

Abstract
HL60 and U937 (acute myeloid leukemia (AML) cell lines) were assessed for sensitivity to YM155, and found to have distinct sensitive and resistant phenotypes, respectively. In HL60 cells, YM155 inhibition of growth proliferation was due to apoptosis which was measured by annexin V/PI staining. YM155 induced apoptosis through activation of intrinsic and extrinsic pathways that also culminated in caspase-3 activity and PARP cleavage. YM155 sensitivity was partially associated with this compound’s ability to downregulate survivin transcription since this was more pronounced in the HL60 cell line. However, marked differences were also observed in XIAP, Bcl-2, and Mcl-1L, and Mcl-1s. Furthermore, YM155 treatment completely inhibited production of total Akt protein in HL60, but not U937 cells. Importantly, Akt activity (pAkt-Ser473) levels were maintained in YM155 treated U937 cells which may help stabilize other anti-apoptotic proteins. Combination treatments with an Akt inhibitor, MK-2206, reduced levels of pAkt-Ser473 in U937 cells and synergistically sensitized them to YM155 cytotoxicity. Collectively our results indicate that Akt signaling may be an important factor mediating YM155 response in AML, and combinatorial therapies with Akt inhibitors could improve treatment efficacy in YM155-resistant cells.
Keywords: YM155, Survivin, Akt, XIAP, Bcl-2, Mcl-1, AML
Introduction
Acute myeloid leukemia (AML) is the most common type of acute leukemia in adults [1]. AML is especially prevalent in older adults, with a median age of diagnosis of approximately 65 years [2]. Standard treatment for AML usually involves intensive induction chemotherapy with anthracycline and cytarabine [3]. While many patients experience an initial remission response, therapies are associated with high rates of toxicity, morbidity, and mortality [3]. Most patients relapse and limited treatment options in such a patient population clearly demonstrate a need to develop new therapeutic strategies [4].
Survivin is a member of the Inhibitor of Apopotosis (IAP) protein family that is highly expressed in many cancers and considered an important candidate for targeted cancer therapy [5]. YM155, a small molecule inhibitor of survivin, potently inhibits the growth of many different cancers at low nanomolar concentrations and is currently in clinical development as a cancer therapeutic [6]. This compound has been investigated as both a monotherapy and combination therapy in several phase II clinical trials for the treatment of both solid and hematological tumors [7–10]. Although no clinical trial has yet been conducted to evaluate YM155 treatment in AML, extensive data indicate that survivin may be an important target in this malignancy [11]. Survivin expression levels are significantly associated with the presence of AML stem cell populations [12], cell proliferation and chemoresistance [13], and adverse patient survival outcomes [14]. Previous research in our lab found that increased expression of survivin in AML cells was an important mechanism of developed resistance to FLT3 inhibitors [15].
YM155’s mechanism of action on survivin is through interference of transcription, which occurs either by disrupting an IL3/p54 complex needed for gene expression [16] or by preventing the transcription factor Sp1 from binding to the survivin promoter [17]. Intriguingly, the growth inhibitory effects of YM155 tested on more than 100 cancer cell lines were found to be only weakly correlated with endogenous levels of survivin expression [6]. In other words, tumor cell sensitivity to YM155 treatment may be only weakly associated with cell dependence on survivin expression. While survivin was initially identified as a primary and specific target of YM155 [18], many subsequent studies have shown that YM155 has other cellular targets including other IAPs and Bcl-2 family members [10].
Mcl-1 is another critical target of YM155 which can inhibit expression of this anti-apoptotic protein in multiple cancer cell lines [19]. In multiple myeloma cells, Mcl-1, rather than survivin, was found to be the target most important to YM155 efficacy [20]. However, in some cancer cell lines such as pancreatic cancer, Mcl-1 levels are unaffected by YM155 treatment and cell death is associated with repression of other anti-apoptotic proteins including XIAP [21].
In order to understand what cells will respond to YM155 therapy, it is important to identify underlying molecular mechanisms relevant to YM155 response. In pediatric acute lymphocytic leukemia (ALL), survivin inhibition due to YM155 treatment depended on p53 activity [22], yet in most cancer cell lines YM155 efficacy was found to be independent of p53 status [6]. In this study we assess the effects of YM155 treatment on AML cell lines that are p53-deficient finding they have distinct response phenotypes. These p53-deficient cell lines, HL60 (YM155-sensitive) and U937 (YM155-resistant), are examined in detail to identify molecular mechanisms associated with YM155 response. We identified differences in several anti-apoptotic proteins including XIAP, Mcl-1, and Bcl-2, as well as Akt signaling mechanisms that are likely to influence treatment outcomes. The results of this study help to identify key regulatory molecules and signaling pathways in p53-deficient AML cells that are associated with responsiveness to YM155 therapy.
Materials and methods
Cell culture and reagents
HL60 and U937 leukemia cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in RPMI 1640 medium supplemented with 10% heat inactivated fetal bovine serum, 10 mM of L-glutamine, 10 U/ml penicillin, and 10 μg/ml streptomycin. Cell cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO2 and exponentially growing cells were used for drug exposure assays. YM155 was obtained from Selleck Chemicals (Houston, TX, USA).
Cell viability assays
Cells were seeded in triplicate, 40,000/well, in a 96-well plate and treated with various doses of YM155 (0 nM, 0.78 nM, 3.12 nM, 12.5 nM, 50 nM, and 200 nM) for different lengths of time (24, 48 and 72 hrs). After treatment, cell viability was assessed using the CellTiter 96 Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI, USA) according to the manufacturer’s protocol. The plates were read on a microplate reader (Bio-Rad Model 3550). The GI50 (50% growth inhibition value) was calculated for each cell line and displayed as the average values obtained from 3 independent experiments.
Caspase activity assays
Approximately 25,000 cells were seeded in triplicate in 96-well plates and treated with the same concentrations of YM155 as used in the cell viability assays. After an 8 hour exposure, caspase activity was measured using either the Caspase-Glo 9 (Promega), Caspase-Glo 8 (Promega), or Caspase-Glo 3/7 (Promega) luminescent assay. Caspase activity was measured in whole cell samples following the manufacturer’s instructions. Briefly, freshly prepared luminescent reagent was added to each cell sample in a 1:1 volume and the luminescent output was measured 30–60 minutes later on the MicroLumat Plus LB96V (Berthold). The relative luminescent units obtained from each reading were transformed into percentage values relative to the untreated control. Results shown are the average values ± SD obtained from three independent experiments.
Western blot analysis
Twenty-four hours after YM155 treatment, HL60 and U937 cells were collected, washed and resuspended in NP-40 lysis buffer. Whole cell lysates (40 μg) were separated through 4–20% sodium dodecyl sulfate (SDS)–polyacrylamide gels under denaturing conditions and transferred to polyvinylidene difluoride (PVDF) membranes (Invitrogen, Carlsbad, CA). The membranes were blocked and incubated with the following antibodies: PARP (cat#9532), Akt (cat#9272), pAkt-Ser473 (cat#92715), β-actin (cat#5125), XIAP (cat#2042), GAPDH (cat#3683S) (Cell Signaling Technologies, Beverly, MA); Bcl-2 (cat#sc-783), Mcl-1 (cat#sc-20679), p21 (cat#sc-397, sc-469) (Santa Cruz Biotechnology, Santa Cruz, CA); and an HRP-conjugated anti-rabbit IgG antibody (cat#7074) (Cell Signaling Technologies, Beverly, MA). Data were normalized to corresponding values of β-actin or GAPDH densitometry.
Cell cycle analysis
HL60 and U937 cells (5 × 105) were exposed to distinct concentrations of YM155 for 24 hours. These cells were collected, fixed with 75% ethanol, then treated with propidium iodide (PI) and ribonuclease staining buffer (BD Pharmingen) according to the manufacturer’s protocol. Samples were analyzed by flow cytometry (FACSCalibur™; Becton Dickinson, Franklin Lakes, NJ).
Apoptosis
Apoptotic cell death was evaluated with an Annexin V and PI apoptosis detection kit (BD Biosciences, San Jose, CA). YM155 treated cells were stained with FITC-conjugated Annexin V in the presence of PI and analyzed by flow cytometry. Annexin V+ cells were scored as apoptotic cells.
Real-time polymerase chain reaction
Total RNA was extracted from HL60 and U937 cells using the Quick RNA mini-prep kit (Zymo Research) according to the manufacturer’s instructions. cDNA was synthesized with a random hex primer using the Verso cDNA kit (Thermoscientific). Quantitative PCR was performed with gene specific primers for survivin, XIAP, Bcl-2, Mcl1L, Mcl1s, AKT, p21, and 18s RNA using the SYBR Green Supermix (Bio-Rad) on a CFX96 Real-time System (Bio-Rad). The PCR conditions were as follows: 95 °C, 15 min; then 35 cycles of 94 °C, 15 s; with 60 °C, 45 s (two-step PCR). The measured transcript abundance was normalized to the expression level of 18s RNA for all samples. At least two independent analyses were performed for each culture condition.
Drug synergy analysis
The growth inhibitory effects of YM155 and MK-2206 [23] combined treatments of the U937 cell line were assessed using the CompuSyn (1.0) software based on the Combination Index (CI) Theorem developed by Chou and Talalay [24]. As previously described [25], CI values <1 are synergistic, =1 additive, and >1 antagonistic.
Results
YM155 inhibits AML cell growth in a time- and dose-dependent manner
HL60 and U937 cells were exposed to different concentrations of YM155 for 24, 48, and 72 hours (Fig. 1). The growth inhibitory effects of this compound were found to differ by more than 100× in these cell lines, with the calculated IC50 = 0.9 nM for HL60 cells vs. IC50 = 133.5 nM for U937 cells at 72 hours. As these observations differed from those of a recent publication [26], we purchased a second set of HL60 and U937 cell lines from ATCC to verify cellular identity and confirm these disparate YM155 exposure outcomes. We then performed additional experiments to identify differences in molecular mechanisms of action associated with YM155 treatment responses.
Fig. 1.
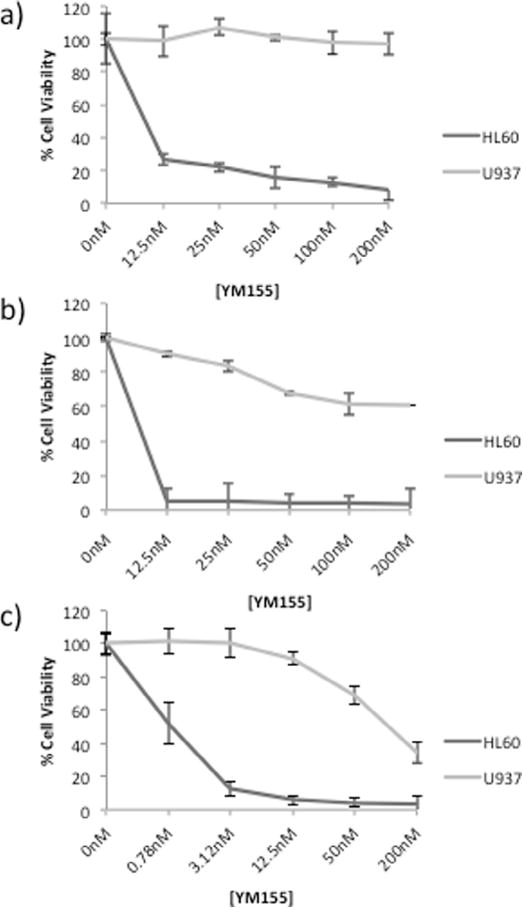
YM155 causes growth inhibition of HL60 and U937 cells in a dose- and time-dependent manner. AML cells were treated with YM155 for a) 24 hours, b) 48 hours, and 72 hours. Data represent mean ± SE from three independent experiments.
YM155 inhibits survivin in a time- and dose-dependent manner
It has been well established that YM155 inhibits expression of survivin in cancer cells [6]. In order to demonstrate down-regulation of survivin using our culture system, the effect of YM155 exposure on the expression of survivin in HL60 and U937 cells was examined. YM155 down-regulated survivin transcript expression in a dose- and time-dependent manner in both cell lines, although the effect was more pronounced and at a lower dose in HL60 cells (Supplementary Fig. S1). To identify other anti-apoptotic factors associated with YM155 response, the effects of this compound on anti-apoptotic proteins Mcl-1, Bcl-2, and XIAP were also analyzed.
YM155 growth inhibition of the HL60 cell line is due to intrinsic and extrinsic apoptosis
To determine if YM155 exposure could induce apoptosis in either HL60 or U937 cells, identical cultures were treated with 0, 12.5, 50, and 200 nM concentrations of YM155 for a period of 24 hours. We found that increasing concentrations of YM155 induced apoptosis in the HL60 cell line in a dose-dependent manner, but had no effect on U937 (Fig. 2a). YM155 exposure activated both intrinsic and extrinsic apoptotic signaling pathways in HL60, but not U937 cells. In HL60 cells we observed a dose-dependent induction of both caspase 8 and caspase 9 activities that paralleled an increase in caspase 3/7 activity (Fig. 2b). Accordingly, cleavage of the caspase-3 target, PARP, is only observed in YM155 treated HL60 cells, but not U937 cells (Fig. 2c).
Fig. 2.
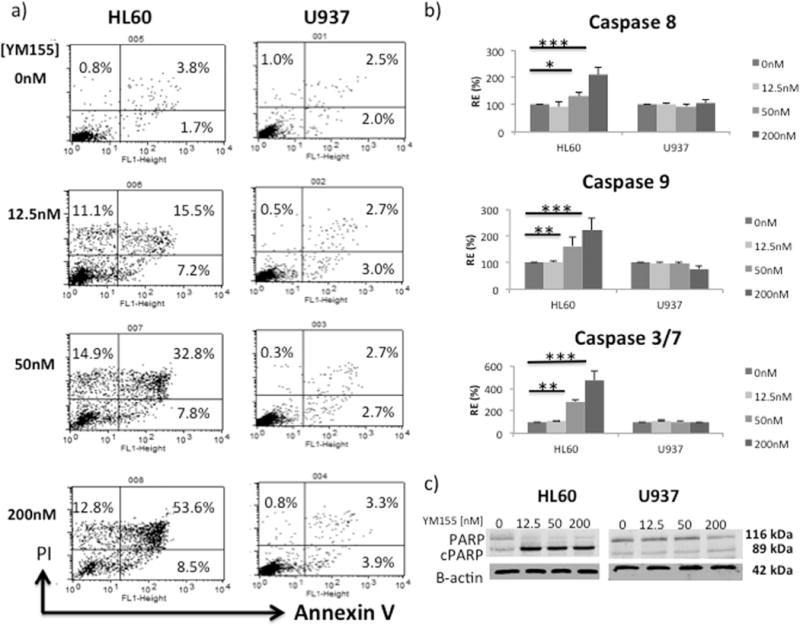
HL60 cells are more susceptible to YM155 induced apoptosis than U937. Both intrinsic and extrinsic apoptoses were evaluated using caspase activity assays. (a) Annexin V/PI apoptotic assay of cells treated with YM155 for 24 hours; (b) activity of caspase 8, 9 and 3/7 in cells exposed to different doses of YM155 for 8 hours (RE = Relative Expression). Data represent the mean ± SD of three independent experiments (*p < .05, ** p < .01, *** p < .001). (c) Western blot analysis of PARP cleavage after 24 hour YM155 treatment.
Cell cycle analysis
The cytotoxic effects of YM155 on HL60 cells were further confirmed by cell cycle analysis showing that treatment caused a dramatic increase in the sub G1/G0 population (Fig. 3a). We found that YM155 exposure up-regulated the expression of the p21 cell cycle inhibitor both at the transcript (Fig. 3b) and protein (Fig. 3c) levels in HL60 cells, yet no cell cycle changes were detected in U937 cells undergoing the same treatments.
Fig. 3.
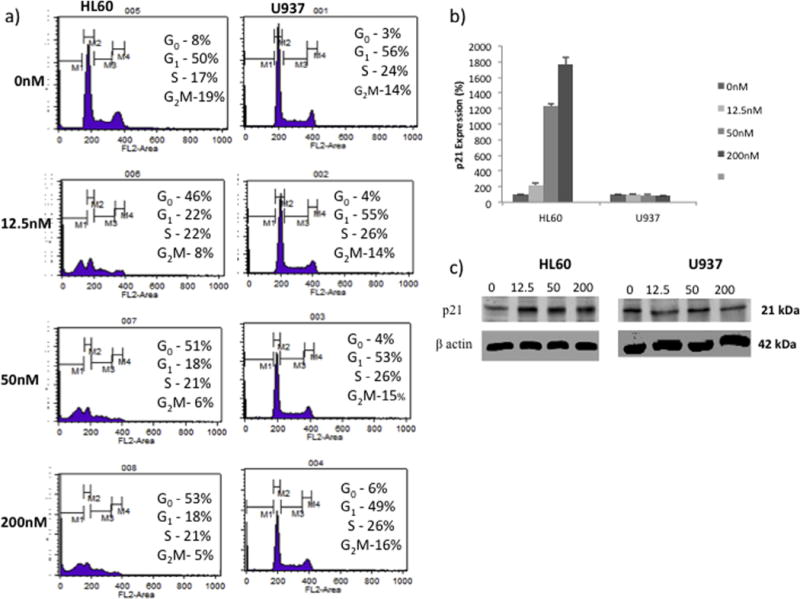
YM155 treatment up-regulates expression of the p21 cell cycle inhibitor and increases in the sub G0/G1 population in HL60 cells. (a) Cell cycle analysis of cells treated with YM155 for 24 hours. (b) Expression of p21 transcript in HL60 and U937 cells treated with YM155 for 8 hours. Data represent the mean ± SE from at least two independent experiments. (c) Western Blot analysis of p21 protein in cells treated with YM155 for 24 hours.
YM155 affects expression of anti- and pro-apoptotic Mcl-1 isoforms
To determine if YM155 treatment affected Mcl-1 expression levels, HL60 and U937 cells were exposed to concentrations of YM155 ≤200 nM (Fig. 4). YM155 inhibited anti-apoptotic Mcl-1L protein expression in both cell lines, and surprisingly also upregulated expression of pro-apoptotic Mcl-1s in HL60 cells (Fig. 4a). HL60, but not U937, cells exposed to YM155 demonstrated a dramatic upregulation in transcript expression of pro-apoptotic Mcl-1s (Fig. 4b). In contrast, the reduction in expression of Mcl-1L protein was not found to be regulated by YM155 at the transcriptional level (Fig. 4c).
Fig. 4.
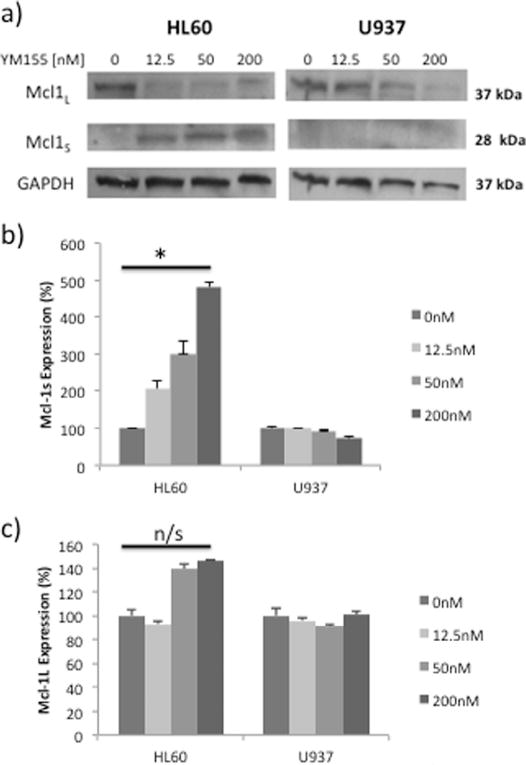
YM155 differentially modulates pro-apoptotic Mcl-1L and anti-apoptotic Mcl-1S in HL60 and U937 cells. (a) Western Blot analysis of Mcl-1L and Mcl-1S in cells treated with YM155 for 24 hours. (b) Expression of Mcl-1S and (c) Mcl-1L transcripts in cells treated with YM155 for 8 hours. Data represent the mean ± SE from at least two independent experiments (*p < .05, n/s = not significant).
Expression of Bcl-2 and XIAP in cells treated with YM155
The effect of YM155 exposure on the expression of Bcl-2 and XIAP proteins in HL60 and U937 cell lines was assessed (Fig. 5). YM155 inhibits both Bcl-2 protein (Fig. 5a) and transcript (Fig. 5b) expression levels in the HL60 cell line, while no changes are observed in U937 cells. Similarly, there is a strong dose dependent reduction of XIAP protein in YM155 treated HL60 cells which is less evident in U937 cells (Fig. 5c). Changes in XIAP protein expression were not found to be regulated by YM155 at the transcriptional level (Fig. 5d).
Fig. 5.
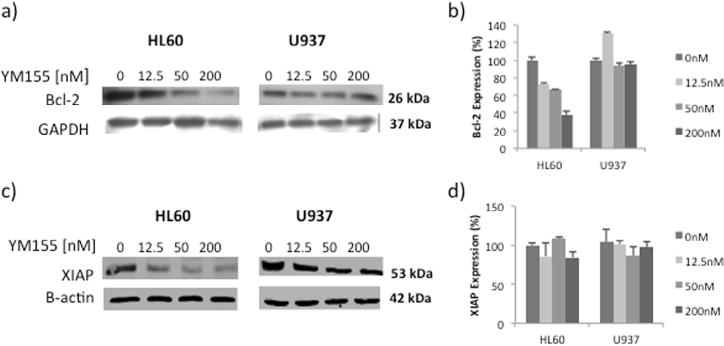
YM155 causes a dose-dependent reduction of Bcl-2 and XIAP in HL60 cells. (a) Western Blot analysis of Bcl-2 protein in cells treated for 24 hours. (b) Expression of Bcl-2 transcripts in cells treated with YM155 for 8 hours. Data represent the mean ± SE from at least two independent experiments. (c) Western Blot analysis of XIAP protein in cells treated for 24 hours. (d) Expression of XIAP transcripts in cells treated with YM155 for 8 hours. Data represent the mean ± SE from at least two independent experiments.
Potential upstream Akt signaling pathway
Since Akt activity has been shown to enhance stability of both Bcl-2 [27] and XIAP proteins [28], the effect of YM155 exposure on Akt expression and activity in HL60 and U937 cell lines was examined (Fig. 6). Total Akt protein was reduced to undetectable levels in HL60 cells exposed to doses of YM155 ≥50 nM, with no measureable change in U937 cells (Fig. 6b). More importantly, fully activated Akt (pAKT-Ser473) could not be detected in protein extracts from HL60 cells treated with YM155 while Akt activity was not affected in U937 cells (Fig. 6b). Intriguingly, Akt gene expression levels were found to be about 20× greater in YM155 resistant U937 cells compared with sensitive HL60 cells (Fig. 6b). Reduction of Akt protein and activity in HL60 cells was only partially explained by YM155 regulation of transcription (Fig. 6b).
Fig. 6.
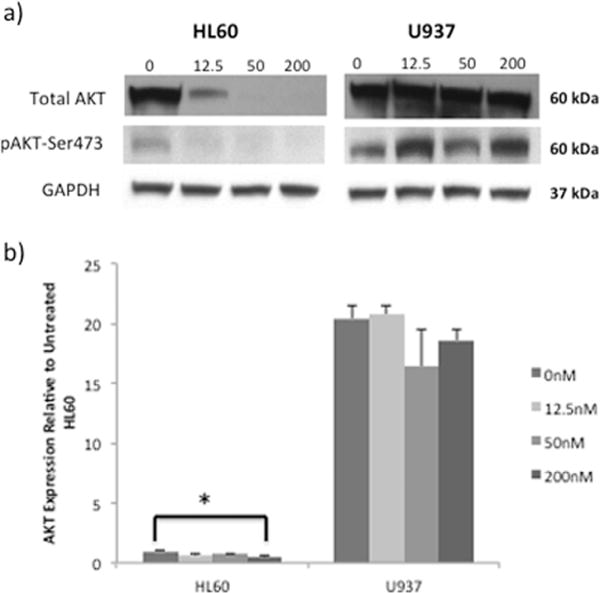
AKT expression and activity are highest in U937 cells and not inhibited by YM155 treatment. (a) Western Blot analysis of AKT activity (pAKT-Ser473) and total AKT protein in cells treated with YM155 for 24 hours. This representative Blot is selected from three independent experiments. (b) AKT transcript expression in HL60 and U937 cells treated with YM155 for 8 hours. Transcript levels are relative to those measured in HL60 control (untreated) and represent the mean ± SD of two independent experiments.
YM155 and MK-2206 have a synergistic growth inhibitory effect on U937
To determine if the Akt inhibitor MK-2206 could sensitize U937 cells to YM155 treatment, the combined effect of these compounds was analyzed (Fig. 7). The Combination Index equation is derived from multiple drug effect interactions that can be used to quantitatively assess drug synergy or antagonism [25]. At most concentrations tested, MK-2206 in combination with YM155 synergistically inhibited the growth proliferation of U937 cells as shown by a Combination Index (CI) value <1. Among multiple drug combination treatments, the most synergistic conditions (CI = 0.285) were found to be 1000 nM of MK-2206 combined with 50 nM of YM155 for 72 hours (Fig. 7a) which reduced cell viability by 52.3% (Fig. 7b). This outcome was associated with inhibition of Akt activity, as levels of pAkt-Ser473 were reduced by more than 80% in U937 cultures containing 1000 nM of the MK-2206 inhibitor (Fig. 7c and d).
Fig. 7.
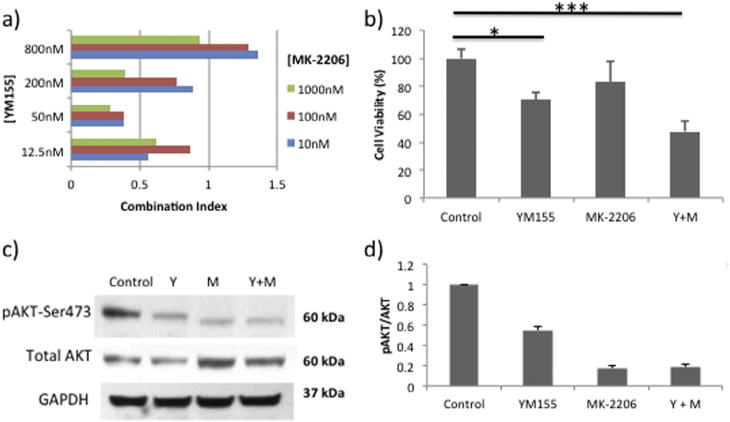
Resistant U937 cells are sensitized to YM155 using an AKT inhibitor (MK-2206). (a) Synergistic growth inhibitory effects of the combination YM155+MK-2206 treatments for 72 hrs. (b) MTT assay of cell viability in cells exposed to YM155 (50 nM), MK-2206 (1000 nM) or both compounds for 72 hours. Data represent the mean ± SD of three independent experiments (*p < .05,*** p < .001). (c) Western Blot analysis of AKT activity (pAKT-Ser473) and total AKT protein in U937 cells treated for 72 hours (Y = 50 nM YM155; M = 1000 nM MK-2206). (d) Graphical representation of the ratio of pAKT-Ser473/AKT for each treatment condition.
Discussion
While YM155 has been in clinical development, its molecular targets remain elusive. We performed analysis of cellular mechanisms associated with YM155 response in two AML cell lines known to be p53-deficient [29]. In other hematological malignancies, YM155 cytotoxicity functioned through a p53 dependent mechanism [22], yet in this study disparate treatment responses were found in AML cells lacking p53 activity, indicating that other targets and signaling pathways are responsible for YM155 treatment outcomes. HL60 cells were sensitive to YM155 exposure and found to be more than 100× more susceptible than U937 cells to the growth inhibitory effects of this compound.
At concentrations ≤200 nM, YM155 was found to induce apoptosis in HL60 in a dose-dependent manner, but not affect U937. A dose-dependent increase in both caspase-8 and caspase-9 activity was measured in treated HL60 cells indicating that both extrinsic and intrinsic pathways are involved in YM155 cytotoxicity. Survivin is able to inhibit both of these apoptotic pathways in vitro [30], so YM155 inhibition of survivin may partially explain this effect. However, many other YM155 targets can also influence treatment outcomes. The molecular analyses performed in this study have helped us to characterize several other YM155 targets and a signaling pathway involved in YM155 response.
Mcl-1 has been described as the critical target of YM155 in other hematological malignancies [20]. In several cancer cell lines, Mcl-1 down-regulation by YM155 was observed to be regulated at a transcriptional level, and to be independent of survivin inhibition or caspase activation [19]. In this study, we found a YM155 dose-dependent down-regulation of the anti-apoptotic Mcl-1L isoform in both cell lines at the protein, but not transcript, level. Yet surprisingly, we found transcriptional regulation of Mcl-1 in the form of a dramatic and significant up-regulation of pro-apoptotic Mcl-1s in treated HL60 cells. This Mcl-1s splice variant is known to resemble the pro-apoptotic BH3 family proteins and promote cell death [31]. In fact, a targeted approach shifting the Mcl-1 splicing pattern from production of Mcl-1L to Mcl-1s is sufficient to induce apoptosis and has therefore been suggested as a therapeutic strategy in the treatment of some cancers [32]. The results of this study indicate that YM155 has the ability to modulate the Mcl-1 splicing process in HL60 cells, and preference for the pro-apoptotic Mcl-1S variant may be a mechanism important for YM155 sensitivity. It is possible that the non-significant up-regulation of anti-apoptotic Mcl-1L transcript in treated HL60 cells is associated with YM155-induced expression of Mcl-1s which may overwhelm the elements necessary for a strictly selective splicing process. Nonetheless, YM155 exposure was capable of reducing anti-apoptotic Mcl-1L protein levels in both HL60 and U937, suggesting that this compound activates a cellular process resulting in preferential elimination of this protein isoform while Mcl-1s levels are not affected. The fact that U937 cells were found to be resistant to YM155 treatment indicates that reduction of Mcl-1L, in the absence of Mcl-1s up-regulation, may be insufficient for induction of apoptosis. Another possibility is that modulation of additional apoptotic molecules is necessary to achieve this cytotoxic outcome, since apoptosis is the cumulative result of a shift in the balance of many pro- and anti-apoptotic molecules [33].
As we observed in this study, YM155 treatment ≤200 nM was able to reduce expression of the anti-apoptotic proteins XIAP and Bcl-2 in HL60 cells which would help establish a cellular environment favorable to apoptosis. In contrast, we found very little change in the levels of both of these anti-apoptotic proteins in treated U937 cells. XIAP expression can inhibit both intrinsic and extrinsic apoptotic pathways by inhibiting effector caspases and preventing SmaC release from the mitochondria [34]. Bcl-2 expression also inhibits apoptosis by preventing permeabilization of the mitochondrial outer membrane [35]. Our findings suggest that YM155-resistant U937 cells have an intracellular environment favorable to the stability of these anti-apoptotic proteins.
It was previously shown that Akt activity can enhance the stability of XIAP by phosphorylating one of its residues (Ser87), which then protects it from ubiquitination and degradation caused by cisplatin treatment [28]. Similarly, Akt activity can protect Bcl-2 from ubiquitination and degradation in response to cytotoxic stimuli [27]. When we examined the transcript expression levels of Akt in HL60 and U937 cell lines, we found them to be 20× greater in U937, suggesting a greater potential for Akt signaling in these cells, although total Akt protein did not differ noticeably among untreated cells. Rather, it was YM155 exposure that completely abolished expression of the total Akt protein in HL60 cells, which remained unaffected in YM155 treated U937 cells. More importantly, functionally active and phosphorylated Akt (pAkt-Ser473) expression was also unchanged in YM155 treated U937 cells, yet disappeared in HL60. Results from a previous study suggest that autocrine insulin-like growth factor production is necessary for activation of Akt in HL60 cells [36], which may have been disrupted in treated cultures. Importantly, differences in Akt activity are a key factor that distinguishes HL60 and U937 cell lines and appear to be directly associated with the ability to resist YM155. In general, Akt activity is known to promote cell proliferation, enhance genomic stability, help cells resist cytotoxic stimuli, and often has a critical role in cancer cell survival [37]. Although Akt has an extensive cell signaling network and dozens of downstream cell survival targets, XIAP and Bcl-2 may be instrumental to YM155 resistance in U937 cells.
It is possible that U937 constitutively expresses Akt due to an alteration in the PTEN regulatory network. PTEN, the second most commonly mutated gene in human cancer, controls Akt activation by negatively regulating PIP3 which recruits and correctly positions Akt in the membrane so that it can be phosphorylated [38]. U937 has a frameshift mutation in the PTEN gene that results in an early termination codon and likely a complete loss of function [39]. Loss of PTEN function in tumor cells leads to increased Akt signaling which frequently results in resistance to apoptosis [37]. In at least one other study, lack of PTEN expression and Akt phosphorylation in U937 was a critical factor in resistance to the cytotoxic compound WEB-2170, which induced apoptosis in other AML cell lines. Although we did not examine PTEN expression levels in this study, a PTEN deficiency could explain the higher levels of Akt activity and resistance to YM155 induced apoptosis in U937 cells.
The Akt inhibitor MK-2206 disrupts Akt signaling mechanisms by suppressing phosphorylation of Akt [23]. In this study, MK-2206 and YM155 combination treatments had synergistic growth inhibitory effects on U937 cells, which were associated with a reduction in levels of phosphorylated Akt. This provides further evidence that Akt signaling has an important role in YM155 resistance in these cells. Collectively, our study suggests that Akt signaling mechanisms in U937 help promote stability of anti-apoptotic proteins and cell proliferation signals important for YM155 resistance (Fig. 8). Although this is the first study to investigate the combination of YM155 with MK-2206, both of these compounds are in clinical trials for the treatment of hematological malignancies. MK-2206 is currently in clinical development for AML [43], while YM155 therapies are being evaluated for the treatment of T-cell leukemia [44] and B-cell lymphoma [45]. Future therapies in AML cells with YM155 resistant phenotypes may benefit from combination treatments that include Akt inhibitors.
Fig. 8.
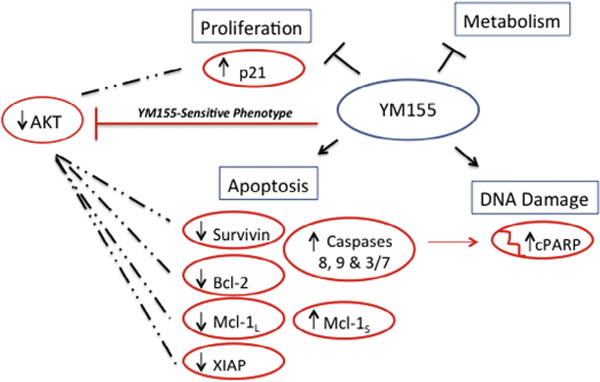
Schematic diagram of 4 major cellular pathways targeted by YM155 treatment in tumor cells [10]. Red molecules were shown to be differentially affected by YM155 exposure in this study, and represent the changes observed in YM155-sensitive HL60 cells (↑ = up-regulation; ↓ = down-regulation). Dashed lines represent a hypothetical mechanism by which constitutive AKT signaling in U937 cells may help protect specific YM155 targets [27,28,40–42] resulting in a treatment resistant phenotype. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Supplementary Material
Acknowledgments
The author’s would like to thank the Center for Health Disparities & Molecular Medicine for resource support of this project and the Graduate Students that it involved. Research reported in this publication was supported by the National Institute of Health Disparities and Minority Health of the National Institutes of Health (NRW & CSC) under award numbers p20MD0016321, p20MD006988 and 2R25 GM060507 and also from the Division of Medical Oncology/Hematology research fund (CSC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Appendix: Supplementary material
Supplementary data to this article can be found online at doi:10.1016/j.canlet.2015.05.034.
Footnotes
Conflict of interest
All authors declare that they have no conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Yanada M, Naoe T. Acute myeloid leukemia in older adults. Int J Hematol. 2012;96:186–193. doi: 10.1007/s12185-012-1137-3. [DOI] [PubMed] [Google Scholar]
- 3.Fernandez HF. New trends in the standard of care for initial therapy of acute myeloid leukemia. Hematology Am Soc Hematol Educ Program. 2010;2010:56–61. doi: 10.1182/asheducation-2010.1.56. [DOI] [PubMed] [Google Scholar]
- 4.Forman SJ, Rowe JM. The myth of the second remission of acute leukemia in the adult. Blood. 2013;121:1077–1082. doi: 10.1182/blood-2012-08-234492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Necochea-Campion R, Chen CS, Mirshahidi S, Howard FD, Wall NR. Clinicopathologic relevance of survivin splice variant expression in cancer. Cancer Lett. 2013;339:167–174. doi: 10.1016/j.canlet.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, et al. Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer Sci. 2011;102:614–621. doi: 10.1111/j.1349-7006.2010.01834.x. [DOI] [PubMed] [Google Scholar]
- 7.Cheson BD, Bartlett NL, Vose JM, Lopez-Hernandez A, Seiz AL, Keating AT, et al. A phase II study of the survivin suppressant YM155 in patients with refractory diffuse large B-cell lymphoma. Cancer. 2012;118:3128–3134. doi: 10.1002/cncr.26510. [DOI] [PubMed] [Google Scholar]
- 8.Giaccone G, Zatloukal P, Roubec J, Floor K, Musil J, Kuta M, et al. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J Clin Oncol. 2009;27:4481–4486. doi: 10.1200/JCO.2008.21.1862. [DOI] [PubMed] [Google Scholar]
- 9.Kelly RJ, Thomas A, Rajan A, Chun G, Lopez-Chavez A, Szabo E, et al. A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann Oncol. 2013;24:2601–2606. doi: 10.1093/annonc/mdt249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rauch A, Hennig D, Schafer C, Wirth M, Marx C, Heinzel T, et al. Survivin and YM155: how faithful is the liaison? Biochim Biophys Acta. 1845;2014:202–220. doi: 10.1016/j.bbcan.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Smolewski P, Robak T. Inhibitors of apoptosis proteins (IAPs) as potential molecular targets for therapy of hematological malignancies. Curr Mol Med. 2011;11:633–649. doi: 10.2174/156652411797536723. [DOI] [PubMed] [Google Scholar]
- 12.Carter BZ, Qiu Y, Huang X, Diao L, Zhang N, Coombes KR, et al. Survivin is highly expressed in CD34(+)38(−) leukemic stem/progenitor cells and predicts poor clinical outcomes in AML. Blood. 2012;120:173–180. doi: 10.1182/blood-2012-02-409888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karami H, Baradaran B, Esfahani A, Estiar MA, Naghavi-Behzad M, Sakhinia M, et al. siRNA-mediated silencing of survivin inhibits proliferation and enhances etoposide chemosensitivity in acute myeloid leukemia cells. Asian Pac J Cancer Prev. 2013;14:7719–7724. doi: 10.7314/apjcp.2013.14.12.7719. [DOI] [PubMed] [Google Scholar]
- 14.Adida C, Recher C, Raffoux E, Daniel MT, Taksin AL, Rousselot P, et al. Expression and prognostic significance of survivin in de novo acute myeloid leukaemia. Br J Haematol. 2000;111:196–203. doi: 10.1046/j.1365-2141.2000.02328.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J, Bi C, Janakakumara JV, Liu SC, Chng WJ, Tay KG, et al. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood. 2009;113:4052–4062. doi: 10.1182/blood-2008-05-156422. [DOI] [PubMed] [Google Scholar]
- 16.Yamauchi T, Nakamura N, Hiramoto M, Yuri M, Yokota H, Naitou M, et al. Sepantronium bromide (YM155) induces disruption of the ILF3/p54(nrb) complex, which is required for survivin expression. Biochem Biophys Res Commun. 2012;425:711–716. doi: 10.1016/j.bbrc.2012.07.103. [DOI] [PubMed] [Google Scholar]
- 17.Cheng Q, Ling X, Haller A, Nakahara T, Yamanaka K, Kita A, et al. Suppression of survivin promoter activity by YM155 involves disruption of Sp1-DNA interaction in the survivin core promoter. Int J Biochem Mol Biol. 2012;3:179–197. [PMC free article] [PubMed] [Google Scholar]
- 18.Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, et al. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67:8014–8021. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- 19.Tang H, Shao H, Yu C, Hou J. Mcl-1 downregulation by YM155 contributes to its synergistic anti-tumor activities with ABT-263. Biochem Pharmacol. 2011;82:1066–1072. doi: 10.1016/j.bcp.2011.07.064. [DOI] [PubMed] [Google Scholar]
- 20.Wagner V, Hose D, Seckinger A, Weiz L, Meissner T, Reme T, et al. Preclinical efficacy of sepantronium bromide (YM155) in multiple myeloma is conferred by down regulation of Mcl-1. Oncotarget. 2014;5(21):10237–10250. doi: 10.18632/oncotarget.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Na YS, Yang SJ, Kim SM, Jung KA, Moon JH, Shin JS, et al. YM155 induces EGFR suppression in pancreatic cancer cells. PLoS ONE. 2012;7:e38625. doi: 10.1371/journal.pone.0038625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tyner JW, Jemal AM, Thayer M, Druker BJ, Chang BH. Targeting survivin and p53 in pediatric acute lymphoblastic leukemia. Leukemia. 2012;26:623–632. doi: 10.1038/leu.2011.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 24.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 25.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 26.Feng W, Yoshida A, Ueda T. YM155 induces caspase-8 dependent apoptosis through downregulation of survivin and Mcl-1 in human leukemia cells. Biochem Biophys Res Commun. 2013;435:52–57. doi: 10.1016/j.bbrc.2013.04.036. [DOI] [PubMed] [Google Scholar]
- 27.Nishioka T, Luo LY, Shen L, He H, Mariyannis A, Dai W, et al. Nicotine increases the resistance of lung cancer cells to cisplatin through enhancing Bcl-2 stability. Br J Cancer. 2014;110:1785–1792. doi: 10.1038/bjc.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dan HC, Sun M, Kaneko S, Feldman RI, Nicosia SV, Wang HG, et al. Akt phosphorylation and stabilization of X-linked inhibitor of apoptosis protein (XIAP) J Biol Chem. 2004;279:5405–5412. doi: 10.1074/jbc.M312044200. [DOI] [PubMed] [Google Scholar]
- 29.Sugimoto K, Toyoshima H, Sakai R, Miyagawa K, Hagiwara K, Ishikawa F, et al. Frequent mutations in the p53 gene in human myeloid leukemia cell lines. Blood. 1992;79:2378–2383. [PubMed] [Google Scholar]
- 30.Satoh T, Okamoto I, Miyazaki M, Morinaga R, Tsuya A, Hasegawa Y, et al. Phase I study of YM155, a novel survivin suppressant, in patients with advanced solid tumors. Clin Cancer Res. 2009;15:3872–3880. doi: 10.1158/1078-0432.CCR-08-1946. [DOI] [PubMed] [Google Scholar]
- 31.Bingle CD, Craig RW, Swales BM, Singleton V, Zhou P, Whyte MK. Exon skipping in Mcl-1 results in a bcl-2 homology domain 3 only gene product that promotes cell death. J Biol Chem. 2000;275:22136–22146. doi: 10.1074/jbc.M909572199. [DOI] [PubMed] [Google Scholar]
- 32.Shieh JJ, Liu KT, Huang SW, Chen YJ, Hsieh TY. Modification of alternative splicing of Mcl-1 pre-mRNA using antisense morpholino oligonucleotides induces apoptosis in basal cell carcinoma cells. J Invest Dermatol. 2009;129:2497–2506. doi: 10.1038/jid.2009.83. [DOI] [PubMed] [Google Scholar]
- 33.Fulda S. Shifting the balance of mitochondrial apoptosis: therapeutic perspectives. Front Oncol. 2012;2:121. doi: 10.3389/fonc.2012.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flanagan L, Sebastia J, Tuffy LP, Spring A, Lichawska A, Devocelle M, et al. XIAP impairs Smac release from the mitochondria during apoptosis. Cell Death Dis. 2010;1:e49. doi: 10.1038/cddis.2010.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ola MS, Nawaz M, Ahsan H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem. 2011;351:41–58. doi: 10.1007/s11010-010-0709-x. [DOI] [PubMed] [Google Scholar]
- 36.Neri LM, Borgatti P, Tazzari PL, Bortul R, Cappellini A, Tabellini G, et al. The phosphoinositide 3-kinase/AKT1 pathway involvement in drug and all-trans-retinoic acid resistance of leukemia cells. Mol Cancer Res. 2003;1:234–246. [PubMed] [Google Scholar]
- 37.Xu N, Lao Y, Zhang Y, Gillespie DA. Akt: a double-edged sword in cell proliferation and genome stability. J Oncol. 2012;2012:951724. doi: 10.1155/2012/951724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27:5443–5453. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- 39.Liu TC, Lin PM, Chang JG, Lee JP, Chen TP, Lin SF. Mutation analysis of PTEN/MMAC1 in acute myeloid leukemia. Am J Hematol. 2000;63:170–175. doi: 10.1002/(sici)1096-8652(200004)63:4<170::aid-ajh2>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Dowbenko D, Lasky LA. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J Biol Chem. 2002;277:11352–11361. doi: 10.1074/jbc.M109062200. [DOI] [PubMed] [Google Scholar]
- 41.Longo PG, Laurenti L, Gobessi S, Sica S, Leone G, Efremov DG. The Akt/Mcl-1 pathway plays a prominent role in mediating antiapoptotic signals downstream of the B-cell receptor in chronic lymphocytic leukemia B cells. Blood. 2008;111:846–855. doi: 10.1182/blood-2007-05-089037. [DOI] [PubMed] [Google Scholar]
- 42.Zhao P, Meng Q, Liu LZ, You YP, Liu N, Jiang BH. Regulation of survivin by PI3K/Akt/p70S6K1 pathway. Biochem Biophys Res Commun. 2010;395:219–224. doi: 10.1016/j.bbrc.2010.03.165. [DOI] [PubMed] [Google Scholar]
- 43.Konopleva MY, Walter RB, Faderl SH, Jabbour EJ, Zeng Z, Borthakur G, et al. Preclinical and early clinical evaluation of the oral AKT inhibitor, MK-2206, for the treatment of acute myelogenous leukemia. Clin Cancer Res. 2014;20:2226–2235. doi: 10.1158/1078-0432.CCR-13-1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J, Pise-Masison CA, Shih JH, Morris JC, Janik JE, Conlon KC, et al. Markedly additive antitumor activity with the combination of a selective survivin suppressant YM155 and alemtuzumab in adult T-cell leukemia. Blood. 2013;121:2029–2037. doi: 10.1182/blood-2012-05-427773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaneko N, Mitsuoka K, Amino N, Yamanaka K, Kita A, Mori M, et al. Combination of YM155, a survivin suppressant, with bendamustine and rituximab: a new combination therapy to treat relapsed/refractory diffuse large B-cell lymphoma. Clin Cancer Res. 2014;20:1814–1822. doi: 10.1158/1078-0432.CCR-13-2707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.