film using Mayer rod on EZ Coater EC-200 (ChemInstruments) which is then used to fill the donut-shaped micron-sized molds. The filling process was carried out using a commercially available hot roll laminator (ChemInstruments) at 414 kPa and 138 °C as measured by Thermax Range C (TMC Hallcrest) temperature indicator films. Lactose was added to the formulation to lower the softening temperature of BSA and to make it moldable in the PRINT process. Glycerol merely serves as a processing aid during the filling stage. The particles were then transferred to a harvesting film made from Luvitec VA 64 (BASF) in a heated laminator (total lamination time of less than 30 s). Particles were freed from the harvesting film by dissolving the adhesive in extra-dry ACN. Due to the fact that the solubility of BSA in ACN is minimal (0.02 mg/mL),41 it was possible to separate these particles from the Luvitec layer through consecutive washing without any recognizable changes to the structural integrity of the particles. Furthermore, the secondary structure of the protein was expected to remain intact in the absence of water when using extra-dry ACN.42
Particle Composition
The composition of particles was determined using a high-performance liquid chromatography (HPLC) instrument (1260 Infinity by Agilent) equipped with an evaporative light scattering detector. The HPLC method used was based on separating BSA from lactose and glycerol using an Ultracel-30K Amicon Ultra centrifugal filter (Millipore) and then running them on two different columns based on reversed phase chromatography using a Poroshell 300SB-C8 column (Agilent) for the BSA in the mixture and ion exchange chromatography using a Hi-Plex Ca column (Agilent) for lactose and glycerol (Table S1, Supporting Information).
Functionalization (Silylation)
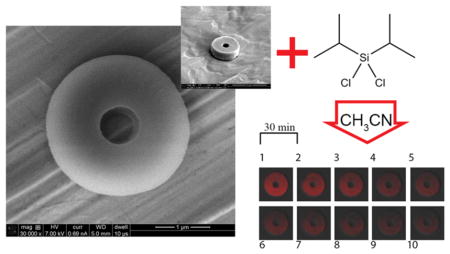
In this work, silylation based on chlorosilanes was carried out on particles after harvesting them in ACN. The mass of the harvested particles was determined using thermogravimetric analysis (TGA Q5000-TA Instruments). Particles (5 mg) were dispersed in ACN (4.7 mL), and then DIDCS (1.27 mL) was added while allowing the reaction (rx) to proceed for 6, 12, and 24 hour (hr) in separate vials. At the end of the treatment with the chlorosilane, the particles were washed with IPA which quenched the reaction at the desired time point and enabled the removal of unreacted DIDCS. The silylation process on the particles and introduction of a silyl group in place of the labile hydrogen atoms upon exposure to DIDCS result in the formation of cross-links (Figure S1, Supporting Information). The amino acid composition of BSA43,44 contains multiple residues with labile hydrogen atoms (e.g., Tyr, Ser) that can serve as an anchor point for attachment of diisopropyl groups. Therefore, it is expected that multiple residues participate in the reaction. The byproduct of the reaction is hydrochloric acid which is expected to behave as a weak acid in pure ACN (acid dissociation constant (pKHA) of 8.9).45
Chemical Characterization
Mass Spectrometry (MS)
To understand the interaction of chlorosilane with the components of the particles, mass spectrometric analysis using electrospray ionization (ESI) was performed on the individual ingredients of the particles. BSA powder was pulverized in an agate pestle and mortar combination and passed through a 400 mesh sieve (38 μm nominal opening) to get a fine dry powder. This powder (20 mg) was then dispersed in extra-dry ACN (12 mL), and DIDCS (5 mL) was later added to the mixture. The silylation reaction was allowed to proceed for 12–24 hr and then neutralized with IPA at the end of the experiment. BSA crystals were then spun down and washed with extra IPA to remove unreacted and physisorbed DIDCS. Intact and silylated samples were all purified and isolated by centrifugal filtration of the aqueous solution through Amicon Ultracel-30K filters. Silylated samples were dialyzed (Slide-A-Lyzer dialysis cassette, Pierce, 20 000 MWCO) overnight in HPLC grade water. Samples (5 mg/mL) were then directly injected into the LTQ-FT mass spectrometer (Thermo Fisher Scientific) and analyzed in positive ion trap mode (600–2000 m/z). Typically, the sample (0.5 mL) was mixed with HPLC grade ACN (1 mL) and FA in HPLC grade water (3 v/v%, 0.5 mL). The instrument was externally calibrated with a resolution setting of 100 000. ProMass (Novatia) software was used for deconvolution of the intact and modified protein spectra to obtain the molecular weights. High baseline removal (1.2) was used in the deconvolution algorithm, and the output range was set to 40 000–100 000 Daltons (Da).
Reaction products of glycerol with DIDCS were analyzed using the same mass spectrometer in Fourier transform (FT) mode. Glycerol (1 mL) was mixed with DIDCS (1 mL), and the reaction was allowed to proceed for 3 days when it was finally neutralized with IPA (3 mL). The reaction mixture was then directly injected to the mass spectrometer without addition of water to prevent desilylation of the reaction products.
EDS/FIB/XPS
The nature of functionalization at the individual particle level was analyzed by energy dispersive spectroscopy (EDS) using INCA (Oxford Instruments) on the FEI Helios 600 Nanolab Dual Beam System which was also used for cross sectional study. Individual particle analysis was done by drying the particles over the surface of copper tape and then coating them with 3 nm gold (Au)/ palladium (Pd) (Cressington sputter coater 108 auto). Three different random particles per each sample were analyzed to quantify the amount of silicon. For cross sectional analysis using focused ion beam (FIB), particles were dried over the surface of a carbon planchet (Electron Microscopy Services) and coated with carbon [10.5 nm] in a high resolution ion beam coater (Gatan 681) to prevent redepositioning of Au/Pd upon cutting the particle at 30.0 kV with 9.7 pA Gallium ion (Ga+) current. Linescans at 5.0 kV were then performed on the particle to measure the variation of elements across the particle cross section.
X-ray photoelectron spectroscopy (XPS) using the monochromatic Al Kα source (1486.6 eV) of Kratos Axis Ultra (150 W) was used to investigate the structural changes and to follow the cross-linking chemistry. Particles were deposited on Au [65 nm]/titanium (Ti) [15 nm] covered silicon wafers, and two different points on each sample were analyzed; then, the measured values were averaged. During these experiments, the charge neutralizer was on. Four sweeps of 800 ms dwelling time were used for sulfur (S 2p), silicon (Si 2p, Si 2s), and chlorine (Cl 2p), while for carbon (C 1s), oxygen (O 1s), and nitrogen (N 1s) only one sweep of 800 ms was performed for each peak. Individual peaks were fitted with Gaussian–Lorentzian shape (GL[30]) curves, and locations of the components were compared against the values reported in the literature after shifting the peaks for +0.24 eV to fix the aliphatic carbon peak at 285.0 eV.
SEM/TEM/Optical Microscopy
Scanning electron microscopy (SEM) images were obtained on a Hitachi S-4700 Cold Cathode Field Emission Scanning Electron Microscope by coating the samples with 3 nm of Au/Pd alloy. Transmission electron microscopy (TEM) was carried by first staining the particles with osmium tetroxide through vapor deposition, and then, an image was obtained using JEOL 2010F-FasTEM on a copper grid. Postfabrication particle size measurements were carried on a dry sample by performing 28 different measurements on SEM images using ImageJ.46 Optical images of the particles were obtained with a LSM 700 (Zeiss) microscope in fluorescent channel.
Circular Dichroism
Circular dichroism (CD) spectra were obtained with Chirascan Plus spectrometer (Applied Photophysics) in a cell with a path length (d) of 0.1 cm at room temperature from 190 to 240 nm. Coomassie plus colorimetric assay (Thermo Scientific) was used to determine the protein concentration (c g/L). Solutions (15 μL) were mixed with assay reagent (300 μL), and the absorbance was measured at 595 nm (25–500 μg/mL). Concentrations obtained from the protein assay were then used to convert the CD data (θλ millidegrees at specific wavelength λ) to Δε molar differential extinction coefficient per residue (M−1cm−1) assuming a mean residue weight (MRW) of 114.15 Da for BSA prior to and postmodification (Δε = ((MRW × θλ)/(c × 10 × 3298 × d))). CDPro software (Colorado State University) was used to analyze the CD spectra and quantify the structure. The spectra were fitted with CONTINLL subroutine using 7 bases, and the secondary structure of the protein was estimated by deconvoluting the peaks and calculating the percentage of helical, beta, turn, and random components of the molecule.47
Modified particles reacted for 24 h, and unmodified control particles were dissolved in pH 7.4 buffer for a period of 18 h at 4 °C and then diluted in 10 mM K2HPO4. For control particles (202 μg/mL) and those reacted for 24 h (82.5 μg/mL), concentrations were determined with coomassie plus assay. Melting experiment of BSA was done by heating a solution (350 μg/mL) using QNW temperature control from 25 to 95 °C to determine the structure of the heat denatured molecule.
Alexa Fluor 555 BSA Conjugate Loading
Alexa Fluor 555 conjugated BSA (5 mg) was loaded into the preparticle solution (per 250 mg of BSA) to fabricate fluorescent particles for studying the dissolution mechanism and kinetics. To understand the dissolution/ desilylation process, particles were reacted with DIDCS for 6, 12, and 24 h, and then, the resulting material was exposed to pH 7.4 buffer to measure the fractional release of Alexa Fluor 555 conjugated BSA. Several different aliquots containing particles (3.13 mg) were prepared for each specific time point (15, 60, 150, 300, and 600 min). Each aliquot was then reconstituted in pH 7.4 buffer (600 μL), and at each time point, the particles were spun down and the supernatant (100 μL) was analyzed in triplicate by measuring the fluorescence intensity (Excitation: 545 nm; Emission: 575 nm) using SpectraMax M5 (Molecular Devices) in Nunc 96 microwell plates (Thermo Scientific). The intensity of soluble unfunctionalized particles at 600 min served as the control reference in calculation of the fractional dye release from the particles.
Fitting and Statistical Analysis
All the model fittings were done by JMP software (SAS Institute), and p values for statistical significances were calculated by QuickCalcs (GraphPad) online statistical calculator.
RESULTS AND DISCUSSION
Particle Fabrication and Composition
A SEM image of sieved BSA powder is shown in Figure 1A while that of the particles as fabricated with PRINT on the surface of the harvesting layer is displayed in Figure 1B. Harvested free particles of Figure 1C are obtained by washing them with ACN. A TEM image of the free particles is presented in Figure 1E. The composition of the particles postfabrication was determined to be 55.7 wt % BSA, 43.1 wt % lactose, and 1.2 wt % glycerol as schematically shown in Figure 1D. These particles were further loaded with Alexa Fluor 555 conjugated BSA as seen in Figure 1G. The mean external and internal diameters plus the thickness of the particles were measured as 2.88 ± 0.21, 0.7 ± 0.06, 0.56 ± 0.17 μm, respectively (Figure 1F).
Figure 1.

(A) BSA powder ground and sieved through a 400 mesh screen. (B) SEM of the particles obtained on the harvesting layer. (C) SEM of free particles after washing with ACN. (D) Particle composition postfabrication (blue: lactose; red: glycerol; green: BSA). (E) TEM of the particles. (F) Particle size as measured by image analysis using ImageJ (n = 28). (G) Particles loaded with Alexa Fluor 555 conjugated BSA on the surface of the harvesting layer (Excitation: 555 nm; Long pass filter: 560 nm).
Component Reactivity
Silylation is extensively used in organic chemistry to block certain reaction centers by introducing a silyl group in place of a labile hydrogen atom. Silylation reactions are reversible and proceed via a mechanism involving bimolecular nucleophilic substitution at the silicon atom. In many cases, the mechanism and rate of silylation are determined by the steric factors associated with the silylating agent or the compound being silylated.48 Rogozhin et al.49 have derivatized amino acids (Pro, Tyr, Trp, Val, Asp, Lys) and dipeptides (Gly-Ala, Val-Val, Gly-Tyr, Gly-Trp) with different silylating agents (trimethylsilyldiethylamine, bis(trimethylsilyl) acetamide, N-trimethylsilyl-N-methylacetamide, and hexamethyldisilazane) in organic solvents at 20 °C.
Silylation with chlorosilanes has been previously used by several authors on nonprotein/peptide based materials to alter the surface properties or incorporate drugs into polymeric matrices. Chlorosilanes have been used on multiwall carbon nanotubes to functionalize their surface.50 Gousse et al.51 modified cellulose whisker dispersed in organic solvents and concluded that it is possible to partially silylate the whiskers where the surface of the whiskers is silylated but the core is kept almost intact. Silylether chemistry has also been used for incorporating drugs into the matrix of particles as a pro-drug strategy with hydroxyl ethyl acrylate.52
Figure 2A compares the spectra of dialyzed BSA post-functionalization (12–24 h) with intact material. After 12 hr of functionalization, mass over charge (m/z) peaks for BSA are shifted toward higher values, which serves as the evidence of functionalization with DIDCS (Figure 2B). Exposure of BSA to DIDCS for 24 h causes formation of visible precipitates. However, due to low signal intensity after 24 h of silylation, no conclusions could be drawn regarding this sample. Once the spectra for intact and 12 hr modified samples are deconvoluted, an increase in the molecular weight of the BSA due to silylation is observed as illustrated in Figure 2C.
Figure 2.

Mass spectra of BSA prior to (black) and after functionalization (red: 12 hr; green: 24 hr of silylation): (A) m/z range 1000–1600 and (B) m/z range 1400–1500. (C) Deconvoluted spectra of intact and modified BSA samples using ProMass software.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) evaluation of the structure of trypsin digested 12 hr functionalized BSA molecules did not reveal any detectable modification of the recovered peptidic sequence. This can be attributed to the susceptibility of the bonds formed to hydrolysis during the digestion period.
Glycerol is capable of forming a variety of combinations with DIDCS under the reaction conditions (Figure S2, Supporting Information). The proposed structures are shown in Figure 3 for compounds A through H. The most abundant component is compound E formed from the reaction of two equivalents of DIDCS with one equivalent of glycerol. Although less abundant compared to compound E, multiple glycerol molecules can also be linked together due to the multifunctionality of both DIDCS (2 × chlorine [Cl]) and glycerol (3 × hydroxyl [OH]) as confirmed by the presence of compounds D, G, and H. Furthermore, the reaction conditions are conducive to the formation of larger fragments. Nevertheless, assigning structures to these peaks is not straightforward due to a multitude of combinations. Errors involved between the observed and theoretical masses of the identified peaks are presented in Table S2, Supporting Information.
Figure 3.

Identified reaction compounds of glycerol with DIDCS. (A) C9H20O3Si, (B) C12H28O4Si, (C) C12H30O3Si2, (D) C12H28O6Si, (E) C15H34O4Si2, (F) C18H42O5Si2, (G) C18H40O6Si2, and (H) C18H42O7Si2.
No evidence of reactivity of lactose with DIDCS could be detected under the reaction conditions. Although lactose bears multiple hydroxyl groups, we believe that insolubility of the material makes it immune to solid state functionalization carried in extra-dry ACN. Lactose is not soluble in acetonitrile–water mixtures containing less than 15% water.53
Particle Ensemble Analysis
XPS is a powerful technique for detecting compositional variations on the surface that are induced by silanization.54 The amount of silicon on the surface of the particles was measured with XPS, and relative intensities of the peaks are plotted in Figure 4A. The sulfur (S 2p) peak serves as a protein marker (disulfide bond-cysteine) and therefore was used as the appropriate reference element for comparing and quantifying the relative intensities of each peak. Both silicon (Si 2s) and (Si 2p) peaks increase with respect to sulfur (S 2p) as a function of reaction time. However, the data for Si 2s is not shown for brevity. High resolution peaks of silicon (Si 2s, Si 2p) and sulfur (S 2p) for unmodified and reacted samples along with the survey scans are compared in Figure S3, Supporting Information. High resolution scans of oxygen (O 1s), carbon (C 1s), and nitrogen (N 1s) peaks were performed to detect any chemical changes associated with functionalization. Figure 4B,C compares oxygen (O 1s) scans for unmodified particles and the sample silylated for 24 hr. The shifted values of the binding energies for the deconvoluted components are listed in Table S3, Supporting Information.
Figure 4.

(A) Average ratio of silicon to sulfur (Si 2p/S 2p) for particle ensemble as a function of reaction time (n = 2). High resolution XPS peaks (experimental: square, predicted/fitted: black line) and corresponding components (O 1sA: red line; O 1sB: green line; O 1sC: blue line) of (B) O 1s unmodified and (C) O 1s 24 hr silylated samples (cps: counts per second).
Lopez and co-workers55 reported the value of 532.8 eV for the oxygen (O 1s) peak of 1,10-decanediol and 532.7 eV for poly(vinyl alcohol) and poly(propylene glycol) with reference to an aliphatic carbon peak at 285.0 eV. Therefore, it can be concluded that the reduction in the intensity of the oxygen (O 1s) peak (labeled B) is consistent with consumption of hydroxyl groups on the surface due to the functionalization after 24 hr. The oxygen (O 1s) peak (labeled A) of 531.4 eV is very close to peptidic oxygen of BSA as reported by Iucci et al.56 at 531.6 eV. Carbon (C 1s) and nitrogen (N 1s) scans for the same samples are also presented in Figure S4, Supporting Information.
Individual Particle Analysis
A representative particle prior to exposure to the electron beam of EDS is shown in Figure 5A. Post-EDS analysis, the particle retains its shape, although there is minor structural damage due to the electron beam (Figure 5B). Elemental mapping at 20.0 kV over the top surface of the particle qualitatively confirmed the presence of silicon (Si) in the particle as illustrated in Figure 5C. Elemental maps of oxygen (O), sulfur (S), chlorine (Cl), nitrogen (N), and carbon (C) are also shown in Figure 5D–H for comparison. The corresponding spectrum obtained from elemental mapping of the particle in Figure 5 is shown in Figure S5, Supporting Information.
Figure 5.

Elemental mapping (20.0 kV) of the particle silylated for 24 hr. (A) Particle prior to EDS beam exposure. (B) Particle after exposure to EDS beam. (C) Si Kα, (D) O Kα, (E) S Kα, (F) Cl Kα, (G) N Kα, and (H) C Kα. (I) EDS analysis of individual particles and calculation of Si/O (red), Si/C (blue), and Si/S (green) ratios as a function of reaction time. (J) The scanned area on top of an individual particle is shown with a pink rectangle (n = 3).
Quantification of the degree of silylation on the particles is done by analyzing an area on the top surface of the particles (pink rectangle in Figure 5J). Three different particles of silylated samples are analyzed, and the result is averaged and presented in Figure 5I. The statistical significances and silicon content variation among these distinct sets of samples/particles are also shown in this figure for comparison. It can be concluded that longer exposure to DIDCS results in a higher ratio of Si/S, Si/O, and Si/C in the particles.
Cross Sectional Analysis
Figure 6C,D shows the top and side views of a particle after being cross sectioned with the FIB, respectively. The distribution of the elements along the yellow line depicted in Figure 6D is plotted in Figure 6B as a function of transverse distance. The sum spectrum of the linescan is also compared with the spectrum of the background to confirm that no silicon signal is detected on the carbon background (Figure 6A). No variation could be observed in the silicon content along the scanned line of Figure 6D, which suggests that DIDCS penetrates all the way through the particle and the reaction is not limited only to the surface of the particle.
Figure 6.

(A) Sum of the elemental spectrum along the EDS linescan on the particle sectioned using FIB compared to the spectrum of the carbon background (5.0 kV). (B) Distribution of the elements along the linescan (red: silicon; blue: sulfur; green: chlorine; black: oxygen; burgundy: gallium). (C) Top view of the FIB sectioned particle. (D) Side view of the same particle (yellow line indicates the position of the line used to perform the EDS analysis).
Mechanism and Dissolution Profile
To study the dissolution of particles, silylated particles were exposed to pH 7.4 buffer and the fractional release of Alexa Fluor 555 conjugated BSA was measured (Figure 7A, symbols). The data demonstrates that the longer the treatment of particles with DIDCS, the slower is the dissolution rate of the particles. This is consistent with the higher silicon content of the particles as a function of reaction time, obtained from EDS and XPS experiments.
Figure 7.

(A) Fractional dye release (n = 3, 0–600 min) from the particles at pH 7.4 as a function of reaction time (6 h: diamond; 12 hr: rectangle; 24 hr: triangle) and corresponding fits to Korsmeyer-Peppas (continuous line) and Weibull models (dashed line). (B) Time lapse images (every 30 min) of a 12 hr silylated particle containing Alexa Fluor 555 conjugated BSA placed in a vat of pH 7.4 buffer showing the gradual migration of the dye from the particle (Sequence starts at image 1 and ends at image 10; Excitation: 555 nm; Long pass filter: 560 nm). Particles silylated for 24 hr. (C) 0.1 v/v% TFA in water, approximate pH = 2.0, (D) dry, and (E) pH 10.0 buffer (Excitation: 555 nm; Long pass filter: 560 nm).
Numerous mathematical models exist in the literature for describing the dissolution behavior.57,58 Weibull59 and Korsmeyer-Peppas60 models are fitted to the experimental data (Figure 7A, lines), and corresponding model parameters are listed in Table S4, Supporting Information. The diffusional exponent in the Korsmeyer-Peppas model is indicative of a release mechanism (mode of transport of solute) and is shape dependent.61
To visualize the dissolution process for particles post-functionalization, the behavior of a single 12 hr silylated particle is tracked in pH 7.4 buffer as a function of time. As observed in Figure 7B, the intensity of Alexa Fluor 555 conjugated BSA in the particle gradually diminishes due to dissolution of the fluorophore from the particle into the surrounding environment. Effects of both acidic (0.1 v/v% TFA, approximate pH = 2.0) and basic (pH 10.0 buffer) conditions on the size of particles are illustrated in Figure 7C,E for particles functionalized for 24 hr, respectively. At these pH extremes, particles significantly swell to 4–5 times their original size of Figure 7D. Shirai et al.62 have studied the effect of different pHs on the hydrolytic removal of silyl groups from trialkyl silyl ethers. Their results indicate that there exists a pH where the observed hydrolysis rate constants are minimized. They demonstrated that this rate of hydrolysis is also affected by the electronegativity and steric size of the substituents on both silicon and oxygen atoms. The extreme swelling of these particles in both acidic and basic conditions suggests that the prevailing mechanism of dissolution is desilylation (removal of silyl groups).
The extreme swelling of the particles in acidic pH as observed in Figure 7C leaves the protein content of the particles prone to degradation in the harsh acidic environment of the gastrointestinal tract, and therefore, an oral delivery route for these suspensions is not envisioned. A measure for the stability of the suspension of 24 hr functionalized particles in pH 7.4 buffer is presented in Figure S6, Supporting Information.
Structural Evaluation
CD signal can determine structural changes associated with processing or functionalization of the protein in the particles. An examination of the melting behavior of intact BSA was done to evaluate the structural changes associated with heat denaturation of the molecule, which will enable us to compare the structures of processed BSA to that of the intact or denatured molecule (Figure 8C). It can be seen that the intensity of the peaks at 209 and 222 nm is greatly reduced upon heating the sample. Figure 8C merely serves as a reference on how the structure of thermally denatured BSA evolves and illustrates the significant reduction in the helical structure upon thermal denaturation.
Figure 8.

(A) CD spectra for heat denatured BSA (circle), intact BSA (diamond), unfunctionalized control particles (square), and functionalized particles for 24 hr (triangle). Corresponding fits using CDPro software are also shown (lines). (B) Contribution of each structure after deconvolution (green: helical; yellow: beta; blue: turn; red: random). (C) CD signal at 222 nm (triangle) and 209 nm (circle) as a function of temperature (°C) for BSA solution and the associated heat denaturation of the molecule.
CD data for unmodified and modified particles are presented in Figure 8A and compared to intact and heat denatured BSA. Houen et al.63 showed that denatured BSA has limited solubility in water, and thus, structural changes postdenaturation are expected to be detectable with CD. Figure 8B shows the corresponding structural components obtained from deconvoluting/fitting the CD spectra while accuracy of the fits to the experimental data is examined in Figure 8A. The data in Figure 8B suggests that the silylation process used herein did not perturb the secondary structure of the fraction of BSA molecules that were dissolved in the buffer through the course of the dissolution experiment. Moreover, no evidence of thermal denaturation post particle fabrication could be obtained, primarily due to the fact that the denaturation mechanism is a time dependent process.64 This method of analysis had been used by Harn et al.2 to analyze the structure of recombinant humanized IgG1κ and also by Estey et al.65 to study the degradation of BSA under acidic conditions.
Aggregates of denatured protein can induce antibody, but they are not expected to induce neutralizing antibody due to lost protein conformation on denaturation. Formation of natively conformed aggregates on the other hand can be detrimental for therapeutic proteins due to the generation of neutralizing antibody response.66 Although subvisible particles less than 10 μm are not being evaluated in the USP 〈788〉 test, they can potentially impact clinical performance.67 Nevertheless, Singh et al.68 emphasize the uncertainty around the potential immunogenicity risk of subvisible particles in pharmaceutical formulations and conclude that the direct link between aggregation and clinical immunogenicity is not well established. They also assert that preclinical studies are not yet capable of evaluating the immunogenicity potential of product attributes such as aggregates and particles smaller than 10 μm. Experiments of van Beers et al.69 with different structures of recombinant human interferon Beta-1a proved that large unreacted aggregates (3.2 μm) and guanidine treated aggregated molecules (0.35 μm) are not necessarily immunogenic in transgenic immune-tolerant mice. The relation between structure and immunogenicity of recombinant human interferon Alpha-2b was investigated in wild-type and transgenic mice by Hermeling et al.,70 and no antibody responses for GA cross-linked molecule were detected. Moreover, boiled/denatured molecules (1–3 μm) proved to be poor immunogens in transgenic mice.
CONCLUSIONS
We have demonstrated that the combination of PRINT technology and silylation provides a valuable tool for tuning the solubility of the protein particles while controlling their size and shape. EDS and XPS confirm the modification at the particle and ensemble levels, respectively. The silylation process renders the solubility of the particles to be tunable, eventually dissolving in aqueous media while not perturbing the secondary structure of the fraction of BSA released from the particles. The rate of dissolution can be controlled by the time particles spend in the functionalization media consisting of extra-dry acetonitrile and diisopropyldichlorosilane. Extreme swelling of the particles in low and high pH confirms that desilylation is the mechanism of particle dissolution. To further understand the nature of functionalization, EDS cross sectional analysis confirmed that the chlorosilane diffuses all the way through the particles and is not just limited to the surface.
Lowering the temperature used in the filling/harvesting stages of the particle fabrication enables us to better protect the sensitive contents of the particles against various modes of instabilities. Fabricating particles from homologous albumin instead of a heterologous one is recommended as it allows us to prepare particles that are made from nonimmunogenic ingredients. Immunogenicity of our subvisible functionalized particles loaded with a therapeutic protein will be thoroughly investigated in future, to elucidate the role of functionalization and fabrication parameters. In vivo behavior of these functionalized particles and their potential in extending protein retention is a subject of a separate study.
Supplementary Material
Acknowledgments
We acknowledge the National Institutes of Health (NIH) Director’s Pioneer Award and Liquidia Technologies for support. Characterization of the particles was done in the Chapel Hill Analytical and Nanofabrication Laboratory (CHANL), and microscopic evaluation of the particles was done at Microscopy Services Laboratory (MSL) of the University of North Carolina at Chapel Hill.
Footnotes
The authors declare the following competing financial interest(s): Joseph M. DeSimone is a founder and maintains a financial interest in Liquidia Technologies.
(1) HPLC analysis of the particles postfabrication. (2) Schematic of particle silylation. (3) FT-MS analysis of reaction products of glycerol and DIDCS. (4) Estimated error involved in the structure assignment for the reaction products of glycerol and DIDCS. (5) High resolution XPS scans of silicon and sulfur. (6) High resolution XPS scans and peak locations. (7) EDS elemental mapping performed on an individual particle. (8) Parameters for Korsmeyer-Peppas and Weibull models. (9) Optical density measurement for the suspension of functionalized particles. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Almeida AJ, Souto E. Solid Lipid Nanoparticles as a Drug Delivery System for Peptides and Proteins. Adv Drug Delivery Rev. 2007;59:478–490. doi: 10.1016/j.addr.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Harn N, Allan C, Oliver C, Middaugh CR. Highly Concentrated Monoclonal Antibody Solutions: Direct Analysis of Physical Structure and Thermal Stability. J Pharm Sci. 2007;96:532–546. doi: 10.1002/jps.20753. [DOI] [PubMed] [Google Scholar]
- 3.Antosova Z, Mackova M, Kral V, Macek T. Therapeutic Application of Peptides and Proteins: Parenteral Forever? Trends Biotechnol. 2009;27:628–635. doi: 10.1016/j.tibtech.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 4.Vermonden T, Censi R, Hennink WE. Hydrogels for Protein Delivery. Chem Rev. 2012;112:2853–2888. doi: 10.1021/cr200157d. [DOI] [PubMed] [Google Scholar]
- 5.Manning MC, Chou DK, Murphy BM, Payne RW, Katayama DS. Stability of Protein Pharmaceuticals: An Update. Pharm Res. 2010;27:544–575. doi: 10.1007/s11095-009-0045-6. [DOI] [PubMed] [Google Scholar]
- 6.Pechenov S, Shenoy B, Yang MX, Basu SK, Margolin AL. Injectable Controlled Release Formulations Incorporating Protein Crystals. J Controlled Release. 2004;96:149–158. doi: 10.1016/j.jconrel.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 7.Lee VHL, Dodda-Kashi S, Grass GM, Rubas W. In: Peptide and Protein Drug Delivery. Lee VHL, editor. Chapter 16. Marcel Dekker; New York, NY: 1991. pp. 691–738. [Google Scholar]
- 8.Brown LR. Commercial Challenges of Protein Drug Delivery. Expert Opin Drug Delivery. 2005;2:29–42. doi: 10.1517/17425247.2.1.29. [DOI] [PubMed] [Google Scholar]
- 9.Riva P, Arista A, Sturiale C, Moscatelli G, Tison V, Mariani M, Seccamani E, Lazzari S, Fagioli L, Franceschi G, Sarti G, Riva N, Natali PG, Zardi L, Scassellati GA. Treatment of Intracranial Human Glioblastoma by Direct Intratumoral Administration of I-131-Labeled Anti-Tenascin Monoclonal-Antibody BC-2. Int J Cancer. 1992;51:7–13. doi: 10.1002/ijc.2910510103. [DOI] [PubMed] [Google Scholar]
- 10.Kitamura K, Takahashi T, Kotani T, Miyagaki T, Yamaoka N, Tsurumi H, Noguchi A, Yamaguchi T. Local-Administration of Monoclonal Antibody-Drug Conjugate - A New Strategy to Reduce the Local Recurrence of Colorectal-Cancer. Cancer Res. 1992;52:6323–6328. [PubMed] [Google Scholar]
- 11.Abrishami M, Ganavati SZ, Soroush D, Rouhbakhsh M, Jaafari MR, Malaekeh-Nikouei B. Preparation, Characterization, and in Vivo Evaluation of Nanoliposomes-Encapsulated Bevacizumab (Avastin) for Intravitreal Administration. Retina. 2009;29:699–703. doi: 10.1097/IAE.0b013e3181a2f42a. [DOI] [PubMed] [Google Scholar]
- 12.Ohtori S, Miyagi M, Eguchi Y, Inoue G, Orita S, Ochiai N, Kishida S, Kuniyoshi K, Nakamura J, Aoki Y, Ishikawa T, Arai G, Kamoda H, Suzuki M, Takaso M, Furuya T, Kubota G, Sakuma Y, Oikawa Y, Toyone T, Takahashi K. Efficacy of Epidural Administration of Anti-Interleukin-6 Receptor Antibody onto Spinal Nerve for Treatment of Sciatica. Eur Spine J. 2012;21:2079–2084. doi: 10.1007/s00586-012-2183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nikas S, Temekonidis T, Zikou A, Argyropoulou M, Efremidis S, Drosos A. Treatment of Resistant Rheumatoid Arthritis by Intra-Articular Infliximab Injections: A Pilot Study. Ann Rheum Dis. 2004;63:102–103. doi: 10.1136/ard.2003.006981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jorgensen L, Nielsen HM. Delivery Technologies for Biopharmaceuticals: Peptides, Proteins, Nucleic Acids, and Vaccines. 1. John Wiley & Sons; West Sussex: 2009. [Google Scholar]
- 15.Banerjee PS, Hosny EA, Robinson JR. In: Peptide and Protein Drug Delivery. Lee VHL, editor. Chapter 10. Marcel Dekker; New York, NY: 1991. pp. 487–543. [Google Scholar]
- 16.Lee KY, Yuk SH. Polymeric Protein Delivery Systems. Prog Polym Sci. 2007;32:669–697. [Google Scholar]
- 17.Yang MX, Shenoy B, Disttler M, Patel R, McGrath M, Pechenov S, Margolin AL. Crystalline Monoclonal Antibodies for Subcutaneous Delivery. Proc Natl Acad Sci US A. 2003;100:6934–6939. doi: 10.1073/pnas.1131899100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller MA, Engstrom JD, Ludher BS, Johnston KP. Low Viscosity Highly Concentrated Injectable Nonaqueous Suspensions of Lysozyme Microparticles. Langmuir. 2010;26:1067–1074. doi: 10.1021/la9023426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arshady R. Albumin Microspheres and Microcapsules -Methodology of Manufacturing Techniques. J Controlled Release. 1990;14:111–131. [Google Scholar]
- 20.Patil GV. Biopolymer Albumin for Diagnosis and in Drug Delivery. Drug Dev Res. 2003;58:219–247. [Google Scholar]
- 21.Tong W, Gao C, Moehwald H. PH-Responsive Protein Microcapsules Fabricated via Glutaraldehyde Mediated Covalent Layer-by-Layer Assembly. Colloid Polym Sci. 2008;286:1103–1109. [Google Scholar]
- 22.Zhou Z, Anselmo AC, Mitragotri S. Synthesis of Protein-Based, Rod-Shaped Particles from Spherical Templates Using Layer-by-Layer Assembly. Adv Mater. 2013;25:2723–2727. doi: 10.1002/adma.201300220. [DOI] [PubMed] [Google Scholar]
- 23.Nettey H, Haswani D, Oettinger CW, D’Souza MJ. Formulation and Testing of Vancomycin Loaded Albumin Microspheres Prepared by Spray-Drying. J Microencapsulation. 2006;23:632–642. doi: 10.1080/02652040600776564. [DOI] [PubMed] [Google Scholar]
- 24.Langer K, Balthasar S, Vogel V, Dinauer N, Von Briesen H, Schubert D. Optimization of the Preparation Process for Human Serum Albumin (HSA) Nanoparticles. Int J Pharm. 2003;257:169–180. doi: 10.1016/s0378-5173(03)00134-0. [DOI] [PubMed] [Google Scholar]
- 25.Lin W, Coombes AG, Davies MC, Davis SS, Illum L. Preparation of Sub-100 nm Human Serum Albumin Nanospheres using a pH-Coacervation Method. J Drug Targeting. 1993;1:237–243. doi: 10.3109/10611869308996081. [DOI] [PubMed] [Google Scholar]
- 26.Bogdansky S. In: Biodegradable Polymers as Drug Delivery Systems. Chasin M, Langer R, editors. Chapter 7. Marcel Dekker; New York, NY: 1990. pp. 231–259. [Google Scholar]
- 27.Bhargava K, Ando HY. Immobilization of Active Urokinase on Albumin Microspheres: Use of a Chemical Dehydrant and Process Monitoring. Pharm Res. 1992;9:776–781. doi: 10.1023/a:1015855622459. [DOI] [PubMed] [Google Scholar]
- 28.Goosen MF, Leung YF, O’Shea GM, Chou S, Sun AM. Slow Release of Insulin from a Biodegradable Matrix Implanted in Diabetic Rats. Diabetes. 1983;32:478–481. doi: 10.2337/diab.32.5.478. [DOI] [PubMed] [Google Scholar]
- 29.Rollett A, Reiter T, Nogueira P, Cardinale M, Loureiro A, Gomes A, Cavaco-Paulo A, Moreira A, Carmo AM, Guebitz GM. Folic Acid-Functionalized Human Serum Albumin Nanocapsules for Targeted Drug Delivery to Chronically Activated Macrophages. Int J Pharm. 2012;427:460–466. doi: 10.1016/j.ijpharm.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 30.Oettinger CW, D’Souza MJ. Microencapsulated Drug Delivery: A New Approach to Pro-Inflammatory Cytokine Inhibition. J Microencapsulation. 2012;29:455–462. doi: 10.3109/02652048.2012.658443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kratz F. Albumin as a Drug Carrier: Design of Prodrugs, Drug Conjugates and Nanoparticles. J Controlled Release. 2008;132:171–183. doi: 10.1016/j.jconrel.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 32.Doshi N, Mitragotri S. Macrophages Recognize Size and Shape of Their Targets. PLoS One. 2010;5(e10051) doi: 10.1371/journal.pone.0010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee TK, Sokoloski TD, Royer GP. Serum Albumin Beads: An Injectable, Biodegradable System for the Sustained Release of Drugs. Science. 1981;213:233–235. doi: 10.1126/science.6787705. [DOI] [PubMed] [Google Scholar]
- 34.Ratcliffe JH, Hunneyball IM, Smith A, Wilson CG, Davis SS. Preparation and Evaluation of Biodegradable Polymeric Systems for the Intra-Articular Delivery of Drugs. J Pharm Pharmacol. 1984;36:431–436. doi: 10.1111/j.2042-7158.1984.tb04419.x. [DOI] [PubMed] [Google Scholar]
- 35.Wong SS. Chemistry of Protein Conjugation and Cross-Linking. 1. CRC Press; Boca Raton, FL: 1991. [Google Scholar]
- 36.Srinivasachar K, Neville D. New Protein Cross-Linking Reagents that Are Cleaved by Mild Acid. Biochemistry. 1989;28:2501–2509. doi: 10.1021/bi00432a023. [DOI] [PubMed] [Google Scholar]
- 37.Benesch RE, Kwong S. Bis-Pyridoxal Polyphosphates - A New Class of Specific Intramolecular Crosslinking Agents for Hemoglobin. Biochem Biophys Res Commun. 1988;156:9–14. doi: 10.1016/s0006-291x(88)80798-8. [DOI] [PubMed] [Google Scholar]
- 38.Blattler WA, Kuenzi BS, Lambert JM, Senter PD. New Heterobifunctional Protein Cross-Linking Reagent that Forms an Acid-Labile Link. Biochemistry. 1985;24:1517–1524. [Google Scholar]
- 39.Parrott MC, Luft JC, Byrne JD, Fain JH, Napier ME, DeSimone JM. Tunable Bifunctional Silyl Ether Cross-Linkers for the Design of Acid-Sensitive Biomaterials. J Am Chem Soc. 2010;132:17928–17932. doi: 10.1021/ja108568g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelly JY, DeSimone JM. Shape-Specific, Monodisperse Nano-Molding of Protein Particles. J Am Chem Soc. 2008;130:5438–5439. doi: 10.1021/ja8014428. [DOI] [PubMed] [Google Scholar]
- 41.Houen G. The Solubility of Proteins in Organic Solvents. Acta Chem Scand. 1996;50:68–70. [Google Scholar]
- 42.Griebenow K, Klibanov AM. On Protein Denaturation in Aqueous-Organic Mixtures but not in Pure Organic Solvents. J Am Chem Soc. 1996;118:11695–11700. [Google Scholar]
- 43.Spahr P, Edsall J. Amino Acid Composition of Human + Bovine Serum Mercaptalbumins. J Biol Chem. 1964;239:850–854. [PubMed] [Google Scholar]
- 44.Brown JR. Structure of Bovine Serum-Albumin. Fed Proc. 1975;34:591. [Google Scholar]
- 45.Kolthoff IM, Bruckenstein S, Chantooni MK. Acid-base Equilibria in Acetonitrile - Spectrophotometric and Conductometric Determination of Dissociation of Various Acids. J Am Chem Soc. 1961;83:3927–3935. [Google Scholar]
- 46.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 Years of Image Analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sreerama N, Woody RW. Estimation of Protein Secondary Structure from Circular Dichroism Spectra: Comparison of CONTIN, SELCON, and CDSSTR Methods with an Expanded Reference Set. Anal Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 48.Kashutina MV, Ioffe SL, Tartakovskii VA. Silylation of Organic Compounds. Russ Chem Rev. 1975;44:733–747. [Google Scholar]
- 49.Rogozhin SV, Davidovich YA, Andreev SM, Mironova NV, Yurtanov AI. Preparation of Trimethylsilyl Derivatives of Amino-Acids and Peptides for Peptide-Synthesis. Bull Acad Sci USSR, Div Chem Sci (Engl Transl) 1974;23:1789–1792. [Google Scholar]
- 50.Vast L, Mekhalif Z, Fonseca A, Nagy JB, Delhalle J. Preparation and Electrical Characterization of a Silicone Elastomer Composite Charged with Multi-Wall Carbon Nanotubes Functionalized with 7-Octenyltrichlorosilane. Compos Sci Technol. 2007;67:880–889. [Google Scholar]
- 51.Gousse C, Chanzy H, Excoffier G, Soubeyrand L, Fleury E. Stable Suspensions of Partially Silylated Cellulose Whiskers Dispersed in Organic Solvents. Polymer. 2002;43:2645–2651. [Google Scholar]
- 52.Parrott MC, Finniss M, Luft JC, Pandya A, Gullapalli A, Napier ME, Desimone JM. Incorporation and Controlled Release of Silyl Ether Prodrugs from PRINT Nanoparticles. J Am Chem Soc. 2012;134:7978–7982. doi: 10.1021/ja301710z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Folkes DJ, Jordan MA. In: Carbohydrates in Food. Eliasson AC, editor. Chapter 1. Taylor & Francis; Boca Raton, FL: 2006. pp. 1–40. [Google Scholar]
- 54.Puglisi O, Torrisi A, Marletta G. XPS Investigation of the Effects Induced by the Silanization on Real Glass Surfaces. J Non-Cryst Solids. 1984;68:219–230. [Google Scholar]
- 55.Lopez GP, Castner DG, Ratner BD. XPS O 1s Binding-Energies for Polymers Containing Hydroxyl, Ether, Ketone and Ester Groups. Surf Interface Anal. 1991;17:267–272. [Google Scholar]
- 56.Iucci G, Polzonetti G, Infante G, Rossi L. XPS and FT-IR Spectroscopy Study of Albumin Adsorption on the Surface of a Pi-Conjugated Polymer Film. Surf Interface Anal. 2004;36:724–728. [Google Scholar]
- 57.Arifin DY, Lee LY, Wang C. Mathematical Modeling and Simulation of Drug Release from Microspheres: Implications to Drug Delivery Systems. Adv Drug Delivery Rev. 2006;58:1274–1325. doi: 10.1016/j.addr.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 58.Costa P, Manuel J, Lobo S. Modeling and Comparison of Dissolution Profiles. Eur J Pharm Sci. 2001;13:123–133. doi: 10.1016/s0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 59.Weibull W. A Statistical Distribution Function of Wide Applicability. J Appl Mech. 1951;18:293–297. [Google Scholar]
- 60.Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of Solute Release from Porous Hydrophilic Polymers. Int J Pharm. 1983;15:25–35. doi: 10.1002/jps.2600721021. [DOI] [PubMed] [Google Scholar]
- 61.Ritger PL, Peppas NA. A Simple Equation for Description of Solute Release II. Fickian and Anomalous Release from Swellable Devices. J Controlled Release. 1987;5:37–42. [PubMed] [Google Scholar]
- 62.Shirai N, Moriya K, Kawazoe Y. pH-Dependence of Hydrolytic Removal of Silyl Group from Trialkylsilyl Ethers. Tetrahedron. 1986;42:2211–2214. [Google Scholar]
- 63.Houen G, Svaerke C, Barkholt V. The Solubilities of Denatured Proteins in Different Organic Solvents. Acta Chem Scand. 1999;53:1122–1126. doi: 10.3891/acta.chem.scand.53-1122. [DOI] [PubMed] [Google Scholar]
- 64.Nakagaki M, Sano Y. Light Scattering Studies on the Thermal Denaturation of Bovine Serum Albumin. Bull Chem Soc Jpn. 1973;46:791–797. doi: 10.1248/yakushi1947.93.7_902. [DOI] [PubMed] [Google Scholar]
- 65.Estey T, Kang J, Schwendeman SP, Carpenter JF. BSA Degradation under Acidic Conditions: A Model for Protein Instability during Release from PLGA Delivery Systems. J Pharm Sci. 2006;95:1626–1639. doi: 10.1002/jps.20625. [DOI] [PubMed] [Google Scholar]
- 66.Rosenberg AS. Effects of Protein Aggregates: An Immunologic Perspective. AAPS J. 2006;8:E501–E507. doi: 10.1208/aapsj080359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carpenter JF, Randolph TW, Jiskoot W, Crommelin DJ, Middaugh CR, Winter G, Fan YX, Kirshner S, Verthelyi D, Kozlowski S, Clouse KA, Swann PG, Rosenberg A, Cherney B. Overlooking Subvisible Particles in Therapeutic Protein Products: Gaps That May Compromise Product Quality. J Pharm Sci. 2009;98:1201–1205. doi: 10.1002/jps.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singh SK, Afonina N, Awwad M, Bechtold-Peters K, Blue JT, Chou D, Cromwell M, Krause HJ, Mahler HC, Meyer BK, Narhi L, Nesta DP, Spitznagel T. An Industry Perspective on the Monitoring of Subvisible Particles as a Quality Attribute for Protein Therapeutics. J Pharm Sci. 2010;99:3302–3321. doi: 10.1002/jps.22097. [DOI] [PubMed] [Google Scholar]
- 69.van Beers MM, Sauerborn M, Gilli F, Brinks V, Schellekens H, Jiskoot W. Oxidized and Aggregated Recombinant Human Interferon Beta is Immunogenic in Human Interferon Beta Transgenic Mice. Pharm Res. 2011;28:2393–2402. doi: 10.1007/s11095-011-0451-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hermeling S, Aranha L, Damen JM, Slijper M, Schellekens H, Crommelin DJ, Jiskoot W. Structural Characterization and Immunogenicity in Wild-Type and Immune Tolerant Mice of Degraded Recombinant Human Interferon Alpha2b. Pharm Res. 2005;22:1997–2006. doi: 10.1007/s11095-005-8177-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.