

Abstract
The incidence and prevalence of pathological fibrosis increase with advancing age, although mechanisms for this association are unclear. We assessed the capacity for repair of lung injury in young (2 months) and aged (18 months) mice. While the severity of fibrosis was not significantly different between these groups, aged mice demonstrated an impaired capacity for fibrosis resolution. Persistent fibrosis in lungs of aged mice is characterized by the accumulation of senescent and apoptosis-resistant myofibroblasts. These cellular phenotypes are sustained by alterations in cellular redox homeostasis resulting from elevated expression of the reactive oxygen species (ROS)-generating enzyme, NADPH oxidase-4 (Nox4), and an impaired capacity to induce the NFE2-related factor 2 (Nrf2) antioxidant response. Lung tissues from human subjects with idiopathic pulmonary fibrosis (IPF), a progressive and fatal lung disease, also demonstrate this Nox4-Nrf2 imbalance. Nox4 mediates senescence and apoptosis resistance in IPF fibroblasts. Genetic and pharmacologic targeting of Nox4 in aged mice with established fibrosis attenuated the senescent, anti-apoptotic myofibroblast phenotype and led to a reversal of persistent fibrosis. These studies support the concept that loss of cellular redox homeostasis promotes pro-fibrotic myofibroblast phenotypes that result in persistent fibrosis associated with aging. Importantly, our studies suggest that restoration of Nox4-Nrf2 redox balance in myofibroblasts may be an effective therapeutic strategy in age-associated fibrotic disorders, potentially to resolve persistent fibrosis or even reverse its progression.
Introduction
Regenerative capacity varies widely across the animal kingdom, and is largely limited by the propensity for fibrosis (1, 2). Reduced regenerative capacity in humans is associated with pathological fibrosis of vital internal organs, such as the heart, lung, central nervous system and kidneys. Human fibrotic disorders are estimated to contribute to 45% of all-cause mortality in the United States (3). Idiopathic pulmonary fibrosis (IPF) is a progressive and fatal lung disease with no effective treatment or cure. The loss of cellular homeostasis in IPF lungs is characterized by accumulating clusters of myofibroblasts (fibroblastic foci), the profusion of which portends decreased survival (4). The myofibroblast is a key effector cell in diverse fibrotic disorders (5); this cell type is primarily responsible for extracellular matrix (ECM) synthesis and tissue remodeling in progressive fibrosis (6). The inability to terminate the host reparative response, specifically myofibroblast activation/accumulation, may underlie the progressive nature of fibrotic reactions in injured tissues (7).
Aging is a risk factor for fibrotic disease (6), including IPF (8). The incidence and prevalence of IPF increase with age, with a mean age greater than 65 years at the time of diagnosis (8, 9). Despite the strong association between aging and IPF, few studies have investigated cellular/molecular mechanisms that account for this age-associated predilection. Additionally, while oxidative stress is associated with age-associated diseases such as IPF (10), it remains unclear how oxidative stress in aging contributes to the pathogenesis of fibrosis.
In this study, we evaluated the reparative response to lung injury in young and aged mice. Our results demonstrate that the capacity for fibrosis resolution in aged mice is markedly impaired. Persistent fibrosis in aged mice is associated with the emergence of a senescent and apoptosis-resistant myofibroblast phenotype mediated, at least in part, by elevated expression of the reactive oxygen species (ROS)-generating enzyme, NADPH oxidase-4 (Nox4), and an impaired capacity to induce the NFE2-related factor 2 (Nrf2) antioxidant responses. Lung tissues from human subjects with IPF confirm high expression of Nox4 in fibroblastic foci, where Nrf2 expression is reduced. In vivo knockdown of Nox4 and pharmacologic targeting of Nox4 during the persistent phase of lung fibrosis in aged mice restores the capacity for fibrosis resolution. These studies support the concept that loss of redox homeostasis in aging promotes the emergence/persistence of a senescent and apoptosis-resistant myofibroblast phenotype that sustains persistent/progressive fibrotic disorders.
Results
Severity of fibrosis following lung injury is similar in young and aged mice
Previous studies indicate an association between age and severity of fibrosis in murine models of lung injury (11–13). However, these studies did not assess dynamic changes over time or the capacity for resolution of established fibrosis. In the current study, we first evaluated fibrotic responses in the lungs of young (2 months) and aged (18 months) mice subjected to the bleomycin lung injury model. In this animal model, intra-tracheal instillation of the chemotherapeutic agent, bleomycin, induces epithelium injury that leads to fibrosis which peaks 2–3 weeks post-injury (14) (Fig. 1A). We observed no significant difference in severity of fibrosis (net increase in total lung hydroxyproline at 3 weeks post-injury) between young and aged mice (Fig. 1B, Masson’s trichrome staining for collagen; Fig. 1C, whole lung hydroxyproline). These data suggest that the higher predilection of fibrosis with aging cannot be explained by a difference in the severity of the initial fibrogenic response to lung injury.
Figure 1.
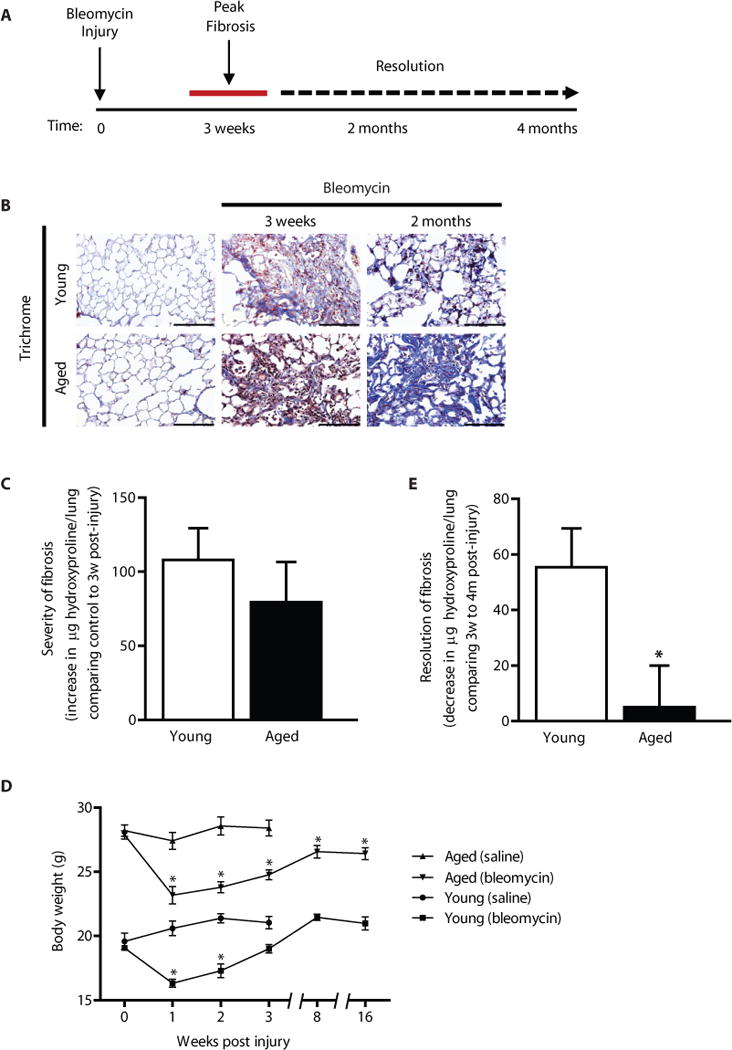
Resolution of fibrosis is impaired in aged mice. C57BL/6 young (2 months) and aged (18 months) mice were subjected to lung injury by airway instillation of intra-tracheal bleomycin (1.25 U/kg). (A) Schematic diagram illustrating the time-course of bleomycin-induced fibrosis and resolution in young mice. (B–D) Lung tissue was harvested at 0 (uninjured), 3, weeks, 2 months, or 4 months post-injury and fibrosis was assessed. Resolution of fibrosis was assessed by Masson’s trichrome blue staining for collagen (B), and whole lung homogenates were analyzed by quantitative hydroxyproline assay (C–D); data are expressed as increase in μg of hydroxyproline/lung comparing control to 3 weeks post-injury (C), or decrease in μg of hydroxyproline/lung comparing 3 weeks to 4 months post-injury (D). Values represent mean ± s.e.m.; n = 5–9 per group; *p value < 0.05, compared with young. (E) Systemic effects in response to bleomycin-induced lung fibrosis or saline control were assessed by a time-course evaluation of body weights. Values represent mean ± s.e.m.; n = 5–20 per group; *p value < 0.05, compared to baseline for each group. Scale bars, 100 μm.
Resolution of fibrosis is impaired in aged mice
We next assessed whether the ability to resolve fibrosis is impaired in aged mice. The net loss of total lung hydroxyproline from peak fibrosis at 3 weeks to 4 months post-injury was evaluated in young and aged mice. Young mice demonstrate a significant decrease in total lung hydroxyproline; in contrast, fibrosis fails to resolve in aged mice (Fig. 1B, histopathology and trichrome staining for collagen; Fig. 1D and Fig. S1, whole lung hydroxyproline). In parallel, we evaluated the body weights of young and aged mice throughout this 4 month time period; body weights of bleomycin-injured mice are known to decrease due to systemic effects of lung injury. We observed that, in both young and aged mice, there was a similar decrease in weights 1–2 weeks post-injury. Recovery to baseline weights was observed in young mice (Fig. 1E); whereas aged mice remained below baseline levels throughout the observation period, up to 4 months (Fig. 1E). These data provide evidence for a deficiency in the capacity for fibrosis resolution in aged mice.
Lack of fibrosis resolution in aged mice is associated with the acquisition of a senescent and apoptosis-resistant phenotype
The myofibroblast is a key effector cell type and a central mediator of fibrosis in diverse fibrotic disorders (15). We evaluated the presence of myofibroblasts in lung tissue sections of young and aged mice at baseline, 3 weeks, and 2 months post-injury. Aged mice exhibited myofibroblast persistence in the fibrotic regions of the lung at 2 months post-injury, as determined by immunohistochemical (IHC) staining for α-SMA, a marker of myofibroblasts (Fig. 2A, top panels). Fibroblast senescence has been proposed as a mechanism to limit the fibrogenic response to tissue injury (16). We evaluated the expression of the senescence-associated tumor suppressor gene, p16, in the lungs of young and aged mice following injury. Fibroblasts isolated from both young and aged mice demonstrated induction of p16 in response to injury (Fig. S2A). However, this induced expression of p16 in the lungs of young mice returned to baseline levels by 2 months post-injury (Fig. 2A, IHC, bottom panels; Fig. S2B, western blot of whole lung homogenates, Fig. 2B, densitometric analysis); in contrast, p16 remained highly expressed in the lungs of aged mice with persistent fibrosis (Fig. 2A–B; Fig. S2B). Similarly, fibroblasts isolated from both young and aged mice exhibit increased senescence, as determined by cellular staining for senescence-associated β-galactosidase (SA-βgal) (Fig. S2C). However, this senescence response was transient in young mice with resolving fibrosis, whereas senescence remains markedly elevated in aged mice at this delayed time point (Fig. 2C, quantitative fluorescence assay for SA-βgal activity; Fig. S2C, cellular staining for SA-βgal). These data indicate that persistent fibrosis in aged mice is associated with the accumulation of senescent myofibroblasts.
Figure 2.
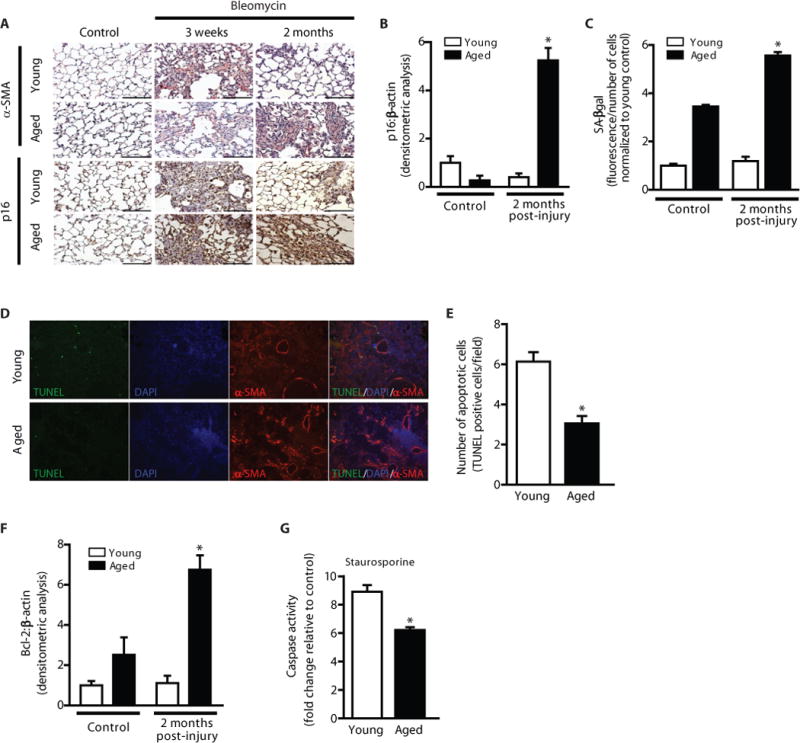
Impaired fibrosis resolution in aged mice is associated with myofibroblast senescence and apoptosis resistance. Young and aged mice were subjected to bleomycin-induced lung fibrosis. Lung tissue was harvested or cells were isolated at various time points following injury. (A) Immunohistochemical analysis of α-SMA expression; myofibroblast marker (upper panels), and p16 expression; senescence marker (lower panels). (B,F) Whole lung tissues were analyzed at the time points indicated by Western blot for protein expression and densitometric analyses of p16 (B) and Bcl-2 (F) was performed; *p value < 0.05, compared with all other groups. (C) Fibroblasts from young and aged mice (uninjured and 2 m post-injury) were isolated and cultured ex vivo. Senescence was evaluated by quantitative measurement of senescence-associated β-galactosidase (SA-βgal) activity; *p value < 0.05, compared with all other groups. (D–E) Lung tissue harvested at 3 weeks post-injury was assessed by immunofluorescence for TUNEL and α-SMA expression (D), and apoptotic cells were quantified by counting the number of TUNEL-positive cells/field in >50 random fields of view (E); *p value < 0.05, compared with young. (G) Fibroblasts were isolated from young and aged mice 6 weeks post-injury and cultured ex vivo. Cells were treated with or without staurosporine (300 nM) for 5 h and caspase activity was assessed. Values represent mean ± s.e.m.; n = 3–5; *p value < 0.05, compared with young. Scale bars, 100 μm.
Apoptosis of tissue myofibroblasts is required for the resolution of fibrosis during normal wound healing (17). We sought to determine the apoptotic fate of senescent fibroblasts in the context of aging and tissue repair. Lung tissue sections from aged mice post-lung injury show lower levels of apoptosis (TUNEL+ cells) in fibrotic regions in comparison to young mice (Fig. 2D–E). Whole lung homogenates from young and aged mice during the early resolving phase were analyzed for expression of the apoptotic marker, DNA fragmentation factor (DFF); lung tissue from aged mice demonstrate decreased levels of DFF as compared to young cohorts (Fig. S2D–E) and elevated levels of Bcl-2 (Fig. S2B, western immunoblotting and Fig. 2F, densitometric analysis). To determine apoptosis susceptibilities of young and aged fibroblasts in the context of lung injury, we isolated fibroblasts from mice 6 weeks post-injury and challenged cells with the apoptosis-inducing agent staurosporine; fibroblasts from aged mice were relatively resistant to apoptosis, as measured by cleaved caspase-3 activity (Fig. 2G). Taken together, these data demonstrate that persistent fibrosis in aging is associated with acquisition of a senescent and apoptosis-resistant fibroblast phenotype.
Nox4 mediates senescence and apoptosis resistance in IPF lung fibroblasts
Although fibrosis is generally thought to be a fibro-“proliferative” process, the relative roles of proliferation and senescence in IPF lung tissues is unknown. First, we determined whether fibroblasts within fibroblastic foci of IPF lungs demonstrate features of senescence. We detected expression of p16 and p21 in fibroblasts within the foci and in the overlying epithelial cells (Fig. 3A, left panels; Fig. S3). Interestingly, cells expressing Ki67, a marker of cell proliferation, were primarily detected at the periphery and were largely absent within fibroblastic foci (Fig. 3A, right panels). These data indicate the presence of a predominantly non-proliferative, senescent cellular phenotype within fibroblastic foci of human IPF lungs. To determine if myofibroblasts within fibroblastic foci manifest apoptosis resistance, we performed terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and immunofluorescence staining for α-SMA on the same IPF lung tissue sections. We detected high levels of apoptosis in epithelial cells lining alveolar spaces, with little evidence of apoptosis in subepithelial α-SMA-positive myofibroblasts (Fig. 3B). These observations support the concept that myofibroblasts within fibroblastic foci of IPF lungs acquire a senescent and apoptosis-resistant phenotype.
Figure 3.
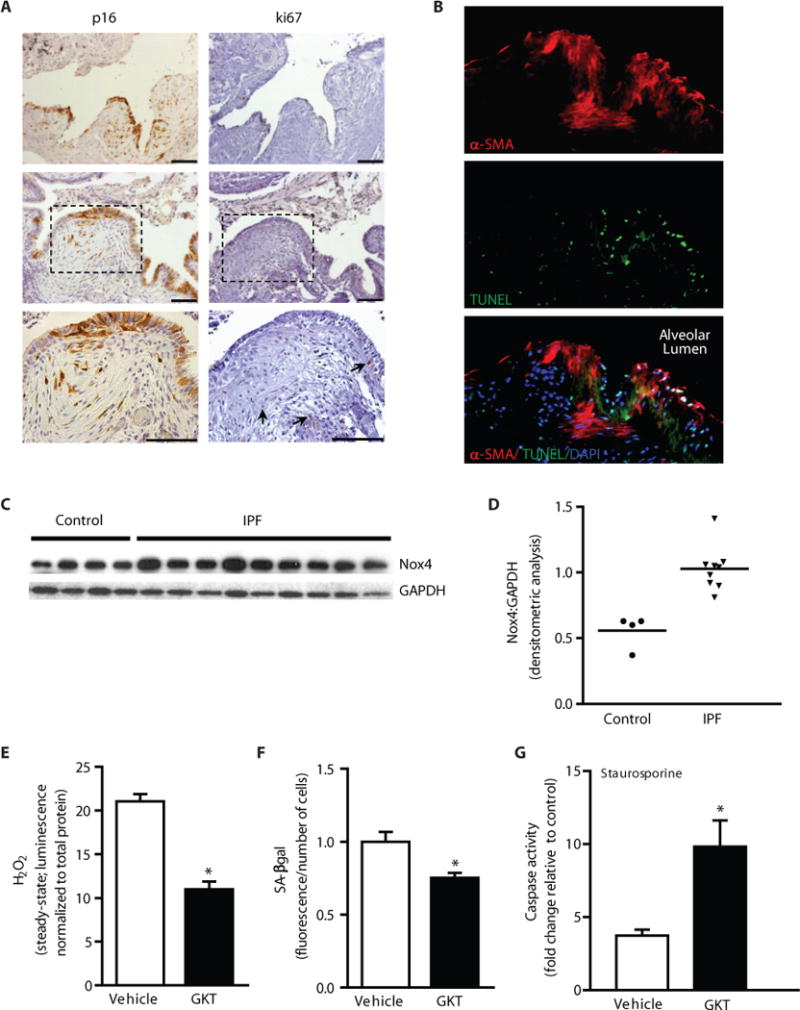
IPF lung myofibroblasts express Nox4 and manifest features of senescence and apoptosis resistance in vitro and in vivo. (A–B) IPF lung tissue sections were analyzed by immunohistochemical analysis (A) and immunofluorescence staining (B). Expression of p16 and Ki67 were evaluated in two different patients (patient 1 in top panels, patient 2 in lower panels). Dashed boxes represent area shown in higher magnification (bottom panels); arrows indicate Ki67+ cells (A). Immunofluorescence labeling for TUNEL, α-SMA, and DAPI in IPF lung tissue sections (B). (C–D) Fibroblasts isolated from the lungs of patients with biopsy-proven IPF (n = 9) and normal-appearing lung tissue from patients undergoing surgical resection for suspected cancer (n=4), and analyzed for protein expression of Nox4 by Western immunoblotting (C); and quantitated by densitometric analysis (D). (E–G) IPF lung fibroblasts were cultured ex vivo and treated with GKT137831 (10 μM) or vehicle (DMSO) for 48 h. H2O2 production was evaluated (E) and senescence was evaluated by quantitative SA-βgal activity assay (F). Cells were treated with or without staurosporine (300 nM) for 8 h and caspase activity was assessed (G). Values represent mean ± s.e.m.; n = 3; *p value < 0.05. Scale bars, 100 μm.
We have previously reported that Nox4 is expressed within fibroblastic foci of IPF lung tissues (18). Here, we examined Nox4 expression in fibroblasts isolated from IPF lungs; constitutive expression of Nox4 protein was significantly higher in fibroblasts isolated from IPF compared to that of control subjects without IPF (Fig. 3C, western blot and Fig. 3D, densitometric analysis). To evaluate the role of Nox4-dependent H2O2 in mediating IPF fibroblast phenotypes, we utilized a first-in-class small-molecule inhibitor of Nox1/4, GKT137831, developed by Genkyotex (Geneva, Switzerland). IPF lung fibroblasts treated with GKT137831 (10 μM) exhibited decreased H2O2 production (Fig. 3E), confirming Nox4 inhibitory activity of this compound. GKT137831 significantly attenuated SA-βgal activity in IPF lung fibroblasts (Fig. 3F), suggesting that Nox4 contributes to cellular senescence of IPF fibroblasts. To evaluate the role of Nox4 in conferring apoptosis resistance, IPF lung fibroblasts were pre-treated with GKT137831 or vehicle and subjected to staurosporine-induced apoptosis; GKT137831 restored IPF fibroblast susceptibility to apoptosis (Fig. 3G; caspase activity). Together, these data support a role for Nox4 in mediating senescence and apoptosis resistance of IPF lung myofibroblasts.
Oxidative stress-induced Nrf2 antioxidant response is impaired in aging
Oxidative stress is defined as an imbalance between ROS production and the antioxidant capacity of cells (19). Oxidative stress has been associated with fibrotic disorders, including IPF (10). In response to oxidative stress, induction of the transcription factor, Nrf2, serves as a master regulator of antioxidant genes. Nrf2 expression has been reported to be decreased in lung tissue homogenates from patients diagnosed with IPF (20). We examined the cellular localization of Nrf2 expression in lung tissues of IPF patients; while Nrf2 expression was expressed in alveolar epithelial cells, it was largely absent in fibroblasts within fibroblastic foci (Fig. 4A, IHC staining).
Figure 4.
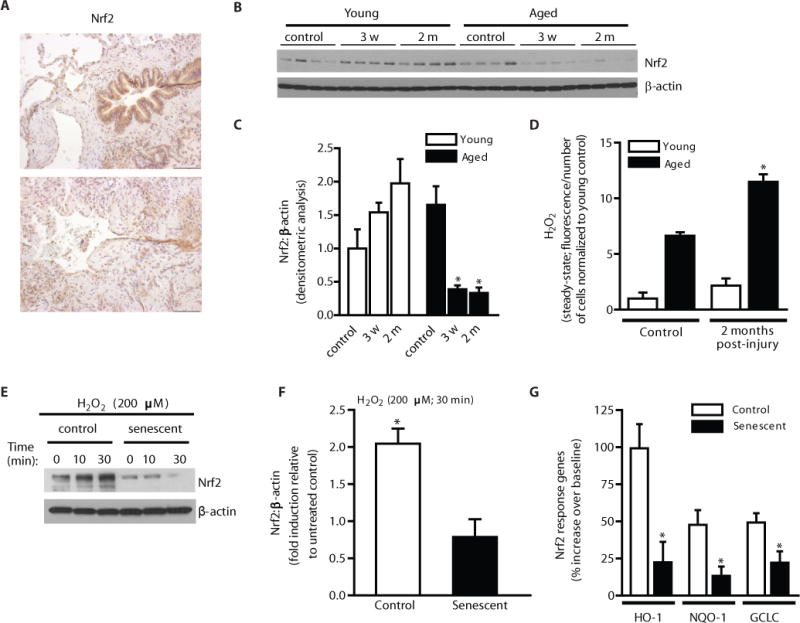
Deficiency in the activation of Nrf2 in fibroblasts from aged mice subjected to lung injury in vivo and in senescent fibroblasts in vitro. (A) IPF lung tissue sections were analyzed for expression of Nrf2 by immunohistochemical analysis. (B–D) Young and aged mice were subjected to bleomycin-induced lung fibrosis. Lung fibroblasts were isolated at the time points indicated and cultured ex vivo. Nrf2 expression was assessed by Western immunoblotting (B) and densitometric analysis (C); *p value < 0.05, compared to aged control. Steady-state H2O2 levels were assessed at the indicated time points by fluorometric assay (D); *p value < 0.05, compared with all other groups. (E–F) Control and senescent IMR90 fibroblasts (at low and high population doublings, respectively) were treated with H2O2 (200 μM). Nrf2 expression was evaluated by Western immunoblotting (E) and quantitated by densitometric analysis (F); *p value < 0.05. Downstream Nrf2-responsive genes were evaluated 16 h post H2O2 treatment by real-time PCR (G); data are expressed as fold increase compared to untreated control (n = 5); *p value < 0.05, compared to non-senescent control. Values represent mean ± s.e.m.; n = 3; *p value < 0.05. Scale bars, 100 μm.
Nrf2 deficient mice develop more severe fibrosis than their wild-type controls (21), although effects of age and reversibility of fibrosis has not been tested. We sought to determine whether fibroblasts from aged mice with persistent fibrosis exhibit alterations in Nrf2 activation. Nrf2 induction was evaluated in fibroblasts isolated from young and aged mice following bleomycin-induced lung injury; Nrf2 was induced in young fibroblasts, whereas its expression is decreased in aged fibroblasts, as determined by western immunoblotting (Fig. 4B) and densitometric analysis (Fig. 4C). In parallel, we evaluated steady-state levels of H2O2 production in lung fibroblasts from young and aged mice pre- and post-injury. H2O2 production by young fibroblasts at 2 months post-injury was not significantly different from control, whereas fibroblasts from aged mice with persistent fibrosis exhibited significantly elevated H2O2 levels (Fig. 4D), consistent with a deficient Nrf2 response.
To determine if Nrf2 responsiveness to oxidative stress is altered in aging, we utilized a cellular model of replicative senescence: IMR90 lung fibroblasts at low and high population doublings (PDL), control and senescent fibroblasts, respectively. Treatment with exogenous H2O2 [200 μM, a dose that is known to induce apoptosis (22)] induced Nrf2 expression in control fibroblasts (Fig. 4E, western immunoblotting and Fig. 4F, densitometric analysis), whereas this induction was absent in senescent fibroblasts (Fig. 4E–F). Similarly, lung fibroblasts isolated from aged mice exhibited deficient Nrf2 induction in response to exogenous H2O2, as compared to young murine fibroblasts (Fig. S4A–B, western immunoblotting and densitometric analysis). To determine if the lack of Nrf2 induction in aged cells affected the capacity to induce Nrf2-responsive target genes, we evaluated mRNA expression of heme oxygenase 1 (HO-1), NAD(P)H:quinone oxidoreductase 1 (NQO-1), and glutamate-cysteine ligase (GCLC) in control and senescent fibroblasts in response to exogenous H2O2. While control fibroblasts demonstrated a robust induction of HO-1, NQO-1, and GCLC, induction of these antioxidant response genes were significantly decreased in senescent fibroblasts (Fig. 4G). These results suggest that, in the context of both age-dependent and replicative senescence, the oxidative stress-induced Nrf2 antioxidant response is impaired.
Nox4-Nrf2 redox balance controls apoptosis susceptibility of fibroblasts
We sought to determine the influence of the Nox4-Nrf2 redox (im)balance on apoptosis susceptibility of lung fibroblasts. We first confirmed that senescent fibroblasts, with deficient Nrf2 responsiveness, demonstrate higher levels of constitutive H2O2 production (Fig. 5A). Apoptosis susceptibilities of control and senescent fibroblasts to staurosporine were evaluated; senescent fibroblast demonstrated relative resistance to apoptosis, as evidenced by decreased caspase activity compared to control (Fig. 5B). We then determined if lung fibroblasts isolated from young mice with an intact Nrf2 response could acquire apoptosis resistance with Nrf2 knockdown; cells were transfected with siRNA targeting Nrf2 or non-targeting control and treated with exogenous H2O2 (200 μM) to induce Nrf2 (as in previous experiments). An inability to induce Nrf2 in young fibroblasts, confirmed by western immunoblotting (Fig. S4C), led to acquisition of apoptosis-resistance (Fig. S4D, caspase activity assay). These studies support the concept that Nrf2 deficiency alters redox balance, which contributes to the acquisition of an apoptosis-resistant fibroblast phenotype.
Figure 5.
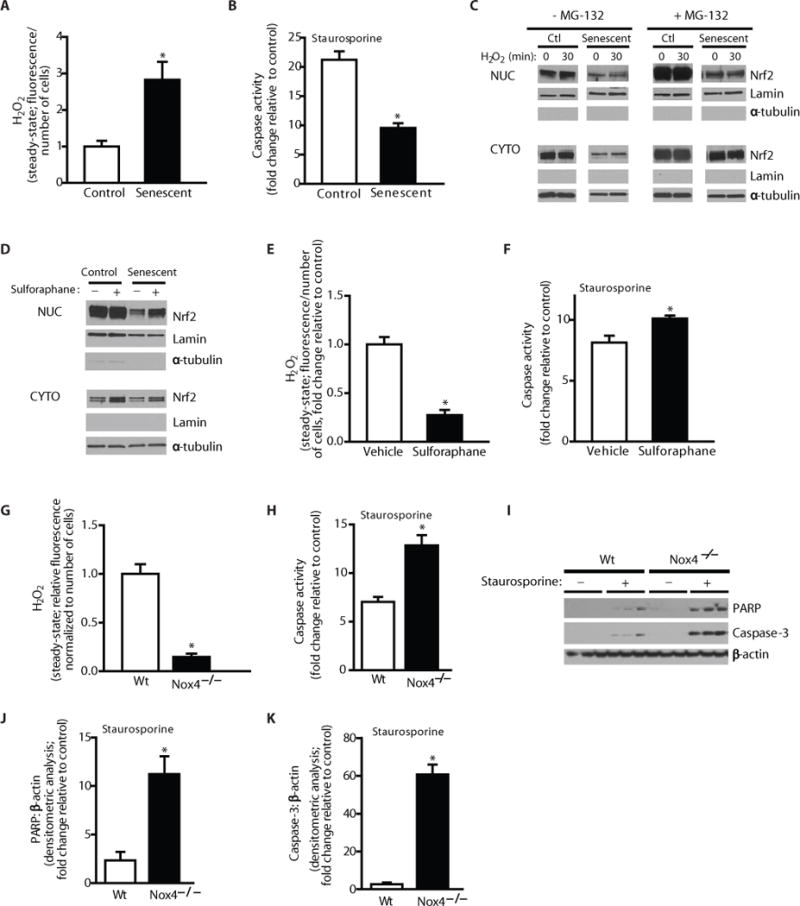
Nox4-Nrf2 imbalance controls redox balance, senescence and apoptosis resistance of fibroblasts. (A–B) Control and senescent cells were evaluated to determine steady-state H2O2 levels (A) or cells were treated with/without staurosporine (300 nM) for 5 h and caspase activity was assessed (B). (C–D) Control and senescent fibroblasts were pre-treated with/without the proteasome inhibitor, MG-132 (25 μM; 2 h), and treated with/without exogenous H2O2 (200 μM) (C) or pre-treated with/without sulforaphane (5 μM; 30 min) (D). Nuclear and cytosolic lysates were evaluated for Nrf2, Lamin and α-tubulin (C–D). (E–F) Fibroblasts were isolated from aged mice at 3 weeks post-injury and cultured ex vivo. Cells were pre-treated with sulforaphane (5 μM) or DMSO for 48 h and Steady-state H2O2 levels were assessed by fluorometric assay (E). Cells were treated with/without staurosporine (300 nM) for 5 h and caspase activity was assessed (F). (G–K) Lung fibroblasts were isolated from Nox4−/− or wild-type mice and cultured ex vivo. Cells were serum starved for 24h, then treated with TGF-β1 (2 ng/ml) for 48 h. (G) H2O2 production was evaluated. (H–K) Cells were treated with or without staurosporine (300 nM) for 8 h and caspase activity was assessed (H). Expression of PARP and caspase-3 were evaluated by western immunoblotting (I) and quantitated by densitometric analysis (J–K). Values represent mean ± s.e.m.; n = 3; *p value < 0.05.
To further characterize the defective Nrf2 response in aging and cellular senescence, we analyzed cytoplasmic-nuclear shuttling by cell fractionation studies in control and senescent cells. Treatment with exogenous H2O2 of non-senescent, control fibroblasts demonstrated increased nuclear Nrf2 translocation, while this was markedly diminished in senescent cells (Fig. 5C). Steady-state levels of Nrf2 are normally controlled by the ubiquitin-proteosomal system (23). To gain further insight into the apparent Nrf2 trafficking defect in senescent cells, we examined nuclear/cytoplasmic levels of Nrf2 in the presence of the ubiquitin proteosomal inhibitor, carbobenzoxy-Leu-Leu-leucinal (MG-132); under these conditions, there was relatively less Nrf2 in the nucleus, despite higher levels in the cytoplasm (Fig. 5C), supporting a primary defect in cytoplasmic-nuclear shuttling as a mechanism for the deficient Nrf2-dependent antioxidant gene response. Treatment with sulforaphane, a known Nrf2 inducer, restores nuclear trafficking of Nrf2 (Fig. 5D, western immunoblotting) and decreases steady-state levels of H2O2 in senescent fibroblasts (Fig. S4E) and in fibroblasts isolated from lungs of injured, aged mice (with decreased Nrf2 expression; Fig. 4B–C) (Fig. 5E). Further, sulforaphane treatment restored apoptosis susceptibility in aged fibroblasts (Fig. 5F, caspase activity). These data suggest that sulforaphane treatment may, at least in part, correct the impairment in Nrf2 nuclear trafficking associated with cellular senescence.
Previous studies indicate that the Nrf2 response is tightly coupled to Nox4 induction or over-expression (24–26), which may in part explain the contextual effects of Nox4. In order to directly evaluate the role of Nox4 in apoptosis resistance, lung fibroblasts isolated from Nox4−/− and wild-type mice were evaluated ex vivo. We confirmed that H2O2 generation was significantly decreased in fibroblasts isolated from Nox4−/− mice (Fig. 5G). Nox4−/− fibroblasts exhibited increased susceptibility to staurosporine-induced apoptosis as compared to wild-type fibroblasts (Fig. 5H, caspase activity assay; Fig. 5I–J, expression of PARP; Fig. 5I,K, expression of cleaved caspase-3). Similarly, Nox4−/− fibroblasts also demonstrated increased susceptibility to H2O2-induced apoptosis (Fig. S5). Together, these data indicate that, in the context of Nrf2 deficiency or Nox4 induction, fibroblasts acquire an apoptosis-resistant phenotype.
Therapeutic targeting of Nox4 in aged mice with persistent fibrosis modulates fibroblast senescence and restores capacity for fibrosis resolution
Previous studies have shown that Nox4 is essential in myofibroblast differentiation (18, 27, 28) and in the induction of a fibrogenic response to lung injury (18, 29, 30). However, these previous studies have not addressed the role of Nox4 in the maintenance of fibrosis or efficacy of targeting this enzyme in age-associated persistent fibrosis. While Nox4 was induced in both young and aged mice at 3 weeks post-injury, its expression remained elevated in fibrogenic regions of aged mice with persistent fibrosis (Fig. 6A, IHC). We evaluated the efficacy of targeting Nox4 in age-associated persistent fibrosis and in modulating myofibroblast senescence in vivo. Nox4 siRNA or a nontargeting control siRNA was administered by intranasal delivery to the lungs of aged mice, specifically during the period of persistent fibrosis; siRNA was administered every other day from weeks 3–6 for a total of 10 treatments (Fig. 6B). Nox4 knockdown in lung fibroblasts from treated mice was confirmed (Fig. S6, western immunoblotting and Fig. 6C, densitometric analysis indicates ~50% reduction). Fibroblasts isolated from mice receiving Nox4 siRNA exhibited reduced senescence, as determined by quantitative assessment of SA-βgal activity (Fig. 6D), cellular staining for SA-βgal (Fig. 6E), and decreased expression of senescence markers p16 and p21 (Fig. S6, western immunoblotting and Fig. 6F–G, densitometric analysis). Fibroblasts from Nox4 siRNA-treated mice also exhibited decreased expression of the anti-apoptotic protein, Bcl-2, and the ECM protein, Col1A1 (Fig. S6, Fig. 6H–I). Importantly, Nox4 knockdown restored the capacity for fibrosis resolution as determined by Masson’s trichrome blue staining for collagen (Fig. 6J), as well as by assessment of total lung collagen by hydroxyproline assay (Fig. 6K).
Figure 6.
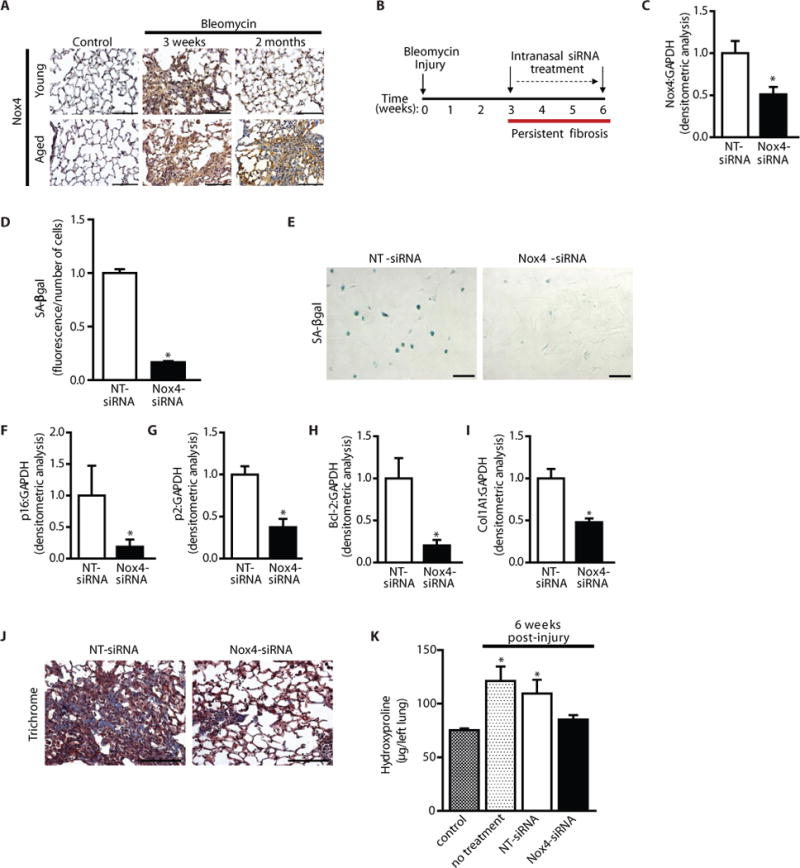
In vivo knockdown of Nox4 restores the capacity for fibrosis resolution in aged mice. (A) Young and aged mice were subjected to bleomycin-induced lung fibrosis. Lung tissues were harvested at 0 (uninjured), 3 weeks, and 2 months post-injury; Nox4 expression was evaluated by immunohistochemical analysis. (B–K) Aged mice subjected to bleomycin-injury were treated with Nox4-targeting or non-targeting siRNA, administered by intranasal delivery every other day for 3 weeks, starting 3 weeks post-injury. A schematic diagram illustrates treatment period following injury (B). (C, F–I) Lung fibroblasts were isolated at the end of treatment (6 weeks post-injury) and cultured ex vivo. Expression of Nox4 (C) p16 (F), p21 (G), Bcl-2 (H), and Col1A1 (I) and were analyzed by Western immunoblotting and quantified by densitometric analyses; values represent mean ± s.e.m.; n = 5–8 per group; *p value < 0.05, compared to NT-siRNA. Fibroblast senescence was evaluated by quantitative measurement of SA-βgal activity (D); n = 3 per group; *p value < 0.05, compared to NT-siRNA; and SA-βgal staining (E). (J–K) Lung tissue was harvested at 6 weeks post-injury; fibrosis was assessed by Masson’s trichrome blue staining for collagen (J), and quantitative hydroxyproline assay (K). Values represent mean ± s.e.m.; n = 6–8 per group; *p value < 0.05, compared to untreated controls. Scale bars, 100 μm.
We also evaluated the in vivo efficacy of GKT137831 in this aging model of persistent fibrosis. GKT137831 or vehicle was administered to aged mice daily by gavage (40 mg/kg body weight) from week 3–6 post-injury for a total of 21 treatments. Recovery to baseline weights was observed in GKT137831 treated mice (Fig. 7A); whereas vehicle treated mice remained below baseline levels throughout the 6 week observation period (Fig. 7A). In parallel, we evaluated total lung weights at the end of the treatment period; lung weights of GKT137831 treated mice were significantly lower, as compared to vehicle treated (Fig. S7A), supporting the reversibility of fibrosis. These data suggest that GKT137831 treatment in aged mice with persistent fibrosis leads to alterations at both organ-specific and systemic levels. We evaluated myofibroblast persistence following GKT137831 treatment; GKT137831 treated mice exhibited decreased myofibroblast accumulation (Fig. 7B, IHC staining for α-SMA). GKT137831 treated mice also showed decreased expression of senescence markers p21 and p16 (Fig. 7B). Importantly, GKT137831 treatment led to a reversal of age-associated persistent fibrosis (Fig. 7B, Masson’s trichrome staining; Fig. 7C and Fig. S7B, hydroxyproline assay) and reduced mortality (Fig. 7D). Together, these data support the feasibility of in vivo targeting of Nox4 in modulating the senescent, anti-apoptotic myofibroblast phenotype and reversal of persistent fibrosis associated with aging (Fig. 8).
Figure 7.
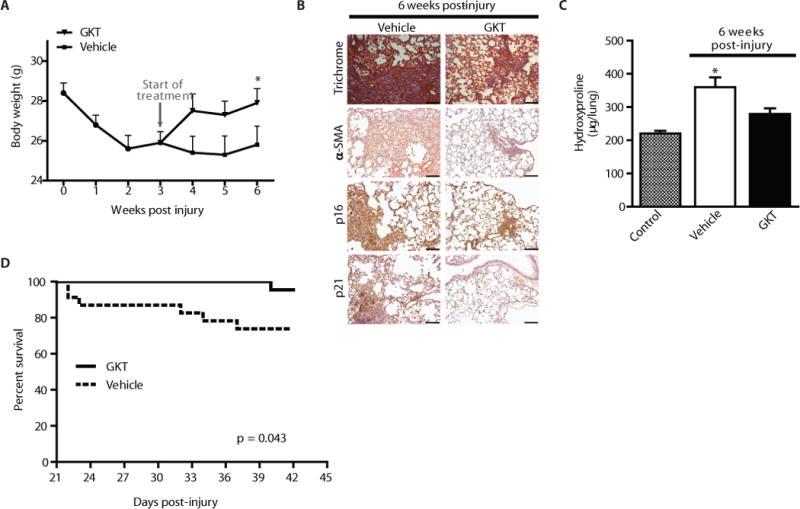
In vivo pharmacologic targeting of Nox4 with GKT137831 leads to reversal of age-associated persistent fibrosis. (A–D) Aged mice (18 m) were subjected to bleomycin-induced lung fibrosis. Starting at 3 weeks post-injury, mice were treated daily with GKT137831 (40 mg/kg) or vehicle by oral gavage through week 6 (21 treatments total). Body weight of the mice was recorded weekly (A); values represent mean ± s.e.m.; n = 17–21 per group; *p value < 0.05, compared to vehicle-treated controls. (B–C) Lung tissues were harvested from control (uninjured) or at 6 weeks post-injury. Tissues were evaluated by Masson’s trichrome blue staining for collagen and by immunohistochemical analyses was performed to evaluate expression of α-SMA and senescence markers (p16 and p21) (B). Whole lung homogenates were analyzed by quantitative hydroxyproline assay (C); data are expressed as total μg of hydroxyproline/whole lung; values represent mean ± s.e.m.; n = 9–10 per group; *p value < 0.05, compared to all other groups. Kaplan-Meier survival curve for GKT137831 (n = 22) and vehicle (n = 23) treated mice (D); log-rank test, p < 0.05. Scale bars, 100 μm.
Figure 8.
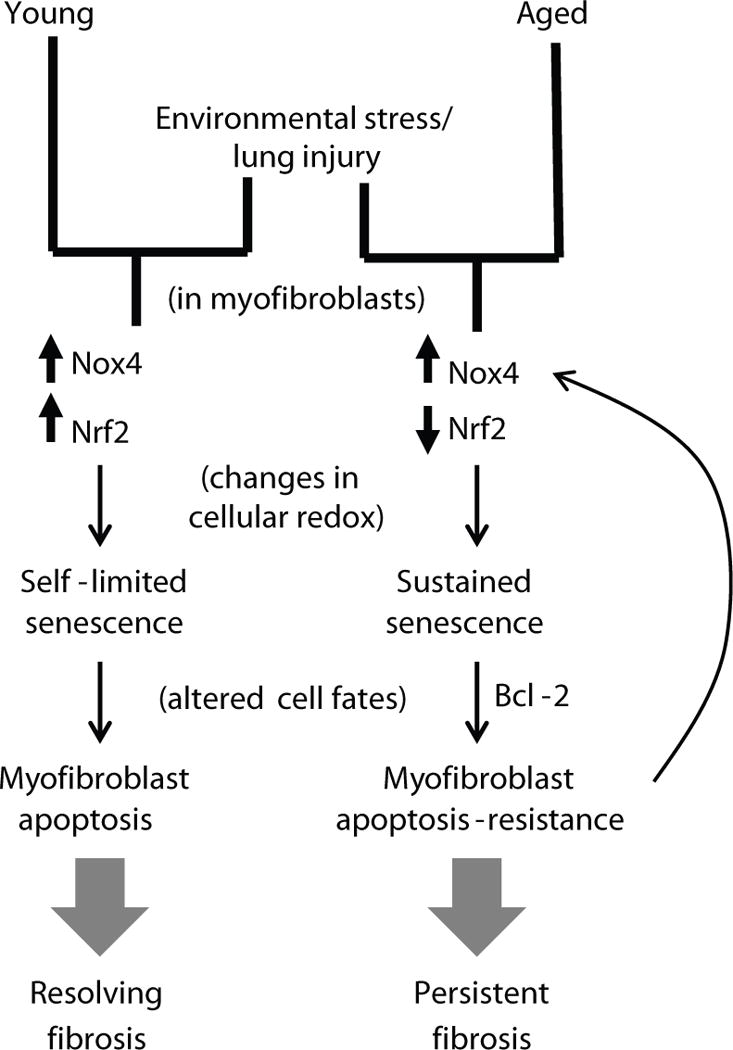
Proposed model for persistent fibrosis in aged mice following lung injury.
Discussion
Fibrotic disorders account for increasing morbidity and mortality worldwide, and aging is a known risk factor (8, 9). Despite the identification of numerous drug candidates and extensive pre-clinical studies, none have translated to effective treatments for patients with lung fibrosis, in particular IPF (31, 32). One potential reason for this lack of clinical translation is the failure to account for the emerging concept of IPF as an age-related disease. Pre-clinical animal models of lung fibrosis are largely employed in young mice, which predominantly results in a self-limited fibrotic response (33, 34). Treatment interventions are largely preventative (dosing before or at the time of injury) rather than curative (delayed drug administration, typically after at least 1 week) (31). Our studies raise the possibility that the lack of translation may not be due to the injury model per se or species differences, but rather the context of age. An age-relevant model of persistent fibrosis, as described in this study, could have important implications for clinical translation of drug candidates (i.e. ability to evaluate reversibility of fibrosis vs. prevention), which may ultimately improve accuracy of predicting therapeutic potential in clinical trials.
The role of oxidative stress in IPF pathogenesis has been well appreciated (10); however, the mechanisms by which oxidative stress contributes to pathogenesis are not well defined. In contrast to indiscriminant oxidative stress-associated damage to tissues, we propose that alterations in redox signaling regulate the pro-fibrotic phenotype of myofibroblasts. Specifically, we show that an aberrant upregulation of the ROS-generating enzyme, Nox4, coupled with a deficiency in Nrf2 induction results in a sustained redox imbalance which promotes persistent myofibroblast senescence that confers an apoptosis-resistant phenotype to these normally reparative cells. While we have shown previously that targeting Nox4 at the time of injury ameliorates the development of injury-induced lung fibrosis in young mice (18), these studies demonstrate that targeting Nox4 in in age-associated persistent fibrosis is sufficient to correct this redox imbalance and promote resolution.
Cellular senescence as a pro-fibrotic process appears, on the surface, to contradict previous studies suggesting senescence as an anti-fibrotic mechanism (16, 35). There may be several reasons for these apparent contradictions. First, the previous studies were conducted in young mice (2–3 months of age); the fate of stress-induced senescent myofibroblasts may be altered with aging, as indicated by our studies. Second, the former studies focused primarily on the initiation/emergence of a fibrotic response to injury, whereas our studies specifically address the capacity for resolution of established fibrosis. Senescence programs in young mice likely reflect a stress-induced response that can be self-limited. Indeed, our studies show that myofibroblasts from young mice manifest transient senescence and apoptosis susceptibility that permits fibrosis resolution, while myofibroblasts in injured tissues of aged mice acquire a sustained senescent and apoptosis-resistant phenotype that impairs the resolution of fibrosis. Finally, the accumulation of senescent cells in most tissues with advancing age is inconsistent with the clinical observation of an increased risk of fibrotic disease with aging. In support of the link between cellular senescence and IPF, we demonstrate that myofibroblasts in fibroblastic foci are relatively non-proliferative and express the tumor suppressor p16, supporting senescence of myofibroblasts in IPF; similar observations have been made in human liver cirrhosis (16). These studies highlight the complexities of cellular senescence which may influence tissue injury repair processes in a pleiotropic manner, determined by factors such as age, phase of repair, and the specific cell types involved.
In response to tissue injury, the local activation of myofibroblasts and ECM deposition are necessary for normal repair. The resolution of fibrosis is preceded by apoptosis of myofibroblasts and clearance of the ECM (17). Thus, as a corollary, the key difference between “physiological” and “pathological” fibrosis is the impaired elimination of myofibroblasts and accumulation of ECM, resembling a persistent wound healing response. Our studies implicate apoptosis resistance as a key mechanism by which myofibroblast senescence contributes to persistent fibrosis in aging. Indeed, studies in our laboratory and others support the acquisition of an apoptosis-resistant phenotype in senescent fibroblasts, in part related to higher expression of Bcl-2 (36, 37). Our current findings of high expression of Bcl-2 in fibroblasts isolated from aged mice subjected to bleomycin injury are consistent with this as one mechanism of apoptosis resistance. We cannot exclude other potential mechanisms by which senescence contributes to persistent fibrosis. For example, immune senescence resulting in impaired recognition, killing and/or clearance of senescent cells may also contribute to the persistence of myofibroblasts in aged, injured tissues (16). Additionally, senescence of epithelial cells may contribute to fibrosis by their diminished capacity for regeneration and/or by their altered secretory profiles.
Our studies provide proof-of-concept for therapeutic modulation of redox imbalance and cell senescence in age-associated persistent fibrosis. Treatment with the small molecule antioxidant, N-acetylcysteine (NAC) has been demonstrated to reverse p21-induced growth arrest, supporting the role of ROS in mediating cellular senescence (38). Anti-oxidant strategies with NAC have been evaluated in a phase III clinical trial for IPF (www.clinicaltrials.gov; NCT00650091: PANTHER). However, despite preclinical and clinical proof-of-concept for therapeutic modulation of redox imbalance, it is not clear whether strategies that enhance antioxidant defenses will be as effective as targeting the source(s) of ROS generation. A Phase III clinical trial of bardoxolone, an Nrf2 activator, in patients with chronic kidney disease was terminated due to lack of efficacy and adverse events, indicating that strategies to further augment this counter-regulatory antioxidant pathway may be ineffective (39, 40). Strategies that more directly target the source(s) of ROS generation may prove to be more specific and effective in comparison to antioxidant interventions for fibrotic diseases. Genkyotex (Geneva, witzerland) recently developed a small-molecule Nox1/Nox4 dual inhibitor (GKT137831), which is currently being developed for treatment of diabetic nephropathy (data presented at the 2013 annual meeting of the American Society of Nephrology, November 7–10, Atlanta, GA). Our studies demonstrate that established fibrosis in lungs of aged mice is, at least partially, reversed by administration of GKT137831, similar to the effects of Nox4-targetted siRNA. In addition to inhibition of fibroblast senescence and apoptosis resistance, targeting of Nox4 may have beneficial effects in fibrosis by preventing excessive epithelial cell apoptosis (29, 41, 42); although other studies suggest an anti-apoptotic role of Nox4, which indicates cell-specific and contextual effects of Nox4. We believe the results of our studies provide convincing pre-clinical data to support therapeutic approaches that target Nox4 in age-associated fibrotic disorders. The studies reported here support the potential efficacy of therapeutic agents that target Nox4 for age-associated fibrotic disorders; additionally, this study provides new insights into redox mechanisms that control pro-fibrotic effects of fibroblast senescence.
Materials and Methods
Mice
Young (2 months) and aged (18 months) female C57BL/6 mice (Jackson Laboratories or National Institute on Aging) were used for in vivo studies. Nox4 knockout mice were a gift from Karl-Heinz Krause, University of Geneva. Mice were sacrificed by CO2 inhalation. All procedures involving animals were approved by the Institutional Animal Care and Use Committees (IACUC) at the University of Alabama at Birmingham.
Study Design
One major objective in this study was to determine the role of aging in the severity and resolution of injury-induced lung fibrosis. Young and aged mice were subjected to lung injury, mice were randomized and fibrosis was assessed at 3 weeks and 4 months post-injury by hydroxyproline assay; aged-matched controls were evaluated in parallel at each time point. Another major objective was to determine the role of Nox4 in age-associated persistent lung fibrosis. The impact of genetic and pharmacologic inhibition of Nox4 on reversal of persistent fibrosis in aged mice was evaluated, where treatments were administered from 3–6 weeks post-injury (during the period of persistent fibrosis); all mice were evaluated at the 6 week endpoint. Mice were randomized to treatment and vehicle groups. Fibrosis was evaluated by hydroxyproline assay, histology, and immunohistochemistry. Mice that died within the treatment period (prior to the 6 week end point) were excluded from analyses of fibrosis. All surviving mice at the designated endpoints were included in the data analyses. Fibroblasts were isolated and evaluated for apoptosis, senescence, and ROS levels by immunofluorescence, immunohistochemistry, and/or biochemical assays. Downstream signaling was evaluated by western immunoblotting. No animals or potential outliers were excluded from the data sets presented.
Human lung tissue and fibroblasts were isolated from the lungs of patients with a confirmed diagnosis of IPF as previously described (18), under an approved protocol by the Institutional Review Board at the University of Alabama at Birmingham (UAB); Informed consent was obtained from all individuals enrolled through the Airway Tissue Procurement program at UAB.
Murine model of bleomycin lung injury
We anesthetized young and aged mice with intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). We administered intra-tracheal bleomycin (1.25 U/kg) to induce lung injury or saline (50 μL total volume) as previously described (18).
In vivo administration of GKT137831
GKT137831 was dissolved in an aqueous solution (0.5% carboxymethylcellulose and 0.25% Tween 20). Treatment was administered to aged mice daily by gavage (40 mg/kg body weight) starting at week 3 through week 6 post-injury, for a total of 21 treatments. Mice received the same volume of vehicle alone as control.
Caspase Activity Assay
Cells were seeded in 35 mm cell culture plates and cultured to ~70–90% confluence in DMEM with 10% fetal bovine serum. Following treatment, cells were lysed using caspase lysis buffer and analyzed for activated caspase 3 using the BioVision Caspase 3 Fluorometric Assay Kit according to the manufacturer’s instructions (BioVision, Inc, Milpitas CA).
Senescence Assays
We used a high-sensitivity substrate (fluorescein di-β-D-galactosidase) for quantitative assessment of cellular senescence (MarkerGene Technologies), according to manufacturer instructions. Cell number was normalized by DAPI (Fluorescent Cell Count Normalization Kit; MarkerGene Technologies). We also used a Senescence Detection Kit designed to histochemically detect SA-β-GAL activity in cultured cells (Abcam).
Detection of H2O2
We assayed extracellular H2O2 release from cultured cells as previously described (43). Cell number was normalized by DAPI (Fluorescent Cell Count Normalization Kit; MarkerGene Technologies).
Hydroxyproline assay
Lung tissues were dried in an oven at 70°C for 48 h, and then hydrolyzed in 6N HCl at 95°C for 20 h. Hydroxyproline assay was performed according to manufacturer instructions (QuickZyme BioSciences) using hydroxyproline as a standard.
Methods for the following are shown in supplemental materials and methods: Reagents, Cell culture, Western immunoblotting, Real-time PCR, Lung histology and immunohistochemical staining, Immunofluorescence labeling, RNA interference, and TUNEL staining.
Statistical Analysis
Graphs were made and statistical analyses were performed using GraphPad Prism (GraphPad Software). Data are expressed as means ± SEM. Differences among groups were assessed using one-way ANOVA with a Bonferroni correction and between pairs using Student’s two-tailed t test. Statistical comparisons of survival were made using the log-rank (Mantel-Cox) test. P < 0.05 is considered statistically significant.
Supplementary Material
Fig. S1: Hydroxyproline content of whole lung homogenates in young and aged mice subjected to bleomycin injury.
Fig. S2: Persistent fibrosis in aging is associated with myofibroblast senescence and apoptosis resistance.
Fig. S3: Fibroblastic Foci in IPF lungs demonstrate high expression of p21.
Fig. S4: Nrf2 deficiency promotes apoptosis resistance in aged fibroblasts.
Fig. S5: Nox4−/− lung fibroblasts demonstrate increased susceptibility to H2O2-induced apoptosis.
Fig. S6: In vivo knockdown of Nox4 leads to decreased pro-fibrotic markers in lung fibroblasts.
Fig. S7: Organ-specific effects and fibrosis resolution resulting from in vivo administration of GKT137831.
Table S1: Primer and siRNA sequences.
Acknowledgments
We thank Ragini Vittal for contributing to immunofluorescence studies of human IPF lung and Cedric Szyndralewiez for technical assistance with protocols related to in vivo administration of GKT137831. We thank Karl-Heinz Krause for providing Nox4 knockout mice.
Funding: Supported by Veterans Administration Health System grant, 1IK2BX001477 (L.H.), and National Institutes of Health grants, P01 HL114470, R01 HL094230 and P50 HL107181 (V.J.T.).
Footnotes
Author contributions: L.H., T.H. and V.J.T. conceived of the project. L.H. and V.J.T. supervised all studies, provided funding, and wrote the manuscript. L.H. and N.J.L. designed, conducted, and/or performed analyses for most of the experiments. D.K. contributed to animal studies and biochemical analyses (western immunoblotting and apoptosis assays). A.K. contributed to immunofluorescence studies of in vivo detection of apoptotic cells. T.H. contributed to immunohistochemistry studies. E.M. helped design animal studies involving administration of GKT137831. All authors contributed to the intellectual input and K.B., T.H., E.M. and Y.Y.S. contributed to manuscript preparation and experimental design.
Competing interests: At the time of these studies, E.M. was employed at Genkyotex as the Chief Scientific Officer. However, all authors declare no conflict of interest and there are no financial relationships related to the design and data presented in this manuscript.
List of Supplementary Materials:
Materials and Methods
References
- 1.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008 May 15;453:314. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 2.Laflamme MA, Murry CE. Heart regeneration. Nature. 2011 May 19;473:326. doi: 10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004 Aug;4:583. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.King TE, Jr, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med. 2001 Sep 15;164:1025. doi: 10.1164/ajrccm.164.6.2001056. [DOI] [PubMed] [Google Scholar]
- 5.Hinz B, et al. The myofibroblast: one function, multiple origins. Am J Pathol. 2007 Jun;170:1807. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host Responses in Tissue Repair and Fibrosis. Annual review of pathology. 2012 Oct 22; doi: 10.1146/annurev-pathol-020712-163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thannickal VJ, Toews GB, White ES, Lynch JP, 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med. 2004;55:395. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- 8.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006 Oct 1;174:810. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 9.Araki T, Katsura H, Sawabe M, Kida K. A clinical study of idiopathic pulmonary fibrosis based on autopsy studies in elderly patients. Intern Med. 2003 Jun;42:483. doi: 10.2169/internalmedicine.42.483. [DOI] [PubMed] [Google Scholar]
- 10.Kinnula VL, Fattman CL, Tan RJ, Oury TD. Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med. 2005 Aug 15;172:417. doi: 10.1164/rccm.200501-017PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sueblinvong V, et al. Predisposition for disrepair in the aged lung. Am J Med Sci. 2012 Jul;344:41. doi: 10.1097/MAJ.0b013e318234c132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naik PN, et al. Pulmonary fibrosis induced by gamma-herpesvirus in aged mice is associated with increased fibroblast responsiveness to transforming growth factor-beta. J Gerontol A Biol Sci Med Sci. 2012 Jun;67:714. doi: 10.1093/gerona/glr211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torres-Gonzalez E, et al. Role of endoplasmic reticulum stress in age-related susceptibility to lung fibrosis. Am J Respir Cell Mol Biol. 2012 Jun;46:748. doi: 10.1165/rcmb.2011-0224OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Izbicki G, Segel MJ, Christensen TG, Conner MW, Breuer R. Time course of bleomycin-induced lung fibrosis. International journal of experimental pathology. 2002 Jun;83:111. doi: 10.1046/j.1365-2613.2002.00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinz B, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012 Apr;180:1340. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krizhanovsky V, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008 Aug 22;134:657. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995 Jan;146:56. [PMC free article] [PubMed] [Google Scholar]
- 18.Hecker L, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 009 Sep 2;15:1077. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000 Dec;279:L1005. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 20.Artaud-Macari E, et al. Nuclear factor erythroid 2-related factor 2 nuclear translocation induces myofibroblastic dedifferentiation in idiopathic pulmonary fibrosis. Antioxid Redox Signal. 2013 Jan 1;18:66. doi: 10.1089/ars.2011.4240. [DOI] [PubMed] [Google Scholar]
- 21.Cho HY, Reddy SP, Yamamoto M, Kleeberger SR. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004 Aug;18:1258. doi: 10.1096/fj.03-1127fje. [DOI] [PubMed] [Google Scholar]
- 22.Chen QM, Liu J, Merrett JB. Apoptosis or senescence-like growth arrest: influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. Biochem J. 2000 Apr 15;347:543. doi: 10.1042/0264-6021:3470543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Villeneuve NF, Lau A, Zhang DD. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: an insight into cullin-ring ubiquitin ligases. Antioxid Redox Signal. 2010 Dec 1;13:1699. doi: 10.1089/ars.2010.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brewer AC, et al. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic Biol Med. 2011 Jul 1;51:205. doi: 10.1016/j.freeradbiomed.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schroder K, et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res. 2012 Apr 27;110:1217. doi: 10.1161/CIRCRESAHA.112.267054. [DOI] [PubMed] [Google Scholar]
- 26.Nlandu Khodo S, et al. NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J Am Soc Nephrol. 2012 Dec;23:1967. doi: 10.1681/ASN.2012040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cucoranu I, et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005 Oct 28;97:900. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 28.Amara N, et al. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax. 2010 Aug;65:733. doi: 10.1136/thx.2009.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carnesecchi S, et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal. 2011 Aug 1;15:607. doi: 10.1089/ars.2010.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jarman ER, et al. An Inhibitor of NADPH Oxidase-4 Attenuates Established Pulmonary Fibrosis in a Rodent Disease Model. Am J Respir Cell Mol Biol. 2013 Aug 26; doi: 10.1165/rcmb.2013-0174OC. [DOI] [PubMed] [Google Scholar]
- 31.Moeller A, Ask K, Warburton D, Gauldie J, Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol. 2008;40:362. doi: 10.1016/j.biocel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thannickal VJ, Roman J. Challenges in translating preclinical studies to effective drug therapies in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010 Mar 15;181:532. doi: 10.1164/rccm.200911-1740ED. [DOI] [PubMed] [Google Scholar]
- 33.Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008 Feb;294:L152. doi: 10.1152/ajplung.00313.2007. [DOI] [PubMed] [Google Scholar]
- 34.Lawson WE, et al. Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am J Pathol. 2005 Nov;167:1267. doi: 10.1016/S0002-9440(10)61214-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nature cell biology. 2010 Jul;12:676. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.L H, Sanders YY, Zhang X, Bernard K, Hecker L, Desai L, Liu G, Thannickal VJ. Histone Modifications in Senescence-Associated Resistance to Apoptosis by Oxidative Stress. Redox Biology. 2013;8 doi: 10.1016/j.redox.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Al-Khalaf HH, Aboussekhra A. Survivin expression increases during aging and enhances the resistance of aged human fibroblasts to genotoxic stress. Age. 2013 Jun;35:549. doi: 10.1007/s11357-011-9378-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Macip S, et al. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002 May 1;21:2180. doi: 10.1093/emboj/21.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Zeeuw D, et al. Bardoxolone Methyl in Type 2 Diabetes and Stage 4 Chronic Kidney Disease. N Engl J Med. 2013 Nov 9; doi: 10.1056/NEJMoa1306033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Himmelfarb J, Tuttle KR. New Therapies for Diabetic Kidney Disease. N Engl J Med. 2013 Nov 9; doi: 10.1056/NEJMe1313104. [DOI] [PubMed] [Google Scholar]
- 41.Eid AA, et al. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J Biol Chem. 2010 Nov 26;285:37503. doi: 10.1074/jbc.M110.136796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sancho P, et al. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS One. 2012;7:e45285. doi: 10.1371/journal.pone.0045285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thannickal VJ, Fanburg BL. Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor beta 1. J Biol Chem. 1995 Dec 22;270:30334. doi: 10.1074/jbc.270.51.30334. [DOI] [PubMed] [Google Scholar]
- 44.Vittal R, et al. Modulation of prosurvival signaling in fibroblasts by a protein kinase inhibitor protects against fibrotic tissue injury. Am J Pathol. 2005 Feb;166:367. doi: 10.1016/S0002-9440(10)62260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thannickal VJ, et al. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003 Apr 4;278:12384. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1: Hydroxyproline content of whole lung homogenates in young and aged mice subjected to bleomycin injury.
Fig. S2: Persistent fibrosis in aging is associated with myofibroblast senescence and apoptosis resistance.
Fig. S3: Fibroblastic Foci in IPF lungs demonstrate high expression of p21.
Fig. S4: Nrf2 deficiency promotes apoptosis resistance in aged fibroblasts.
Fig. S5: Nox4−/− lung fibroblasts demonstrate increased susceptibility to H2O2-induced apoptosis.
Fig. S6: In vivo knockdown of Nox4 leads to decreased pro-fibrotic markers in lung fibroblasts.
Fig. S7: Organ-specific effects and fibrosis resolution resulting from in vivo administration of GKT137831.
Table S1: Primer and siRNA sequences.