

Abstract
Salinipyrones and pacificanones are structurally related polyketides from Salinispora pacifica CNS-237 proposed to arise from the same modular polyketide synthase (PKS) assembly line. Genome sequencing revealed a large macrolide PKS gene cluster that codes for the biosynthesis of rosamicin A and a series of new macrolide antibiotics. Mutagenesis experiments unexpectedly correlated salinipyrone and pacificanone biosynthesis to the rosamicin octamodule Spr PKS. Remarkably, this bifurcated polyketide pathway illuminates a series of enzymatic domain and module skipping reactions that dictate natural polyketide product diversity. Our findings enlarge the growing knowledge of polyketide biochemistry and illuminate potential challenges in PKS bioengineering.
Keywords: biosynthesis, macrolide antibiotic, polyketide synthase, Salinispora
Salinipyrone A (1) and pacificanone A (2) are structurally related polyketides produced by the marine bacterium Salinispora pacifica CNS-237 (Scheme 1).[1] These natural products share identical lipid tails yet differ in the composition of their lipid head groups, γ-hydroxypyrone versus cyclohexanone, respectively. It has been suggested that the associated polyketide synthase (PKS) may skip the penultimate extension module to yield the slightly smaller salinipyrone product.[1] The concept of module skipping by assembly line, modular PKSs is best illustrated with the methymycin/picromycin system in which 12- and 14-membered ring macrolactones are generated by the same PKS complex adopting alternative conformations to enable an early chain termination in the case of the shorter methymycin aglycone.[2] Module skipping, as well as module stuttering, can lead to biosynthetic diversification in nature as remarkably exemplified with the thalassospiramide family of calpain inhibitors.[3] We thus set out to explore the biosynthetic basis for salinipyrone/pacificanone differentiation and were surprised to learn that while module skipping is indeed a contributing factor, these metabolites are in fact byproducts of an even larger PKS system associated with the rosamicin macrolide antibiotics.
Scheme 1.
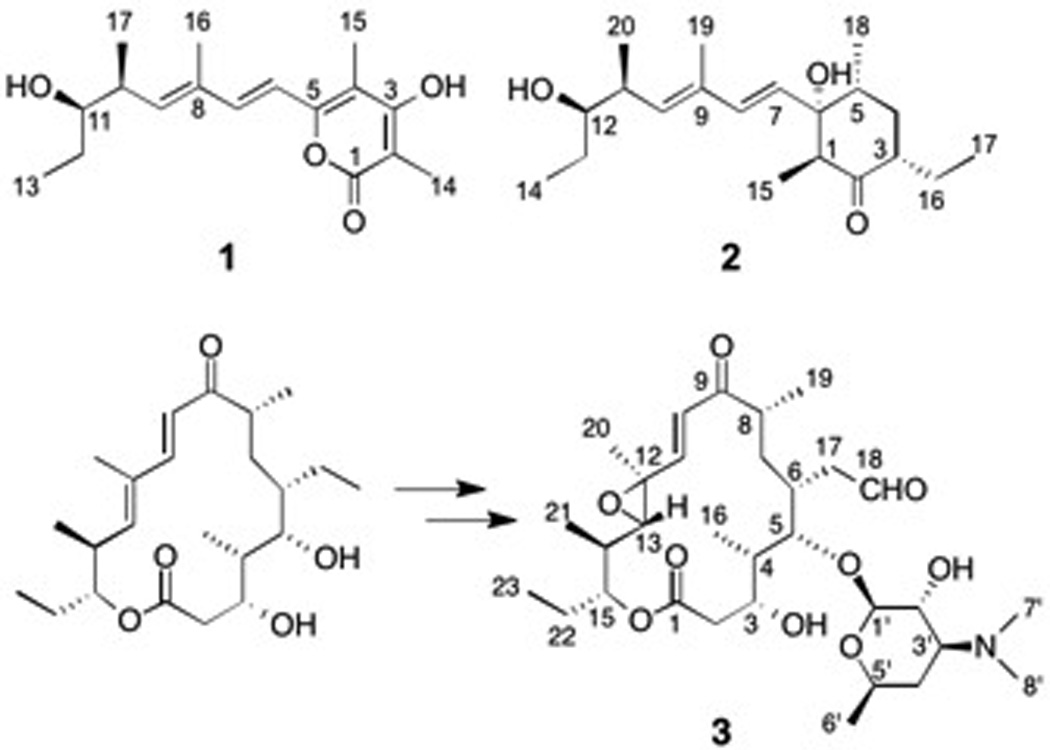
The chemical structures of salinipyrone A (1), pacificanone A (2), and rosamicin A (3)
To define the biosynthetic logic of salinipyrone/pacificanone construction, we examined the recently sequenced draft genome of S. pacifica CNS-237.[4] Since 2 contains an ethyl side chain in the cyclohexanone unit, we anticipated its biosynthetic origin from the PKS substrate ethylmalonyl-CoA[5] and thus specifically searched for ethylmalonate-specifying acyl transferase (AT) domains within modular PKSs. We identified a single genomic example in a biosynthetic gene cluster fragmented across several contigs. Extensive primer walking completed the DNA sequence of the cluster to give the 23 open reading frame spr locus spanning 64.2 kb (GenBank KP997155). This PKS gene cluster shares high sequence homology with that for the macrolide rosamicin A (3) (Scheme 1) in Micromonospora carbonacea subsp. aurentiaca NRRL 2997[6] and contains five PKS genes (spr7–11) consisting of eight PKS modules, three cytochrome P450 genes (spr5–6, -12),[7] one glycosyltransferase (spr13), and genes encoding desosamine biosynthesis (spr4, 14, 19–20, 22–23)[8] (Table S1).
Inspection of the chemical structure of the macrolide 3 revealed striking structural similarities to 1 and 2 (Scheme 1), suggesting that all three structure types may originate from the same PKS assembly line. To evaluate this revised biosynthetic hypothesis, we first inactivated the spr gene cluster by inserting an apramicin resistance gene cassette (aac(3)IV) into the region encoding spr8–11 by intergenic conjugation (Figure S1). As predicted, the inactivation abolished the production of 1 and a previously unreported derivative, dehydrosalinipyrone A (4) (Figure 1A and B), thus confirming that the Spr PKS is responsible for their production (Table S1). Unfortunately, we could not reproduce the production of 2 in the wild-type strain under all fermentation conditions tested, and thus could not positively link its biosynthesis to the spr locus. We did, however, observe the loss of five additional compounds in the spr mutant never before reported from this bacterium (Figure 1A). Upon isolation and complete spectral analysis,compounds included the known rosamicin analogues 3 and 21-hydroxyrosamicin (izenamicin A3,[9] 5) along with three novel derivatives, namely 18-dihydro-14-hydroxyrosamicin (6), 18-dihydro-21-hydroxyrosamicin (7), and 14-hydroxyrosamicin (8). We predict that the conserved stereocenters in the new rosamicin analogues 6–8 are identical to those previously reported for 3 and that the new C-14 tertiary hydroxyl stereocenter in 6 and 8 is S as in the macrolide mycinamicin II.[10] In the latter case, the cytochrome P450 protein MycG[11] installs the analogous 14S hydroxyl group. Of the three cytochrome P450s encoded within the spr gene cluster, Spr6 is the most identical with MycG at 45% and is thus a likely candidate for the C-14 hydroxylation. Rosamicins 5–8 exhibit moderate antibacterial properties (Table S10).
Figure 1.
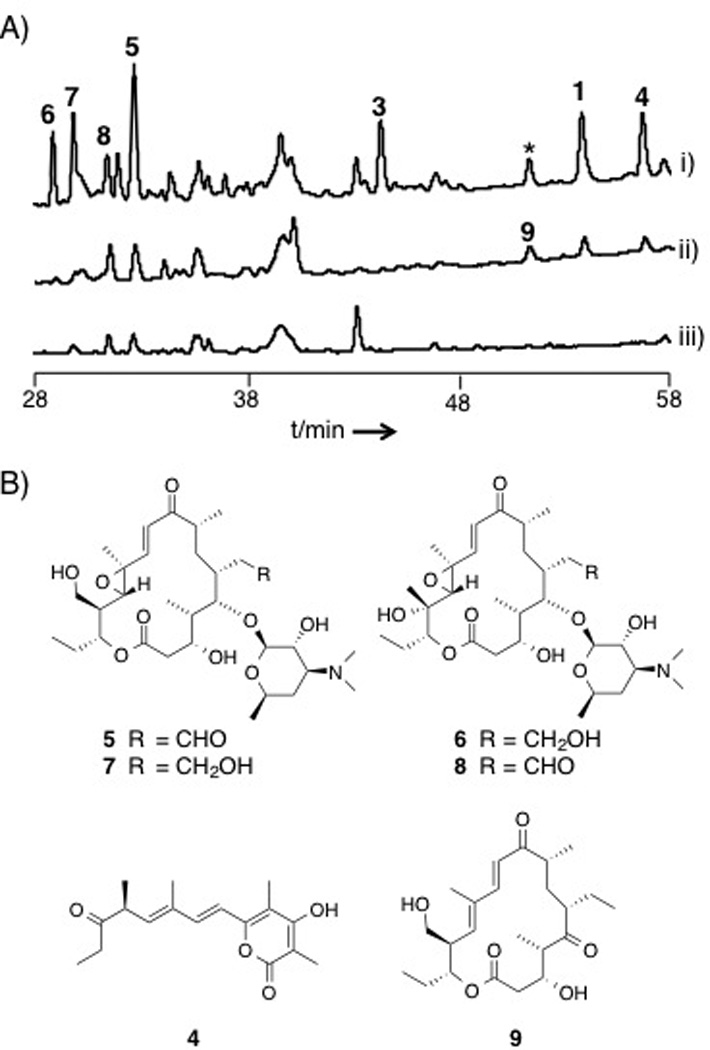
(A) HPLC chromatograms of the metabolites from i) CNS-237 wild-type, ii) spr10Y1290F, and iii) Δspr cultured in A1FeBC medium. The traces represent chromatograms acquired by detection at 254 nm. (* indicates the compound whose MS and UV spectrum are identical to 1.) Though 9 is eluted at the same RT as the compound marked with an asterisk, the peak of 9 is detected at 280 nm only in the Y1290F strain (Figure S2). (B) Chemical structures of 4–9.
The biosynthetic logic of the spr gene cluster is consistent with rosamicin octaketides 3, 5–8 in which all eight PKS modules of Spr7–11 are progressively involved in the linear construction of the macrolide aglycone (Scheme 2A). The smaller salinipyrone hexaketides 1 and 4, however, are consistent with an incomplete processing by the PKS in which modules 6 and 8 are both skipped. Alternatively, the slightly larger heptaketide 2 could derive by the skipping of just the final PKS module 8. In both cases, the truncated products appear to arise from intermediates independent of the ketoreductase (KR) domain of Spr10 (Scheme 2A), suggesting its role, both directly and indirectly, in product dispersal. To explore this concept, we inactivated the spr10-KR domain by introducing a chromosomal point mutation through a single reciprocal recombination event to effectively replace the catalytic tyrosine residue at position 1290 with phenylalanine. Detailed HPLC-MS analyses revealed that the mutant no longer produced the rosamicin octaketides 3, 5–8 but retained 1 and 4, albeit at lower production titers (Figure 1A). The spr10-Y1290F mutant also produced a new product, 18-deoxo-5-deoxy-5-oxotylonolide (9), in which the desosamine sugar, the C12-C13 epoxide, and the C18 hydroxyl groups were missing, suggesting that 9 is not a substrate for post-PKS modifying enzymes. We further identified two new compounds with the same molecular composition as 2 and the epimeric pacificanone B (Figure S3), yet due to poor production yields, we were unable to conclusively elucidate their structures.
Scheme 2.
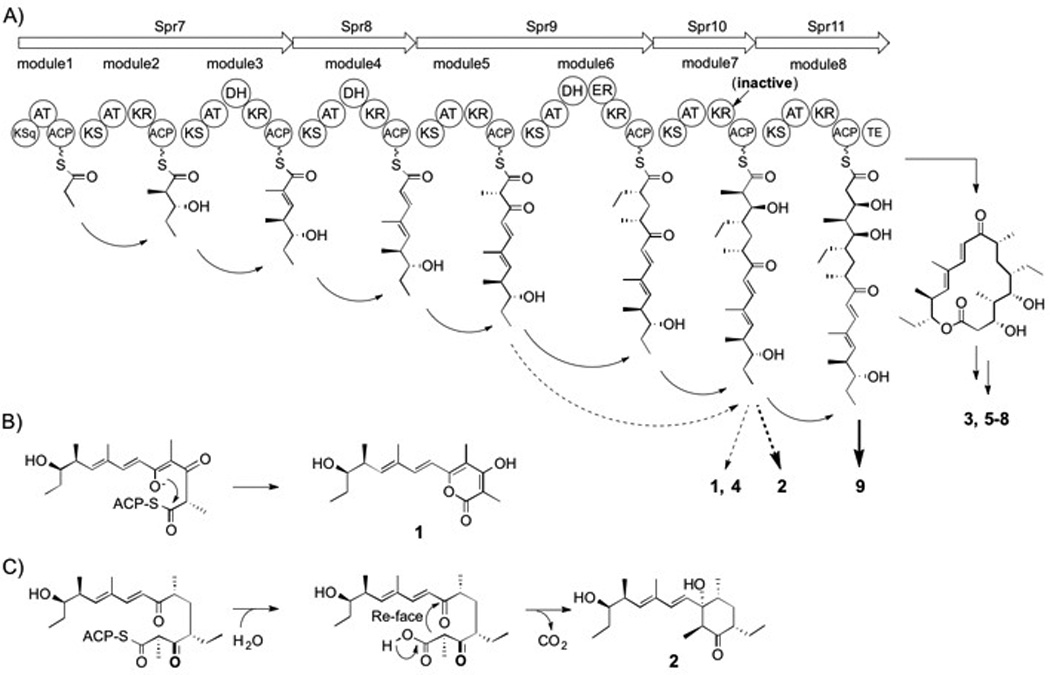
Proposed biosynthesis of 1–9. (A) Spr PKS organization and deduced assembly of 1–9. The arrows depict the sequential synthesis of intermediates leading to the mature products 3 and 5–8. The dotted arrows represent module skipping movements leading to 1 and 4. The bold arrow shows the assembly of 9, while the bold dotted arrow leading to 2 involves the skipping of the final module. In both cases, the KR domain in module 7 is inactive. The formation of 1 (B) and 2 (C) involve reactions on premature ACP-bound intermediates.
Inspection of the wild-type S. pacifica CNS-237 strain in certain growth media also revealed 9 (Figure S4), albeit at lower levels than the spr10-Y1290F mutant strain, suggesting that the Spr10 KR7 domain is naturally partially effective in processing Spr10-linked substrates. We suspect that unreduced β-keto intermediates linked to the module 7-acyl carrier protein (ACP7) also give rise to products in the truncated salinipyrone/pacificanone series, and that their protein offloading may be driven by the innate chemical reactivity of their associated polyketone substrates (Scheme 2B and C). In the case of 1, this proposal combines a rare intermodule skipping mechanism (module 6) and a domain skipping reaction (KR7) to yield the ACP7-bound salinipyrone intermediate that is likely released through a spontaneous enol lactonization reaction. This biosynthetic bifurcation mediated by module and domain skipping explains the different product outcomes of the Spr PKS in yielding the salinipyrone, pacificanone, and rosamicin natural products, and represents an entirely different mechanism than what we previously reported in the related bacterium Salinispora arenicola that co-produces the arenicolide and rifamycin polyketide natural products by off-PKS enzymatic processing.[12] This study further adds to our growing appreciation of the enzymatic complexity of biosynthetic assembly lines[13] and illuminates how much we still need to learn about the structural complexity of these systems to use their full potential in ongoing PKS bioengineering research.
Experimental Section
General
All chemicals were acquired from Fisher Scientific and Sigma–Aldrich and used without further purification. PCR was performed with Takara PrimeStarMax DNA polymerase. Gibson assembly cloning kits were from New England Biolabs. All solvents were of HPLC grade or higher. Analytical HPLC analyses were conducted with an Agilent 1200 HPLC system with diode array detection connected to a Phenomenex Luna C18(2), 250 × 21.5 mm × 10 mm column. LC-MS analyses were conducted with an Agilent 6530 Accurate-Mass Q-TOF MS (MassHunter software, Agilent) equipped with a Dual electrospray ionization source and an Agilent 1260 LC system (ChemStation software, Agilent) with diode array detector. Preparative HPLC was carried out using an Agilent 218 purification system (ChemStation software, Agilent) equipped with a ProStar 410 automatic injector, an Agilent ProStar UV-Vis Dual Wavelength Detector, and a 440-LC fraction collector connected to Phenomenex Luna C18(2), 10.0 × 250 mm, 5 µm column. NMR data were acquired at the UCSD Skaggs School of Pharmacy and Pharmaceutical Sciences NMR Facility on a 600 MHz Varian NMR spectrometer (Topspin 2.1.6 software, Bruker) with a 1.7 mm cryoprobe.
Construction of spr gene inactivation plasmid
A 3.0-kb DNA fragment containing the spr8-coding region was amplified by PCR with primer D1f, 5’-accatgattacgccaagcttGATTCCCGTCGTTAGCAATGTGACC-3’ (overlap sequence boldfaced), and D1r, 5’-cagctccagcctaca GACTGGTAGCCGACTGTCGATCTAG-3’ (overlap sequence boldfaced), and the chromosome of CNT-237; a 1.1-kb DNA fragment containing aac(3)IV and OriT was amplified by PCR with primer AprOriTf, 5’- AGTCGGCTACCAGTCTGTAGGCTGGAGCTGCTTC-3’ (overlap sequence boldfaced), and AprOriTr, 5’-GAAACCCATGGCTGACATTATTCCGGGGATCCGTCGACC-3’ (overlap sequence boldfaced), and pIJ773; and a 3.0-kb DNA fragment containing the Spr11-coding region was amplified by PCR with primer D2f, 5’-ggtcgacggatccccggaatAATGTCAGCCATGGGTTTCATTCGG-3’ (overlap sequence boldfaced), and D2r, 5’-aaaacgacggccagtgaattcCGCGTATTCGGTCATCATCGACAAG-3’ (overlap sequence boldfaced), and the chromosome of CNT-237. The three amplified fragments were assembled with a linear pUC19 which is amplified by PCR with primer pUC19f, 5’-gaattcactggccgtcgttttacaac-3’, and pUC19r, 5’-aagcttggcgtaatcatggtcatagc-3’, resulting in pUC19-Δspr.
Construction of spr10-KR point mutation plasmid
A 3.0-kb DNA fragment containing the Spr10-coding region was amplified by PCR with primer Spr10KR1f, 5’-cccccgggctgcaggaattcCCGGTACGGCGATCAACCAGGAC-3’ (overlap sequence boldfaced), and Spr10KR1r, 5’-GCCCCGAATGCTGATTGCCCGCCGCTACCCCACACGCCCGCGTTTGAG-3’ (overlap sequence boldfaced, the mutated sequence for Y1290F was italicized), and the chromosome of CNT-237; and a 3.0-kb DNA fragment containing the spr10-coding region was amplified by PCR with primer Spr10KR2f, 5’-GGGCAATCAGCATTCGGGGCCGCCAACGCAGCCCTGGACGCAC -3’ (overlap sequence boldfaced, the mutated sequence for Y1290F was italicized), and Spr10KR2r, 5’-ccagcctacacatcgaattcAGCGGTGCGATCTCTTCCTCGCTG-3’ (overlap sequence boldfaced), and the chromosome of CNT-237. The two amplified fragments were assembled with a linear pIJ773 which is amplified by PCR with primer pIJ773f, 5’- gaattcctgcagcccgggggatc-3’, and pIJ773r, 5’-gaattcgatgtgtaggctggag-3’, resulting in pIJ773-spr10.
Introducing plasmids into S. pacifica CNT-237 for the inactivation of spr and the point mutation of the Spr10KR
pUC19-Δspr and pIJ773-spr10 were separately introduced into E. coli S17-1 cells by electroporation and then transferred into S. pacifica CNS-237 by intergeneric conjugation. Mutants, in which the apramycin resistance cassette was integrated into the S. pacifica chromosome, were selected on A1 agar by overlaying plates with 200 µgmL−1 apramycin and 100 µgmL−1 nalidixic acid. Following incubation at 28°C for 7–9 days, exconjugants were randomly selected and those exhibiting an Aprr (for pUC19-Δspr) or Aprs phenotype (for pIJ773-spr10) were evaluated by PCR and DNA sequencing for double crossover mutation, respectively. Primer set-1 (f, 5’-AGACCGCTGTGGAAGAACAGATCG-3’ and r, 5’-gaacttcgaagcagctccagcctac-3’) and primer set-2 (f, 5’-cttattcgcacctggcggtgctcaacg-3’ and r, 5’-CGCTAACGGGGGATGTCATAATCTGC-3’) were used for the PCR for the evaluation of Δspr strain. 595-bp fragment amplified by KRpcrf, 5’-gaacttcgaagcagctccagcctac-3’ and KRpcrr, 5’-CGCGGTGAACAGATCCCAATCCACGTC-3’ was sequenced for the evaluation of spr10-Y1290F strain.
Comparative metabolite profiling between wild type and mutant S. pacifica strains
Wild type S. pacifica CNS-237 and the mutants prepared above were grown in 1.0 L liquid seawater-based A1+BFe medium (10 g starch, 4 g yeast extract, 2 g, peptone, 1 g CaCO3, 40 mg Fe2(SO4)3•4H2O, 100 mg KBr) for 11 days at 28°C with shaking at 230 rpm. 20 mL of the whole culture was extracted by EtOAc, and the resulting extract was concentrated to dryness in vacuo and dissolved with 500 µL CH3OH. HPLC analyses were conducted with a flow rate of 1.5 mLmin−1 and a gradient from 10–100% CH3CN (0.1% TFA)/water (0.1% TFA) for 90 min, and LC-MS analyses were conducted with a flow rate of 0.7 mLmin−1 and mobile phase gradient from 10–100% CH3CN (0.1% formic acid)/water (0.1% formic acid) for 23 min with positive mode ESI.
Preparation of 5–8
S. pacifica CNS-237 was cultured in five 2.8 L Fernbach flasks, each containing 1.0 L of seawater-based A1-BFe medium and shaken at 230 rpm at 27°C. After seven days of cultivation, HP-20 resin was added to adsorb the organic products, and the culture and resin were stirred for 1.5 h. The resin was filtered by a glass column, washed with deionized water, and eluted with CH3OH. The methanol was removed under reduced pressure, and the resulting aqueous layer was extracted with 100:5 CH2Cl2:CH3OH. The extract was subjected to silica-gel column chromatography using a CH2Cl2:CH3OH stepwise gradient system (100:5, 100:7.5, 100:10 and 100:20). The fraction containing 7–8 (100:10) was purified by reversed-phase preparative HPLC using a gradient of 10–55% CH3CN (0.1% TFA)/water (0.1% TFA) for 30 min twice to afford 0.3 mg of 8, and 0.2 mg of 7. The fraction containing 5–6 (100:20) was purified by reversed-phase preparative HPLC using a gradient of 10–50% CH3CN (0.1% TFA)/water (0.1% TFA) for 30 min to afford 0.5 mg of 6, and 0.1 mg of 5.
Preparation of 3
S. pacifica CNS-237 was cultured in three 2.8 L Fernbach flasks, containing 1.0 L of seawater-based A1-BFe medium and shaken at 230 rpm at 27°C. After seven days of cultivation, the whole culture was extracted by EtOAc, and the resulting extract was concentrated to dryness in vacuo. The extract was subjected to silica-gel column chromatography using a CH2Cl2:CH3OH stepwise gradient system (100:0, 100:1, 100:2, 100:3, 100:4 and 100:5). The fraction containing 3 (100:2) was purified by reversed-phase preparative HPLC using a gradient of 10–60% CH3CN (0.1% TFA)/water (0.1% TFA) for 40 min to afford 0.5 mg of 3.
Preparation of 1 and 4
S. pacifica CNS-237 was cultured in a 2.8 L Fernbach flask, containing 1.0 L of seawater-based A1-BFe medium and shaken at 230 rpm at 27°C for 3 days. 25 mL of the culture was transferred into another 2.8 L Fernbach flask, containing 1.0 L of seawater-based A1-BFe medium and shaken at 200 rpm at 27°C. After seven days of cultivation, the whole culture was extracted with EtOAc, and the resulting extract was concentrated to dryness in vacuo. The extract was subjected to silica-gel column chromatography using a CH2Cl2:CH3OH stepwise gradient system (100:0, 100:1, 100:3, 100:4 and 100:5). The fractions containing 1 (100:4 and 100:5) were purified by reversed-phase preparative HPLC using 45% CH3CN (0.1% TFA)/water (0.1% TFA) for 40 min to afford 0.4 mg of 1. The fractions containing 4 (100:3) were purified by reversed-phase preparative HPLC using 50% CH3CN (0.1% TFA)/water (0.1% TFA) for 40 min to afford 0.3 mg of 4.
Preparation of 9
The spr10Y1290F strain was cultured in four 2.8 L Fernbach flasks, each containing 1.0 L of seawater-based A1-production medium (10 g starch, 4 g yeast extract, 2 g, peptone, 1 g CaCO3) and shaken at 230 rpm at 27°C. After seven days of cultivation, the whole culture was extracted with EtOAc, and the resulting extract was concentrated to dryness in vacuo. The extract was subjected to silica-gel column chromatography using a CH2Cl2:CH3OH stepwise gradient system (100:0, 100:1, 100:3 and 100:5). The fraction containing 9 (100:3) was purified by reversed-phase preparative HPLC using 40% CH3CN (0.1% TFA)/water (0.1% TFA) for 40 min to afford 0.8 mg of 9.
Spectroscopic data
Salinipyrone A (1)
yellow oil; [α]D25.9 = −76.2 (c = 0.33, CH3OH); 1H- and 13C-NMR: see Table S2; HR-ESI-MS (positive) m/z 293.1760 [M+H]+ (calcd for C17H25O4 293.1747).
Rosamicin A (3)
white powder; [α]D26.6 = −25.4 (c = 0.30, EtOH); 1H- and 13C-NMR: see Table S7; HR-ESI-MS (positive) m/z 582.3660 [M+H]+ (calcd for C31H52NO9 582.3636).
Dehydrosalinipyrone A (4)
yellow oil; [α]D26.3 = +30.6 (c = 0.33, CH3OH); 1H- and 13C-NMR: see Table S8; HR-ESI-MS (positive) m/z 291.1605 [M+H]+ (calcd for C17H23O4 291.1590).
18-Dihydro-21-hydroxyrosamicin (5)
white powder; [α]D27.4 = −18.8 (c = 0.30, EtOH); 1H- and 13C-NMR: see Table S4; HR-ESI-MS (positive) m/z 600.3751 [M+H]+ (calcd for C31H54NO10 600.3742).
18-Dihydro-14-hydroxyrosamicin (6)
white powder; [α]D27.4 = −27.8 (c = 0.30, EtOH); 1H- and 13C-NMR: see Table S3; HR-ESI-MS (positive) m/z 600.3768 [M+H]+ (calcd for C31H54NO10 600.3742).
21-Hydroxyrosamicin (izenamicin A3) (7)
white powder; [α]D27.4 = −34.9 (c = 0.30, EtOH); 1H- and 13C-NMR: see Table S6; HR-ESI-MS (positive) m/z 598.3580 [M+H]+ (calcd for C31H52NO10 598.3586).
14-Hydroxyrosamicin (8)
white powder; [α]D27.4 = −2.4 (c = 0.30, EtOH); 1H- and 13C-NMR: see Table S5; HR-ESI-MS (positive) m/z 598.3584 [M+H]+ (calcd for C31H52NO10 598.3586).
18-Deoxo-5-deoxy-5-oxotylonolide (9)
white powder; [α]D27.4 = +33.6 (c = 0.30, EtOH); 1H- and 13C-NMR: see Table S9; HR-ESI-MS (positive) m/z 431.2426 [M+Na]+ (calcd for C23H36O6Na 431.2410).
Supplementary Material
Acknowledgements
We thank our UCSD colleagues B. M. Duggan for assistance with NMR data acquisition and analysis, P.R. Jensen for materials and helpful advise, and E. O’Neill, S. Diethelm, K. Murata, and C.A. Kauffman for discussions. Funding was generously provided by NIH grant GM085770 to B.S.M. and the Uehara Memorial Foundation via a postdoctoral fellowship to T.A.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Oh D, Gontang EA, Kauffman CA, Jensen PR, Fenical W. J. Nat. Prod. 2008;71:570–575. doi: 10.1021/np0705155. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jensen PR, Moore BS, Fenical W. Nat. Prod. Rep. 2015 doi: 10.1039/c4np00167b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Xue Y, Sherman DH. Nature. 2000;403:571–575. doi: 10.1038/35000624. [DOI] [PubMed] [Google Scholar]; b) Beck BJ, Yoon YJ, Reynolds KA, Sherman DH. Chem. Biol. 2002;9:575–583. doi: 10.1016/s1074-5521(02)00146-1. [DOI] [PubMed] [Google Scholar]; c) Kittendorf JD, Beck BJ, Buchholz TJ, Seufert W, Sherman DH. Chem. Biol. 2007;14:944–954. doi: 10.1016/j.chembiol.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kittendorf JD, Sherman DH. Bioorg. Med. Chem. 2009;17:2137–2146. doi: 10.1016/j.bmc.2008.10.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ross A, Xu Y, Lu L, Kersten RD, Shao Z, Al-Suwailem AM, Dorrestein PC, Qian P, Moore BS. J. Am. Chem. Soc. 2013;135:1155–1162. doi: 10.1021/ja3119674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ziemert N, Lechner A, Wietz M, Millán-Aguiñaga N, Chavarria KL, Jensen PR. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E1130–E1139. doi: 10.1073/pnas.1324161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson MC, Moore BS. Nat. Prod. Rep. 2012;29:72–86. doi: 10.1039/c1np00082a. [DOI] [PubMed] [Google Scholar]
- 6.Farnet CM, Staffa A, Yang X. 20030113874. United States Patent US. 2002
- 7.Iizaka Y, Higashi N, Ishida M, Oiwa R, Ichikawa Y, Takeda M, Anzai Y, Kato F. Antimicrob. Agents Chemother. 2013;57:1529–1531. doi: 10.1128/AAC.02092-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao L, Sherman DH, Liu H. J. Am. Chem. Soc. 1998;120:10256–10257. [Google Scholar]
- 9.Imai H, Suzuki K, Morioka M, Sakaki T, Tanaka K, Kadota S, Iwanami M, Saito T, Eiki H. J. Antibiot. 1989;42:1000–1002. doi: 10.7164/antibiotics.42.1000. [DOI] [PubMed] [Google Scholar]
- 10.a) Hayashi M, Kinoshita K, Satoi S, Nakatsu K. J. Antibiot. 1982;35:1243–1244. doi: 10.7164/antibiotics.35.1243. [DOI] [PubMed] [Google Scholar]; b) Kinoshita K, Satoi S, Hayashi M, Harada K, Suzuki M, Nakatsu K. J. Antibiot. 1985;38:522–526. doi: 10.7164/antibiotics.38.522. [DOI] [PubMed] [Google Scholar]; c) Kinoshita K, Satoi S, Hayashi M, Nakatsu K. J. Antibiot. 1989;42:1003–1005. doi: 10.7164/antibiotics.42.1003. [DOI] [PubMed] [Google Scholar]
- 11.a) Anzai Y, Li S, Chaulagain MR, Kinoshita K, Kato F, Montgomery J, Sherman DH. Chem. Biol. 2008;15:950–959. doi: 10.1016/j.chembiol.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li S, Tietz DR, Rutaganira FU, Kells PM, Anzai Y, Kato F, Pochapsky TC, Sherman DH, Podust LM. J. Biol. Chem. 2012;287:37880–37890. doi: 10.1074/jbc.M112.410340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson MC, Gulder TAM, Mahmud T, Moore BS. J. Am. Chem. Soc. 2010;132:12757–12765. doi: 10.1021/ja105891a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khosla C, Herschlag D, Cane DE, Walsh CT. Biochemistry. 2014;53:2875–2883. doi: 10.1021/bi500290t. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.