

Abstract
BACKGROUND & AIMS
The kinase Akt mediates resistance of pancreatic cancer (PaCa) cells to death and is constitutively active (phosphorylated) in cancer cells. Whereas the kinases that activate Akt are well characterized, less is known about phosphatases that dephosporylate and thereby inactivate it. We investigated regulation of Akt activity and cell death by the phosphatases PHLPP1 and PHLPP2 in PaCa cells, mouse models of PaCa, and human pancreatic ductal adenocarcinoma (PDAC).
METHODS
We measured the effects of PHLPP overexpression or knockdown with small interfering RNAs on Akt activation and cell death. We examined regulation of PHLPPs by growth factors and reactive oxygen species, as well as associations between PHLPPs and tumorigenesis.
RESULTS
PHLPP overexpression inactivated Akt, whereas PHLPP knockdown increased phosphorylation of Akt in PaCa cells. Levels of PHLPPs were greatly reduced in human PDAC and in mouse genetic and xenograft models of PaCa. PHLPP activities in PaCa cells were down-regulated by growth factors and Nox4 reduced nicotinamide adenine dinucleotide phosphate oxidase. PHLPP1 selectively dephosphorylated Akt2, whereas PHLPP2 selectively dephosphorylated Akt1. Akt2, but not Akt1, was up-regulated in PDAC, and Akt2 levels correlated with mortality. Consistent with these results, high levels of PHLPP1, which dephosphorylates Akt2 (but not PHLPP2, which dephosphorylates Akt1), correlated with longer survival times of patients with PDAC. In mice, xenograft tumors derived from PaCa cells that overexpress PHLPP1 (but not PHLPP2) had inactivated Akt, greater extent of apoptosis, and smaller size.
CONCLUSIONS
PHLPP1 has tumor suppressive activity and might represent a therapeutic or diagnostic tool for PDAC.
Keywords: ROS, NADPH Oxidase, Apoptosis, Tumor Progression
Akt is a serine/threonine kinase that plays a major prosurvival role in cancer cells. Akt is persistently active in pancreatic adenocarcinoma, a very aggressive cancer for which there is no effective treatment available. Akt inhibition stimulates pancreatic cancer (PaCa) cell death and sensitizes PaCa cells to gemcitabine chemotherapy and is therefore considered a promising target for treatment of PaCa.1–4 Among the 3 members of the Akt family, Akt2 has been implicated in several human malignancies, including ovarian, breast, and PaCas.2,5–9
Akt is activated through the receptor-mediated PI3-kinase– dependent signaling pathway. The termination of Akt signal is mediated by phosphatases. For a long time PTEN, which dephosphorylates phosphatidylinositol-3,4,5 triphosphate, was considered the only phosphatase to terminate Akt signaling.10 In many cancers PTEN is inactivated, because of either aberrant expression or mutation. In the past several years, 2 novel serine phosphatases, PHLPP1 and PHLPP2, were discovered to directly dephosphorylate and inactivate Akt.11,12 PHLPPs specifically dephosphorylate Akt at Ser473; phosphorylation at this site is critical for full Akt activity and its antiapoptotic function.12,13 In colon and breast cancers, PHLPP down-regulation is associated with high levels of phosphorylated Akt (pAkt).14–16
Mechanisms through which cells regulate phosphatase activities are, in general, poorly understood. Reactive oxygen species (ROS) are strong inhibitors of protein tyrosine phosphatases (PTPs). We recently showed17 that in PaCa cells, growth factor receptors inhibit PTPs through ROS-dependent mechanisms. However, PHLPPs belong to the family of serine/threonine phosphatases, which are usually insensitive to ROS. The effects of ROS and growth factor receptors on PHLPPs have not been studied.
Here we show that PHLPP1 and PHLPP2 terminate Akt signaling and stimulate cell death both in PaCa cell lines and in vivo in the orthotopic mouse PaCa model. Levels of PHLPPs are greatly reduced in both human pancreatic ductal adenocarcinoma (PDAC) and mouse PaCa models. PHLPP activities in PaCa cells are down-regulated through pathways mediated by growth factors and ROS generated by reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. PHLPPs selectively regulate Akt isoforms; PHLPP1 dephosphorylates Akt2, whereas PHLPP2 dephosphorylates Akt1. Further, the effects of PHLPPs and Akt are isoform specific. In human PDAC, decreased levels of PHLPP1, but not PHLPP2, correlate with shorter median survival. The strong association of PHLPP1 expression with survival of patients with PDAC is likely attributable to the fact that Akt2, but not Akt1, mediates development of PDAC.
Materials and Methods
In Vivo Studies
Human PDAC and normal pancreatic tissue samples, as well as survival data, were collected from patients undergoing surgery at the Ernst-Moritz-Arndt University.
Immunohistochemistry (IHC) for PHLPP1 and PHLPP2 was performed on samples from 40 patients, and analysis of pAkt, Akt1, and Akt2 was performed on samples from 28 patients. PHLPP expression was classified as low (less than normal pancreas) or high (equal or higher than in normal tissue). A Kaplan–Meier survival analysis and a log-rank test were performed to compare the 2 groups.
Transgenic Pdx-1/KrasG12D and Pdx1-Cre/LSL KrasG12D/Trp53R172H mice developing PDAC were described before.18 Tissue samples from these mice were analyzed by IHC.
An orthotopic xenograft model was performed as described before.19 A total of 5 × 106 control or lentiviral-transduced, MIA PaCa-2 cells overexpressing PHLPP1 or PHLPP2 (described in Supplementary Materials and Methods) were injected subcutaneously into the flank of 2 donor nude mice. After 6 weeks, pancreatic tumors grown subcutaneously were harvested, and tumor pieces (1 mm3) were transplanted into the pancreas tail of recipient nude mice.19 Mice were killed 6 weeks after transplant.
In Vitro Studies
The procedures for cell culture, immunoblotting and immunoprecipitation, small interfering RNA (siRNA) and plasmid transfections, immunofluorescence, measurements of apoptosis, necrosis, autophagy, and proliferation were as previously reported20–23 and described in detail in Supplementary Materials and Methods.
PHLPP Activity
Briefly, PHLPP1 and PHLPP2 were immunoprecipitated and their activities were measured using pNPP as a substrate.11,17 All procedures were performed in an anaerobic chamber. To determine the effects of ROS on phosphatase activity, PHLPP1 and PHLPP2 immunoprecipitates were incubated with 1 μmol/L H2O2 or 1 mmol/L dithiothreitol for 5 minutes before adding pNPP.
Statistical Analysis
The values are means ± SE from at least 3 independent experiments. Statistical differences were analyzed using Student t test, Kaplan–Meier survival analysis, log-rank test, and Spearman correlation.
Results
PHLPP1 and PHLPP2 Dephosphorylate Akt in PaCa Cells
Immunofluorescence showed that both PHLPP1 and PHLPP2 colocalize with pAkt in MIA PaCa-2 and PANC-1 cells (Figure 1A). There were differences in the intracellular localization of PHLPP1 and PHLPP2; PHLPP1 localized both to the plasma membrane and perinuclear area, whereas PHLPP2 only localized to the perinuclear area (Figure 1A).
Figure 1.

The phosphatases PHLPP1 and PHLPP2 inactivate Akt in PaCa cells. PHLPP1 dephosphorylates Akt2, and PHLPP2 dephosphorylates Akt1. (A–E) MIA PaCa cells and (A) PANC-1 cells, (A and E) nontransfected or (B–D) transfected, were cultured with 15% FBS, and the (A) cellular localization and (B–E) levels of PHLPP1 and PHLPP2, pAkt (at Ser473), pmTor, pGSK-3α, pGSK-3β, pFoxo3α, and total Akt, Akt1, and Akt2 were measured by (A) immunofluorescence and (B–E) immunoblotting. (A) Cells were immunostained using primary antibodies against PHLPP1, PHLPP2 (green), and pAkt (red), and secondary antibodies were conjugated with Alexa 488 or 594. The sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and analyzed by confocal microscopy. Images are representative of at least 100 cancer cells analyzed. (B–D) Cells were transfected with (B and D) control, PHLPP1, or PHLPP2 siRNAs or (C) control, PHLPP1, or PHLPP2 overexpression plasmids, and the efficiency of transfections was confirmed by immunoblotting. In this and other figures, the blots were reprobed for GAPDH to confirm equal loading. In B, 2 lanes (1a and 1b; 2a and 2b) indicate 2 different siRNAs used for, respectively, each of PHLPP1 and PHLPP2. In D, cells were transfected with PHLPP1 or PHLPP2 siRNA, and lysates were subjected to immunoprecipitation with pAkt antibody that immunoprecipitates all isoforms of pAkt. Immunoprecipitates were probed with isoform-specific antibodies against total Akt1 or total Akt2. The equal levels of Akt1 and Akt2 in the original lysates were confirmed by immunoblotting. The intensities of Akt and pAkt bands were quantified by densitometry (right panels in B–D). (E) Lysates were subjected to immunoprecipitation with antibodies against total Akt1 or Akt2 and probed for Akt1, Akt2, PHLPP1, and PHLPP2. In B–E, the immunoblots and immunoprecipitations are representative of 3 independent experiments, which all gave similar results. In this and other figures, values are means ± SE from at least 3 independent experiments. *P < .05, **P < .01, and ***P < .001 versus control cells (ie, transfected with control siRNA or plasmid).
To determine whether PHLPPs dephosphorylate Akt in PaCa cells, we used PHLPP siRNAs or overexpression plasmids to transfect the cells and measured Akt phosphorylation at Ser473, critical for Akt activation. Two different siRNAs against each of PHLPP1 and PHLPP2 similarly and greatly (>3-fold) increased pAkt level in MIA PaCa-2 cells (Figure 1B). PHLPP1 and PHLPP2 siRNAs also increased pAkt in PANC-1 cells (Supplementary Figure 1). Correspondingly, PHLPP1 and PHLPP2 overexpression decreased the level of pAkt in MIA PaCa-2 (Figure 1C) and PANC-1 cells (Supplementary Figure 1B and C). Of note, overexpression of either PHLPP decreased phosphorylation of the downstream Akt substrates mTor, Foxo3α, and GSK-3β (Figure 1C). Interestingly, overexpression of PHLPP1, but not PHLPP2, decreased pGSK-3α level (Figure 1C).
It was recently reported in other cells that PHLPP1 and PHLPP2 differentially regulate distinct Akt isoforms.11 To determine specificities of PHLPPs toward Akt isoforms in PaCa cells, we measured the effects of PHLPP1 and PHLPP2 knockdowns on the levels of pAkt1 and pAkt2 (Figure 1D). In these experiments, pAkt was immunoprecipitated from MIA PaCa-2 cells transfected with PHLPP1 or PHLPP2 siRNAs, and the pAkt immunoprecipitates were probed with antibodies against Akt1 or Akt2. (Of note, there are no specific antibodies against pAkt1 versus pAkt2 because the Akt isoforms are highly homologous at the phosphorylation site.) We found that PHLPP1 siRNA knockdown specifically increased pAkt2 but not pAkt1, whereas PHLPP2 knockdown increased pAkt1 but not pAkt2 (Figure 1D). Coimmunoprecipitation experiments further showed that PHLPP1 and PHLPP2 selectively interact with the Akt isoforms; PHLPP1 coimmunoprecipitated with Akt2 but not with Akt1 and, in contrast, PHLPP2 coimmunoprecipitated with Akt1 but not Akt2 (Figure 1E). These results indicate that PHLPP1 and PHLPP2 selectively dephosphorylate the 2 Akt isoforms; they also help explain the finding (Figure 1C) that PHLPP1, but not PHLPP2, dephosphorylated GSK-3α, which is a specific substrate of Akt2 but not Akt1.11
We next addressed the specificity of PHLPPs for the Akt relative to other kinases. In addition to Akt, PHLPPs have been reported to dephosphorylate protein kinase C,24 which belongs to the same AGC kinase family as Akt. In neurons, PHLPP1β down-regulated the Ras-MEK/ERK pathway.21 However, in PaCa cells, PHLPPs did not affect the phosphorylation status of either protein kinase C or ERK (Supplementary Figure 2). The data indicate that knockdown of PHLPP1 or PHLPP2 decreases the expression levels of ERKs (Supplementary Figure 2), the mechanism of which remains to be determined.
Growth Factors Decrease PHLPP Activity in PaCa Cells
We next measured the kinetics of fetal bovine serum (FBS)-induced changes in pAkt levels in both control cells and cells depleted of PHLPP1 or PHLPP2 (Figure 2A and B). Knockdown of PHLPPs greatly up-regulated pAkt at all time points, as shown by densitometry. Importantly, PHLPP depletion specifically up-regulated pAkt but not the total Akt levels (Figure 2A and B).
Figure 2.

Serum (FBS) and IGF-I inhibit activities of PHLPP1 and PHLPP2 in PaCa cells. (A–E) MIA PaCa-2 and (F) PANC-1 cells, nontransfected or transfected with control or PHLPP1 or PHLPP2 siRNA, were depleted of growth factors for 48 hours and then cultured with and without 15% FBS or 100 ng/mL IGF-I for (A–E) indicated times or (F) 48 hours. (A, B, and E) Levels of PHLPP1 and PHLPP2, pAkt, and total Akt were measured by immunoblotting. The intensities of Akt and pAkt bands were quantified by densitometry, and pAkt/Akt ratios were calculated (bottom panels in A and B). (C, D, and F) Activities of PHLPP1 and PHLPP2 were measured in their immunoprecipitates and normalized to those in the immunoprecipitates from cells cultured without growth factors (GF). *P < .05, **P < .01, and ***p < .001 versus (C and D) 0 minutes or (F) no growth factors.
Both serum and insulin growth factor I (IGF-I) markedly decreased PHLPP1 activities in both cell lines; PHLPP2 activity was similarly inhibited in MIA PaCa-2 cells and somewhat less in PANC-1 cells (Figure 2C, D, and F). Of note, inhibition of PHLPPs was detected as early as after 15 minutes of incubation with growth factors and persisted for 48 hours of observation (Figure 2C, D, and F). Importantly, growth factors inhibited PHLPP activities without altering levels of PHLPPs (Figure 2A and B). Also, the amount of PHLPP coimmunoprecipitated with Akt isoforms was the same at 25 minutes and 48 hours after addition of FBS (Supplementary Figure 3).
While the PHLPP activities were persistently decreased, the FBS-induced increase in pAkt was maximal at ~15 minutes (Figure 2A, B, and E) due to the transient receptor-mediated stimulation of Akt phosphorylation by upstream kinases.25,26
Nox4 NADPH Oxidase Inhibits PHLPP1 Activity in PaCa Cells
We previously showed17,27–29 that in PaCa cells, growth factors activate the Nox4 NADPH oxidase and that Nox4-generated ROS inactivate PTPs, resulting in sustained activation of the prosurvival Jak/STAT. Here we examined the effect of Nox4 NADPH oxidase on the serine/threonine PHLPP phosphatases. siRNA knockdown of each of the NADPH oxidase subunits Nox4 or p22, as well as the NADPH oxidase inhibitor DPI, all increased PHLPP1 activity in MIA PaCa-2 and PANC-1 cells (Figure 3A–C). Nox4 colocalized with PHLPP1 both in the perinuclear area and on plasma membrane (Figure 3D). Furthermore, knocking down Nox4 or p22 or applying DPI decreased pAkt levels (Figure 3E), in accord with the data from another group30 These findings, together with our published results,17,27–29 indicate that growth factors inhibit PHLPP1 activity in PaCa cells through activating Nox4 NADPH oxidase. Of note, neither the siRNA transfection nor DPI increased PHLPP1 level while increasing PHLPP1 activity (Figure 3E). Thus, Nox4 NADPH oxidase inactivates PHLPP1 in PaCa cells through inhibiting PHLPP1 phosphatase activity rather than affecting its level.
Figure 3.

Nox4 NADPH oxidase negatively regulates PHLPP1 activity in PaCa cells. (A–F) MIA PaCa-2 and (A and F) PANC-1 cells, nontransfected or transfected with control, Nox4, or p22 siRNA, were cultured for 48 hours with (A, C–E) 15% FBS or (B) 100 ng/mL IGF in the presence or absence of 10 μmol/L DPI. (A and B) PHLPP1 activity was measured in PHLPP1 immunoprecipitates as in Figure 2 and normalized to that in cells transfected with control siRNA or cultured without DPI. *P < .05, **P < .01, and ***P < .001 versus cells transfected with control siRNA or cultured without DPI. (C and E) Transfection efficiencies and the levels of pAkt, Akt, and PHLPP1 were measured by immunoblotting. Representative of 3 independent experiments, which all gave similar results. (D) Nontransfected MIA PaCa-2 cells were stained for PHLPP1 (green) and Nox4 (red) and analyzed by confocal microscopy. Each image is representative of at least 50 cancer cells. (F) PHLPP1 was immunoprecipitated from cells cultured without and with FBS, as indicated; 1 μmol/L H2O2 or 1 mmol/L dithiothreitol (DTT) was added to the immunoprecipitates for 5 minutes and phosphatase activities were measured. **P < .01, ***P < .001 versus activity of PHLPP1 without H2O2 (left panel) or without ETT (right panel).
To test whether ROS directly affect PHLPP1 activity, we examined the effect of H2O2 on PHLPP1 immunoprecipitates (Figure 3F). PHLPP1 was immunoprecipitated from cells cultured without and with growth factors, H2O2 was added to the immunoprecipitates, and PHLPP1 activities were measured 5 minutes later. The activity of PHLPP1 immunoprecipitated from cells cultured without growth factors was high and was dramatically (>70%) reduced by the addition of H2O2, indicating a direct inhibitory effect of ROS on PHLPP1 activity. Differently, the activity of PHLPP1 immunoprecipitated from cells cultured with FBS was low and was little affected by H2O2.
The explanation for the observed difference in the effect of H2O2 on PHLPP1 activity in PaCa cells cultured without versus with growth factors is that the cellular ROS levels differ in these conditions, resulting in different levels of PHLPP1 activity. In the absence of growth factors, the activity of NADPH oxidase and thus cellular ROS level are relatively low17,27–29 and not sufficient to inhibit phosphatase activity in PaCa cells. Therefore, in these conditions PHLPP1 activity is relatively high, and the inactivating (oxidizing) effect of exogenous H2O2 on PHLPP1 is quite dramatic (Figure 3F). Differently, in cells cultured with growth factors the NADPH oxidase activity is relatively high, resulting in high ROS level and a markedly decreased phosphatase activity.17,27–29 Therefore, PHLPP1 immunoprecipitated from cells cultured with FBS has low activity, and the addition of H2O2 has little effect (Figure 3F). Correspondingly, addition of the reducing agent dithiothreitol (which restores the active, nonoxidized form of phosphatase17,27–29) to PHLPP1 immunoprecipitates obtained from cells cultured with FBS increased PHLPP1 activity (Figure 3F).
Interestingly, neither Nox4 NADPH oxidase inactivation (Supplementary Figure 4) nor H2O2 (data not shown) had any effect on PHLPP2 activity, indicating that the 2 PHLPP isoforms are differentially regulated by ROS.
PHLPP Overexpression Stimulates PaCa Cell Death
Overexpression of either PHLPP1 or PHLPP2 increased apoptotic DNA fragmentation in both MIA PaCa-2 and PANC-1 cells cultured with and without growth factors (Figure 4A). The stimulation of apoptosis was more pronounced with overexpression of PHLPP1 compared with that of PHLPP2 (Figure 4A). In accord with these data, knockdown of Akt2 potentiated apoptosis to a greater extent than that of Akt1 (Supplementary Figure 5A). Knockdown of both Akt1 and Akt2 isoforms increased DNA fragmentation to the same extent as PHLPP overexpression (Supplementary Figure 5B). The effects of PHLPP overexpression and Akt siRNA knockdown on apoptosis were not additive, indicating a common pathway (Supplementary Figure 5B).
Figure 4.

PHLPP1 and PHLPP2 overexpression increases PaCa cell death. MIA PaCa-2 and PANC-1 cells were transfected with control, PHLPP1, or PHLPP2 plasmids or subjected to double transfection with PHLPP1 or PHLPP2 and LC3-GFP plasmids. Cells were grown for 48 hours without growth factors (no GF) or with 15% FBS or 100 ng/mL IGF-I (IGF). (A) Apoptosis was quantified by measuring DNA fragmentation with the Cell Death ELISA kit. (B) Necrosis was measured as the percentage of propidium iodine (PI)-positive cells. (C) Autophagy was quantified in cells overexpressing LC3-GFP as the percentage of cells with more than 5 LC3 dots per cell. (D) Proliferation was measured by 3H-thymidine uptake. *P < .05 versus cells transfected with control plasmids, #P < .05 versus cells transfected with PHLPP2 plasmid.
In both MIA PaCa-2 (Figure 4B) and PANC-1 (not shown) cells, PHLPP overexpression also increased the percentage of cells permeable for PI, which include AnV+/PI+ cells, representing late apoptosis associated with secondary necrosis, and AnV−/PI+ cells with primary necrosis.22 Both PHLPPs stimulated autophagy in PaCa cells (Figure 4C). In MIA PaCa-2 cells, the effects of PHLPP1 and PHLPP2 on autophagy were of the same magnitude; in PANC-1 cells, PHLPP1 increased autophagic vacuoles to a greater extent than PHLPP2 (Figure 4C).
The results in Figure 4 show that PHLPP overexpression overcomes the resistance of PaCa cells to death, both in the presence and absence of growth factors. Surprisingly, we did not detect any significant effect of PHLPP overexpression on MIA PaCa-2 cell proliferation (Figure 4D).
PHLPP1 Expression in PDAC Negatively Correlates With Patients' Survival
PHLPPs were highly expressed in normal mouse pancreas, and their levels were greatly decreased in the KrasG12D and KrasG12D/Trp53R172H genetic mouse models of PDAC (Figure 5). Further, the results in Figure 5 indicate that PHLPP1 and PHLPP2 levels time-dependently decrease with PanIN development in KrasG12D mice.
Figure 5.
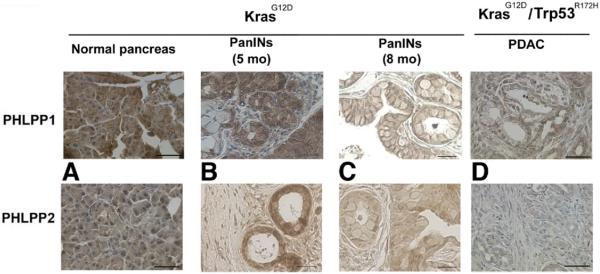
PHLPP1 expression correlates with tumor development in pancreatic mouse cancer models. PHLPP1 and PHLPP2 IHC was performed on pancreatic tissue samples from KrasG12D and KrasG12D/Trp53R172H mice at different stages of tumor development. The images were visualized with light microscopy and are representative of 3 different mice for each condition. Scale bar = 50 μm.
IHC showed that PHLPPs were also highly expressed in normal human pancreas, both in acinar and ductal but not stromal cells (Figure 6A). In human PDAC, PHLPPs were confined to cancer but not to stromal cells (Figure 6A). Intracellular localizations of PHLPP1 and PHLPP2 were different. PHLPP1 showed both membrane and perinuclear staining, whereas PHLPP2 was only localized to the cytosol (Figure 6B). Of note, these differences in PHLPP1 and PHLPP2 cellular localizations were similarly observed in normal human pancreas and PDAC (Figure 6B) and PaCa cell lines (Figure 1A).
Figure 6.

PHLPP1 expression correlates with survival of patients with PDAC. (A, C–F) IHC and (B) immunofluorescence were performed on human normal pancreas and PDAC tissue samples using primary antibodies against PHLPP1, PHLPP2, pAkt, Akt1, or Akt2 and biotinstreptavidin-peroxidase system (A and E; hematoxilin counterstain) or secondary antibodies conjugated with Alexa 488 (B). Scale bar = 50 μm. (C) Forty PDAC samples were grouped as high and low PHLPP1 or PHLPP2 or pAkt expression, and a Kaplan–Meier survival analysis and a log-rank test were performed. (D) Correlations between PHLPP and pAkt levels were calculated on 28 PDAC samples using Spearman rank correlation test. (F) Percentage of tumors with high or low Akt1 and Akt2 expression were quantified in 28 PDAC samples.
The expression of both PHLPP1 and PHLPP2 dramatically decreased in PDAC as compared with normal pancreas (Figure 6A). Differently and in agreement with previous reports,20,23 Akt phosphorylation was barely detectable in normal pancreas but greatly increased in PDAC (Figure 6A). Further, the level of pAkt inversely correlated with survival in the cohort of patients with PDAC we investigated (Figure 6C). We found an inverse correlation between PHLPP expression (for both isoforms) and the level of pAkt in patients with PDAC (Figure 6D). Most strikingly, the decrease in PHLPP1 level in human PDAC highly correlated (P < .009) with shorter survival for patients with PDAC (Figure 6C and Supplementary Table 1); in contrast, there was no correlation between PHLPP2 level and patients' survival (Figure 6C).
An explanation for these different roles of PHLPP1 and PHLPP2 in PDAC is provided by our finding that PHLPP1 and PHLPP2 selectively target the 2 Akt isoforms in PaCa cells; PHLPP1 specifically dephosphorylates Akt2, whereas PHLPP2 dephosphorylates Akt1 (Figure 1). Our data indicate that the roles of Akt1 and Akt2 in PDAC are different; Akt2 was up-regulated in 77% of pancreatic tumors in the PDAC patient cohort we investigated, whereas Akt1 level was elevated in only 18% of tumors (Figure 6E and F). Strikingly, the median survival of patients with PDAC with high Akt2 levels (in the cohort under study) was 16 months versus 44 months for those with low Akt2 expression. In contrast, there was no difference in the median survival time between patients with high and low Akt1 expression (Supplementary Table 2).
Thus, our findings suggest that the strong association of PHLPP1 expression with survival of patients with PDAC (Figure 6C) is due to the fact that Akt2, which is a substrate for PHLPP1 (but not Akt1, which is targeted by PHLPP2), mediates development of PDAC. The role of Akt2 in development of PaCa is also supported by data in the literature.2,5,8,13
PHLPP1, But Not PHLPP2, Overexpression Drastically Reduces Tumor Growth and Stimulates Apoptosis in the Orthotopic Model of PaCa
We next examined the effects of PHLPP1 and PHLPP2 overexpression in the orthotopic mouse model of PaCa (Figure 7).
Figure 7.

PHLPP1, but not PHLPP2, overexpression inhibits tumor growth and stimulates apoptosis in the orthotopic xenograft mouse model of PaCa. Orthotopic xenograft PaCa models were developed using control MIA PaCa-2 cells (WT-MIA) or lentiviral-transduced MIA PaCa-2 cells stably overexpressing PHLPP1 (PHLPP1-MIA) or PHLPP2 (PHLPP2-MIA). Mice were killed 6 weeks after transplant of the xenograft. (A) PHLPP1 and PHLPP2 and pAkt levels were analyzed with IHC using a biotin-streptavidin-peroxidase system. (B) Tumor volume and weight were normalized to those in WT-MIA, the values for which were, respectively, 2.0 ± 0.3 cm3 and 3.5 ± 0.8 g (means ± SE from 14 WT-MIA mice). (C) Apoptosis and proliferation were quantified with terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling and Mib1 staining, correspondingly, in 10 fields per tumor. Values are means ± SE (n = 60). *P < .05 versus ***P < .001; ****P < .0001 WT-MIA (n = 6 mice per group).
We compared the orthotopic xenograft PaCa models that we developed using control MIA PaCa-2 cells (WT-MIA) versus MIA PaCa-2 cells stably overexpressing PHLPP1 (PHLPP1-MIA) or PHLPP2 (PHLPP2-MIA). The cells were generated by lentiviral transduction (Supplementary Materials and Methods). IHC confirmed that the tumors derived from PHLPP1-MIA cells had higher PHLPP1 and lower pAkt levels than tumors in WT-MIA mice (Figure 7A). The first indication of the antitumorigenic effect of PHLPP1 was that subcutaneous engraftment of PHLPP1-overexpressing cells in the donor mice resulted in tumor formation in only 50% of mice, whereas engraftment of control (WT-MIA) cells induced tumor formation in 100% of mice. Further, the tumors in PHLPP1-MIA xenograft mice were >3 times smaller (Figure 7B) and had >5 times more apoptotic cells than tumors in the WT-MIA model (Figure 7C). There was no difference in proliferation of tumor cells between the PHLPP1-MIA and WT-MIA orthotopic models (Figure 7C). Differently, overexpression of PHLPP2 had a lesser inhibitory effect on pAkt and did not affect tumor weight and volume and apoptotic death of tumor cells. These data support our findings that the PHLPP1/Akt2, but not PHLPP2/Akt1, pathway mediates development of pancreatic tumors.
Discussion
Our results establish an important role for the serine/threonine protein phosphatases PHLPP1 and PHLPP2, antagonizing Akt activity in PaCa. We show that both PHLPPs colocalize with and dephosphorylate Akt in PaCa cells. PHLPP1 overexpression down-regulated pAkt in the orthotopic mouse model. Furthermore, expression of PHLPPs in human PDAC and mouse genetic and orthotopic models of PaCa inversely correlated with Akt phosphorylation level.
Growth factors (serum and IGF-I) inhibited activities of PHLPPs. We have previously shown17,27–29 that, in PaCa cells, growth factors inactivate PTPs (such as LMW-PTP17) through a Nox4 NADPH oxidase–dependent mechanism. Specifically, we showed that FBS and IGF-I cause nuclear factor κB-dependent transcriptional up-regulation of the p22 subunit of the Nox4 NADPH oxidase, resulting in its activation and increase in ROS.28 In turn, ROS inactivate PTPs, maintaining the activation of the pro-survival Jak/STAT pathway.17 The present study indicates that Nox4 NADPH oxidase inactivates the serine/threonine phosphatase PHLPP1, suggesting the following chain of events: growth factors up-regulate p22, resulting in NADPH oxidase activation and an increase in cellular ROS; in turn, ROS inactivate PHLPP1, resulting in pAkt up-regulation.
Of note, the current paradigm31,32 is that regulation by ROS is unique to PTPs (such as LMW-PTP17) due to specific structure of their catalytic center. The importance of our results is that they show inactivation of a serine/threonine phosphatase by ROS. Furthermore, the results reveal a novel mechanism through which growth factors activate Akt: by inhibiting PHLPPs, which terminate Akt signaling.
PHLPP isoforms have the same domain structure and ~50% sequence homology.11 PHLPP1 and PHLPP2 both decreased phosphorylation of Akt and its downstream targets mTor, Foxo3α, and GSK-3β, and both stimulated PaCa cell death. However, our data indicate that the properties of PHLPP1 and PHLPP2 are not identical. (1) The PHLPP isoforms have different intracellular localizations in PaCa cells; PHLPP1 localizes to both the perinuclear area and the plasma membrane, whereas PHLPP2 predominantly localizes to the perinuclear area. (2) PHLPP1, but not PHLPP2, is regulated by ROS and the Nox4 NADPH oxidase. (3) PHLPP1 and PHLPP2 regulate distinct Akt isoforms; PHLPP1 selectively dephosphorylates Akt2, whereas PHLPP2 dephosphorylates Akt1. PHLPP1 has a greater proapoptotic effect than PHLPP2.
Compared with normal pancreas, levels of PHLPPs were much lower in both human PDAC and mouse KrasG12D and KrasG12D/Trp53R172H genetic models of PaCa. Thus, PaCa is associated with down-regulation of PHLPP. Furthermore, we found that high PHLPP1 (but not PHLPP2) levels strongly correlated with better survival of patients with PDAC in the cohort under study, suggesting a possible prognostic value for PHLPP1. The strong association of PHLPP1 expression with survival of patients with PDAC is likely due to the fact that Akt2, which is a selective substrate for PHLPP1, mediates development of PDAC. Indeed, Akt2 but not Akt1 was up-regulated in a majority of tumors in the cohort of patients with PDAC. Median survival of the patients with PDAC inversely correlated with tumor Akt2 level, whereas there was no such correlation for Akt1. Our results are in accord with data in the literature2,5,7–9,33 indicating that Akt2 is highly expressed and could be a prognostic marker in PDAC. In contrast, the role of Akt1 in pancreatic tumorigenesis is questionable; recent data34 even suggest that Akt1 expression is associated with favorable prognosis in PaCa. The reasons for the different roles of Akt2 and Akt1 in PDAC, and why up-regulation of Akt2 in PDAC is greater than Akt1, are unknown and require further investigation.
In accord with the results in human PDAC, we found that PHLPP1, but not PHLPP2, overexpression greatly suppressed pancreatic tumorigenesis in the orthotopic mouse model of PaCa. PHLPP1, but not PHLPP2, overexpression dramatically stimulated apoptotic death of tumor cells, providing a mechanism for the in vivo antitumorigenic effect of PHLPP1. Again, the reasons for the different role of PHLPP1/Akt2 and PHLPP2/Akt1 pathways in the orthotopic model remain to be determined.
In summary, PHLPP1 and PHLPP2 are potent negative regulators of Akt activation in PaCa. Activities of PHLPPs are inhibited in PaCa cells through growth factor and ROS-dependent mechanisms. PHLPP1 and PHLPP2 selectively inactivate the 2 Akt isoforms; PHLPP1 dephosphorylates Akt2, whereas PHLPP2 dephosphorylates Akt1. The levels of PHLPPs are much lower in PaCa than in normal pancreas. Low PHLPP1, but not PHLPP2, levels correlate with shorter survival time of patients with PDAC, likely because higher expression of Akt2 (but not Akt1) is associated with PDAC. PHLPP1, but not PHLPP2, overexpression suppresses tumor growth and stimulates apoptosis in the ortothopic mouse model, indicating that the antitumorigenic effect of PHLPP1 is through promoting cancer cell death. The results elucidate the mechanisms by which PaCa cells inactivate PHLPPs, indicate a tumor-suppressive role for PHLPP1, and suggest PHLPP1 as a target for therapeutic approaches and a potential diagnostic tool in PDAC.
Supplementary Material
Acknowledgments
The authors thank Dr Alexandra Newton (University of California at San Diego) for providing the PHLPP plasmids.
Funding Supported by National Cancer Institute grant R01CA119025 and the Department of Veterans Affairs Merit Review (to A.S.G.) and the Alfried Krupp von Bohlen und Halbach Stiftung grant (to C.N.).
Abbreviations used in this paper
- FBS
fetal bovine serum
- IHC
immunohistochemistry
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- PaCa
pancreatic cancer
- pAKT
phosphorylated Akt
- PDAC
pancreatic ductal adenocarcinoma
- PTP
protein tyrosine phosphatase
- ROS
reactive oxygen species
- siRNA
small interfering RNA.
Footnotes
Supplementary Material Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2011.10.026.
Conflicts of interest The authors disclose no conflicts.
References
- 1.Fahy BN, Schlieman M, Virudachalam S, et al. AKT inhibition is associated with chemosensitisation in the pancreatic cancer cell line MIA-PaCa-2. Br J Cancer. 2003;89:391–397. doi: 10.1038/sj.bjc.6601037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng JQ, Ruggeri B, Klein WM, et al. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci U S A. 1996;93:3636–3641. doi: 10.1073/pnas.93.8.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holcomb B, Yip-Schneider MT, Matos JM, et al. Pancreatic cancer cell genetics and signaling response to treatment correlate with efficacy of gemcitabine-based molecular targeting strategies. J Gastrointest Surg. 2008;12:288–296. doi: 10.1007/s11605-007-0406-6. [DOI] [PubMed] [Google Scholar]
- 4.Liu D, Zhang Y, Dang C, et al. siRNA directed against TrkA sensitizes human pancreatic cancer cells to apoptosis induced by gemcitabine through an inactivation of PI3K/Akt-dependent pathway. Oncol Rep. 2007;18:673–677. [PubMed] [Google Scholar]
- 5.Altomare DA, Tanno S, De Rienzo A, et al. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem. 2002;87:470–476. doi: 10.1002/jcb.10287. [DOI] [PubMed] [Google Scholar]
- 6.Bellacosa A, de Feo D, Godwin AK, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64:280–285. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- 7.Pham NA, Schwock J, Iakovlev V, et al. Immunohistochemical analysis of changes in signaling pathway activation downstream of growth factor receptors in pancreatic duct cell carcinogenesis. BMC Cancer. 2008;8:43. doi: 10.1186/1471-2407-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruggeri BA, Huang L, Wood M, et al. Amplification and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol Carcinog. 1998;21:81–86. [PubMed] [Google Scholar]
- 9.Shi XH, Liang ZY, Ren XY, et al. Combined silencing of K-ras and Akt2 oncogenes achieves synergistic effects in inhibiting pancreatic cancer cell growth in vitro and in vivo. Cancer Gene Ther. 2009;16:227–236. doi: 10.1038/cgt.2008.82. [DOI] [PubMed] [Google Scholar]
- 10.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27:5443–5453. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- 11.Brognard J, Sierecki E, Gao T, et al. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;25:917–931. doi: 10.1016/j.molcel.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 12.Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 13.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–395. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Weiss HL, Rychahou P, et al. Loss of PHLPP expression in colon cancer: role in proliferation and tumorigenesis. Oncogene. 2009;28:994–1004. doi: 10.1038/onc.2008.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ouillette P, Erba H, Kujawski L, et al. Integrated genomic profiling of chronic lymphocytic leukemia identifies subtypes of deletion 13q14. Cancer Res. 2008;68:1012–1021. doi: 10.1158/0008-5472.CAN-07-3105. [DOI] [PubMed] [Google Scholar]
- 16.Qiao M, Iglehart JD, Pardee AB. Metastatic potential of 21T human breast cancer cells depends on Akt/protein kinase B activation. Cancer Res. 2007;67:5293–5299. doi: 10.1158/0008-5472.CAN-07-0877. [DOI] [PubMed] [Google Scholar]
- 17.Lee JK, Edderkaoui M, Truong P, et al. NADPH oxidase promotes pancreatic cancer cell survival via inhibiting JAK2 dephosphorylation by tyrosine phosphatases. Gastroenterology. 2007;133:1637–1648. doi: 10.1053/j.gastro.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 18.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 19.Eibl G, Reber HA. A xenograft nude mouse model for perineural invasion and recurrence in pancreatic cancer. Pancreas. 2005;31:258–262. doi: 10.1097/01.mpa.0000175176.40045.0f. [DOI] [PubMed] [Google Scholar]
- 20.Schlieman MG, Fahy BN, Ramsamooj R, et al. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br J Cancer. 2003;89:2110–2115. doi: 10.1038/sj.bjc.6601396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimizu K, Okada M, Nagai K, et al. Suprachiasmatic nucleus circadian oscillatory protein, a novel binding partner of K-Ras in the membrane rafts, negatively regulates MAPK pathway. J Biol Chem. 2003;278:14920–14925. doi: 10.1074/jbc.M213214200. [DOI] [PubMed] [Google Scholar]
- 22.Vaquero EC, Edderkaoui M, Nam KJ, et al. Extracellular matrix proteins protect pancreatic cancer cells from death via mitochondrial and nonmitochondrial pathways. Gastroenterology. 2003;125:1188–1202. doi: 10.1016/s0016-5085(03)01203-4. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto S, Tomita Y, Hoshida Y, et al. Prognostic significance of activated Akt expression in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2004;10:2846–2850. doi: 10.1158/1078-0432.ccr-02-1441. [DOI] [PubMed] [Google Scholar]
- 24.Gao T, Brognard J, Newton AC. The phosphatase PHLPP controls the cellular levels of protein kinase C. J Biol Chem. 2008;283:6300–6311. doi: 10.1074/jbc.M707319200. [DOI] [PubMed] [Google Scholar]
- 25.Galetic I, Andjelkovic M, Meier R, et al. Mechanism of protein kinase B activation by insulin/insulin-like growth factor-1 revealed by specific inhibitors of phosphoinositide 3-kinase–significance for diabetes and cancer. Pharmacol Ther. 1999;82:409–425. doi: 10.1016/s0163-7258(98)00071-0. [DOI] [PubMed] [Google Scholar]
- 26.Sarbassov DD, Guertin DA, Ali SM, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 27.Edderkaoui M, Hong P, Vaquero EC, et al. Extracellular matrix stimulates reactive oxygen species production and increases pancreatic cancer cell survival through 5-lipoxygenase and NADPH oxidase. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1137–1147. doi: 10.1152/ajpgi.00197.2005. [DOI] [PubMed] [Google Scholar]
- 28.Edderkaoui M, Nitsche C, Zheng L, et al. NADPH oxidase activation in pancreatic cancer cells is mediated through Akt dependent up-regulation of p22phox. J Biol Chem. 2011;286:7779–7787. doi: 10.1074/jbc.M110.200063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaquero EC, Edderkaoui M, Pandol SJ, et al. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J Biol Chem. 2004;279:34643–34654. doi: 10.1074/jbc.M400078200. [DOI] [PubMed] [Google Scholar]
- 30.Mochizuki T, Furuta S, Mitsushita J, et al. Inhibition of NADPH oxidase 4 activates apoptosis via the AKT/apoptosis signal-regulating kinase 1 pathway in pancreatic cancer PANC-1 cells. Onco-gene. 2006;25:3699–3707. doi: 10.1038/sj.onc.1209406. [DOI] [PubMed] [Google Scholar]
- 31.Ariño J, Alexander DR. Protein phosphatases. Springer; Berlin: 2004. [Google Scholar]
- 32.Tanner JJ, Parsons ZD, Cummings AH, et al. Redox regulation of protein tyrosine phosphatases: structural and chemical aspects. Antioxid Redox Signal. 2011;15:77–97. doi: 10.1089/ars.2010.3611. [DOI] [PubMed] [Google Scholar]
- 33.Jiang K, Coppola D, Crespo NC, et al. The phosphoinositide 3-OH kinase/AKT2 pathway as a critical target for famesyltransferase inhibitor-induced apoptosis. Mol Cell Biol. 2000;20:139–148. doi: 10.1128/mcb.20.1.139-148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J, Cheng Sun SH, Sun SJ, et al. Phosph-Aktl expression is associated with a favourable prognosis in pancreatic cancer. Ann Acad Med Singapore. 2010;39:548–547. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.