

Abstract
Significance: Traumatic brain injury (TBI) and spinal cord injury (SCI) are major causes of death and long-term disability worldwide. Despite important pathophysiological differences between these disorders, in many respects, mechanisms of injury are similar. During both TBI and SCI, some cells are directly mechanically injured, but more die as a result of injury-induced biochemical changes (secondary injury). Autophagy, a lysosome-dependent cellular degradation pathway with neuroprotective properties, has been implicated both clinically and experimentally in the delayed response to TBI and SCI. However, until recently, its mechanisms and function remained unknown, reflecting in part the difficulty of isolating autophagic processes from ongoing cell death and other cellular events. Recent Advances: Emerging data suggest that depending on the location and severity of traumatic injury, autophagy flux—defined as the progress of cargo through the autophagy system and leading to its degradation—may be either increased or decreased after central nervous system trauma. Critical Issues: While increased autophagy flux may be protective after mild injury, after more severe trauma inhibition of autophagy flux may contribute to neuronal cell death, indicating disruption of autophagy as a part of the secondary injury mechanism. Future Directions: Augmentation and/or restoration of autophagy flux may provide a potential therapeutic target for treatment of TBI and SCI. Development of those treatments will require thorough characterization of changes in autophagy flux, its mechanisms and function over time after injury. Antioxid. Redox Signal. 23, 565–577.
Introduction
Central nervous system (CNS) trauma, including traumatic brain injury (TBI) and spinal cord injury (SCI), is one of the most important causes of death and long-term disability among young adults worldwide (22, 69). Since the patients are often disabled during the most productive periods of their lives, the injuries have an enormous physical, emotional, and economic burden on both the individuals and the society. More than 2.5 million new cases of TBI are reported annually in the United States (1). They can lead to long-term cognitive, sensorimotor, and psychiatric changes, with the extent dependent on the severity of injury (69). TBI may also cause epilepsy and increase the risk of developing neurodegenerative diseases with age.
SCI has an annual incidence of 12,000–20,000 and a prevalence of nearly half a million in the United States (2). It can result in sensorimotor deficits, autonomic changes, and chronic pain (22). Depending on the level and severity of the injury, SCI may also affect respiratory, urinary, or gastrointestinal function. Long-term complications of SCI may additionally include cognitive impairments, depression, and anxiety (21, 33).
Over the years, many animal models have been developed to study the mechanisms and the functional consequences of TBI and SCI [recently reviewed in more detail in (14, 18, 46)]. Although no single model can recapitulate the complexities of human TBI, considered among the most heterogeneous of neurological disorders, they are essential for elucidating pathological mechanisms of injury and evaluating potential therapeutic interventions (14, 39). An example is the commonly used rodent controlled cortical impact model, where craniotomy is performed to remove a part of the skull, followed by a localized contusive impact delivered directly to the exposed dura (25). Additional models have been developed to allow studies of specific types or aspects of injury, such as concussion, blast injury, or remote degeneration, in distant brain regions functionally connected to the lesion area.
Similarly, many animal models have been developed that reflect different components of SCI. Clinically, approximately half of SCI cases result from contusion injuries (45). In rodent models, spinal contusion injuries commonly involve laminectomy to allow direct access to the spinal cord, followed by a localized contusive impact. Additional specialized models include spinal cord transection, chronic compression, and models of neuropathic pain, such as spinal root ligation (18).
Mechanisms of Traumatic Injury to the CNS
There are currently no well-established neuroprotective treatments for either TBI or SCI. This is at least in part due to the complex nature of the injury mechanisms. Trauma causes both direct mechanical tissue damage (primary injury) and biochemical changes that cause more delayed or progressive cell loss (secondary injury) (5, 69). The primary mechanical damage results in cellular strain and plasma membrane rupture or damage, leading, among others, to ionic imbalance, excitatory amino-acid release, and oxidative species formation in the area of injury (3). These events initiate complex secondary changes that collectively spread the injury to the intact neighboring cells. In addition, the initial physical damage to the blood–brain barrier or the blood–spinal cord barrier allows infiltration of macrophages and other immune cells (76). The resulting inflammatory changes involve both infiltrating macrophages and activated resident microglia and contribute to secondary cell damage and loss. Recent studies also demonstrate a potential second peak of inflammation till 2 months after injury as well as chronic neuroinflammatory changes (6, 71). Additional damage can occur at a distance over days and months after injury via Wallerian degeneration and/or demyelination, reflecting CNS connectivity (54, 65).
Thus, the secondary injury can occur over hours, days, and months after initial impact, further intensifying and spreading tissue damage and functional deficits (Fig. 1). Since primary injury is almost instantaneous and cell damage in the directly impacted area is often severe and irreversible, it cannot be treated. However, the secondary injury takes time and involves changes in specific biochemical, cellular, and molecular pathways, thus presenting a therapeutic opportunity.
FIG. 1.
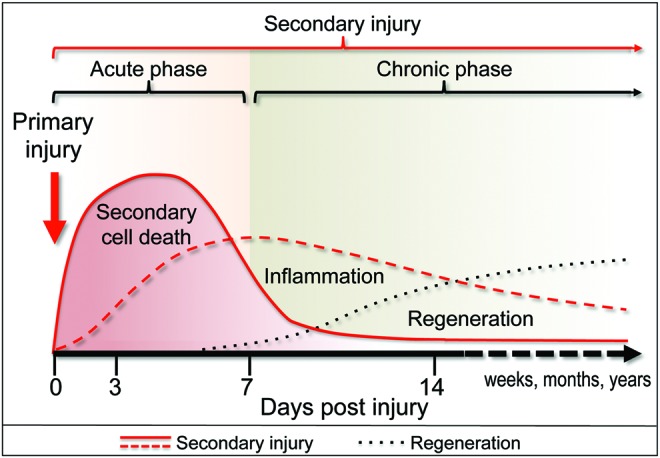
Sequence of events after CNS trauma in rodent models. The initial mechanical impact (primary injury—red arrow) disrupts the structure of the brain or spinal cord and initiates complex secondary changes that collectively spread the damage to the intact neighboring tissue (secondary injury—red lines). The secondary injury includes secondary neuronal cell death as well as long-term inflammatory changes, which contribute to further damage. Cell damage and death resulting from secondary injury are followed by a restorative phase (regeneration—black dotted line) during which the brain or the spinal cord remodels itself in an attempt to compensate for the tissue damage. CNS, central nervous system. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Cell damage and death resulting from secondary injury are followed and often overlap with a restorative phase during which the brain or the spinal cord remodels itself in an attempt to compensate for the tissue damage (54). These repair mechanisms include proliferation and differentiation of endogenous neural precursors as well as stimulation of neurite outgrowth and re-myelination. In addition, increased angiogenesis has been observed after CNS injury and is believed to promote neuroregeneration (29). Together, these processes counter neurodegeneration and contribute to the partial recovery of function commonly observed after both TBI and SCI. Enhancement of these endogenous restorative processes represents another therapeutic target for treatment of TBI and SCI.
The mechanisms of cell death after TBI and SCI include initial necrosis of irreversibly damaged cells in the impact area, followed by delayed secondary cell death in the neighboring penumbral regions (3, 54). Although most studies focus on neuronal cell death induced by CNS trauma, glial and endothelial cell changes are also important determinants of posttraumatic tissue damage. The mechanisms of cell death after CNS trauma are diverse, and experimental data suggest that multiple cell death pathways are involved (54, 58). Markers of classical apoptosis, including pro-apoptotic BCL-2 family proteins and activated caspases that are specific for both intrinsic and extrinsic pathways, are strongly upregulated after neurotrauma. In addition, release of caspase-independent pro-death molecules, such as the apoptosis-inducing factor (AIF)/AIFM1, has been noted after both TBI and SCI (58). The regulated necrosis pathway, necroptosis, may also contribute to cell death during secondary injury (67, 74). Simultaneous involvement of multiple cell death pathways is supported by studies using knockout mice and inhibitors of specific death mechanisms (54, 58). These manipulations are often able to suppress cell death after injury; however, these improvements are only partial and sometimes temporary. This has lead to more recent proposals for therapeutic use of multiple drug cocktail or multifunctional drugs that target upstream biochemical changes and cellular events involved in the initiation of multiple pro-death pathways (58).
Among posttraumatic secondary biochemical responses, signs of macroautophagy (hereafter called autophagy) have also been observed. However, until recently, its mechanisms and the consequences have remained obscure. In this review, we detail recent advances in the study of autophagy after CNS trauma, which have begun to clarify how autophagy levels and flux are affected by injury and how their manipulation may represent a potential novel neuroprotective target.
Autophagy and Autophagy Flux
Autophagy is a lysosome-dependent essential cellular catabolic pathway that serves to degrade cytoplasmic proteins, protein aggregates, and organelles (42). During autophagy, double-membrane vesicles (autophagosomes) sequester cytoplasmic components, including damaged organelles and toxic protein aggregates. Autophagosomes fuse with lysosomes to allow degradation of cargo by lysosomal proteases. The progress of cargo through the autophagy system and leading to its delivery and degradation in the lysosomes is termed autophagy flux (Fig. 2).
FIG. 2.

Autophagy flux under normal and pathological conditions. (A) During autophagy, double-membrane vesicles (autophagosomes) sequester cytoplasmic components, including damaged organelles and toxic protein aggregates, and then fuse with lysosomes to allow degradation of cargo by lysosomal proteases. This progress of cargo through the autophagy system is termed autophagy flux and generally serves a cytoprotective function. (B) Under pathological conditions, autophagy flux may be blocked, for example due to lysosomal defects. This can lead to accumulation of dysfunctional autophagosomes and contribute to cell damage and death. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Although under certain conditions pathologically increased autophagy has been implicated in cell death (44), under most circumstances autophagy is considered cytoprotective. Basal levels of autophagy are important for homeostasis in all cells but are especially crucial in terminally differentiated cells such as neurons and oligodendrocytes. Its importance is underscored by the fact that mice with neural tissue specific knockout of the essential autophagy genes Atg5 or Atg7 develop severe neurodegeneration, leading to abnormal motor function and reflexes. The proposed mechanisms contributing to cell injury and death when autophagy flux is inhibited include accumulation of toxic protein aggregates and defective organelles (11, 44). Accumulated dysfunctional autophagosomes themselves may also serve a pathologic function, for example, by serving as sites for amyloid β generation or platforms for assembly of pro-death signaling complexes (4). Autophagy is also upregulated, and often plays a protective function, in response to cell injury (12, 72). For example, autophagy is activated in response to and can limit effects of homeostasis perturbation in the endoplasmic reticulum (ER) stress. Conversely, defects in autophagy flux can exacerbate ER stress and potentiate ER stress-induced apoptosis (12).
Impaired autophagy flux has been implicated in neurodegenerative disorders such as Parkinson's, Alzheimer's, and Huntington's disease and in lysosomal storage disorders (42, 44). In lysosomal storage diseases, defects in autophagy are secondary to deficiencies in specific lysosomal hydrolases and consequent impairment of the lysosomal function (56). Neurodegenerative diseases are associated with autophagy defects contributing to accumulation of ubiquitin-positive protein aggregates and to neuronal cell dysfunction and death (44). Conversely, increasing efficiency of autophagic flux can improve outcomes in animal models of neurodegenerative diseases (11) and has been proposed as a potential therapeutic approach.
Autophagy and Autophagy Flux After CNS Trauma
Increased markers of autophagy have been observed after both TBI and SCI, but their mechanisms and functions remain controversial. It is clear that accumulation of autophagosomes is an early event during secondary injury, which is initiated within hours after initial impact (28, 31, 37). This has been observed in both rat and mouse models of TBI and SCI, as well as in human TBI autopsy samples (51). Many reports, including those referencing human samples (51), also noted that markers of autophagy remained elevated for weeks to months after injury. This long-term upregulation may reflect potential function for autophagy not only in the acute phase of the secondary injury but also more chronically in association with neurodegenerative and/or restorative processes (Fig. 1). Further long-term studies in animal models will be necessary to elucidate the role of such persistent changes.
Less certain, at least in TBI, is whether accumulation of autophagosomes after injury is due to increased autophagosome biosynthesis and elevation of autophagy flux or to decreased autophagosome degradation and inhibition of flux. Many of the earliest reports described accumulation of autophagosomes based on electron microscope studies and/or accumulation of the autophagosome marker protein LC3-II (MAP1LC3B, Fig. 3A); however, they did not address the issue of flux (19, 20, 23, 37). More recently, autophagy flux has been assessed in several models of TBI based on the levels of the autophagic substrate protein sequestosome 1/p62 (SQSTM1) (30). SQSTM1 is an adaptor protein that directs ubiquitinated cargo to autophagosomes for degradation. As SQSTM1 is degraded along with its cargo, when autophagy flux is increased, its protein levels decrease; conversely, when autophagy flux is inhibited, SQSTM1 levels increase (Fig. 3B, C). It is important to note that Sqstm1/SQSTM1 can also be regulated at the transcriptional level. Therefore, it is necessary to also assess its mRNA levels to distinguish altered rate of degradation from synthesis.
FIG. 3.

Common methods used to assess autophagy and autophagy flux. (A) When autophagy is induced, cytosolic LC3-I protein is covalently conjugated to PE to form LC3-II, which translocates to the autophagosomal membrane. Accumulation of LC3-II can be measured as a marker of autophagosome formation. (B, C) Comparison of LC3-II and SQSTM1 levels under conditions when autophagy flux is increased or inhibited. (B) When flux is induced, numbers of autophagosomes and levels of LC3-II increase but levels of autophagy substrates such as SQSTM1 decrease. When autophagy flux is blocked, both numbers of autophagosomes (LC3-II) and autophagy substrates (SQSTM1) increase. (C) Comparison of levels of LC3 (GFP-LC3 fluorescence—green) and SQSTM1 (immunohistochemistry—red) in the cortex of GFP-Lc3 transgenic autophagy reporter mice under conditions when autophagy flux is induced (in vivo Rapamycin treatment for 48 h) versus when autophagy flux is inhibited (24 h after controlled cortical impact injury). Data adapted from Sarkar et al. (53) with permission of authors. PE, phosphatidylethanolamine.
Decreased levels of SQSTM1 have been reported to accompany LC3-II accumulation in a mouse models of contusive TBI (weight-drop model) (41). These and other reports also noted an increase in BECN1 (Beclin 1), a regulatory subunit of the type III PI3 kinase, which is necessary for autophagosome formation. Increased autophagy flux was also described in a mouse model of acute focal cerebellar lesion, where it was confirmed by in vivo LC3-II flux assay (64). During the flux assay, usually carried out in vitro, LC3-II protein levels are compared in the presence or absence of a lysosomal inhibitor such as chloroquine (CQ, a lysosomotropic compound that neutralizes lysosomal pH), Bafilomycin A (V-ATPase inhibitor), or E-64d/pepstatin A (lysosomal hydrolase inhibitors) (30). When flux is high, CQ (or any other lysosomal inhibitor) leads to additional accumulation of LC3-II. When flux is blocked, CQ cannot further increase LC3-II levels. Molinari and colleagues (64) performed this assay in vivo by treating GFP-Lc3 transgenic autophagy reporter mice with CQ via an intracellebroventricular injection. Accumulation of endogenous LC3-II and numbers of neuronal GFP-LC3 puncta, corresponding to autophagosomes, were elevated after cerebellar lesion and further increased after CQ treatment, indicating that autophagy flux was increased.
In contrast, inhibition of autophagy flux was recently reported after contusion TBI (using pneumatic impactor) in GFP-Lc3 mice (53). Accumulation of autophagosomes near the injury site was accompanied by increased levels of SQSTM1 (Fig. 3C), demonstrated by both Western blot and immunohistochemistry. This was further confirmed by ex vivo LC3-II flux experiment where organotypic brain slices from control and injured animals were incubated in the presence or absence of CQ. CQ increased LC3-II levels in control, but not in injured brains, confirming inhibition of autophagy flux. In this model, inhibition of flux was temporary as in vivo levels of SQSTM1 returned to baseline 1 week after injury.
The reasons for the differences in autophagy flux across these studies are not clear. It is possible that different injury models or injury severity may differentially affect autophagy activation, as well as the ability of autophagy flux to proceed to completion. For example, a relatively mild injury could lead to upstream activation of autophagy flux as a protective mechanism (Fig. 4). This may be the case especially in the cerebellar lesion model (64), which examined autophagy levels not in the direct lesion area but rather in distant sites functionally connected to the cerebellum. In this model, activation of autophagy flux was also shown to be neuroprotective, as levels of cell death were increased and functional outcomes were worsened in injured autophagy-deficient Becn1+/− mice as compared with wild-type controls. On the other hand, more severe injury could alter the ability of cells near the injury area to correctly deliver and degrade autophagosomal cargo, leading to inhibition of flux. This perspective is supported by a recent investigation of human TBI brain autopsy samples (51), where elevated levels of SQSTM1 were found in the majority of samples, with accumulation being the most pronounced in those with shortest survival times, implying more severe TBI.
FIG. 4.

Changes in autophagy flux after mild versus severe CNS trauma. In some models, TBI can lead to an increase in autophagy flux (green line), which likely serves a protective function. In other models, TBI or SCI can inhibit autophagy flux (red line), contributing to secondary injury. At later time points (dashed lines), autophagy flux may be increased in all models as compared with uninjured animals, suggesting a potential beneficial function. SCI, spinal cord injury; TBI, traumatic brain injury. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Increased markers of autophagy have also been observed in SCI models (27, 28, 55, 60). Most reports describe concomitant accumulation of both LC3-II and SQSTM1 after injury, suggesting more uniform inhibition of autophagy flux. This includes diverse injury models such as acute contusion SCI in rat (38) and mouse (Lipinski and Wu, unpublished), chronic spinal cord compression in mouse and rat (17, 60), and a mouse spinal nerve ligation model of neuropathic pain (8). The fact that autophagy flux was inhibited even in the latter mild injury model may suggest that spinal cord neurons may be more sensitive to autophagy flux perturbations on injury than cortical or hippocampal neurons examined in most TBI models. However, studies directly comparing sensitivity of different subtypes of spinal and brain neurons will be needed to test this hypothesis.
Cell Type Specificity of Autophagy After CNS Trauma
Neurons are the cell type most commonly reported to accumulate autophagosomes after both TBI and SCI. An increase in autophagy in neurons occurs early, initiated within 24 h after injury. In brain injury, this includes both cortical and hippocampal neurons in the hemisphere ipsilateral to the injury (19, 37, 53). After SCI, most reports either do not specify the type or location of investigated neurons or focus exclusively on the ventral horn motor neurons. However, a recent paper (38) noted more pronounced accumulation of both LC3 and SQSTM1 in the ventral horn motor neurons as compared with the dorsal horn sensory neurons, despite greater proximity of the latter to the impact area. This suggests that motor neurons may be particularly vulnerable to disruption of autophagy flux. The dorsal sensory neurons can, however, also be affected. For example, spinal nerve ligation in a model of neuropathic pain led to disruption of autophagy flux in the dorsal horn (8, 77). In addition to neuronal cell bodies, autophagosome accumulation has been reported to occur in axons after both TBI and SCI (19, 49).
Increased autophagy has been also reported for other cell types in the CNS. Glial fibrillary acid protein positive astrocytes have been reported to accumulate autophagosomes (20, 23, 28, 60), and increased levels of autophagy have been documented in oligodendrocytes after both TBI and SCI (28, 38, 53, 60). Recent reports also noted increased LC3 levels in microglia after TBI and SCI (38, 53, 60). Only the most activated, CD68-expressing microglia with amoeboid morphology were affected in TBI (53), suggesting potential involvement of autophagy in regulation of microglial activation or function after injury. In all glial sub-types, the extent of autophagosome accumulation generally correlated with the proximity to the injury site. In addition, accumulation of autophagosomes in glial cells was observed later than neurons (starting around day 3 after injury in most reports), suggesting that these cells may have differential sensitivity to perturbation of autophagy or that different mechanisms may be involved than in neurons.
Mechanisms of Autophagy After CNS Trauma
Depending on whether induction or inhibition of autophagy flux was observed, authors have proposed different mechanisms of how autophagy may be regulated after CNS injury. However, in general, evidence for the involvement of specific signaling pathways is limited and mostly circumstantial. This is, in part, due to the fact that there are no accurate in vitro model systems of CNS trauma (43). Therefore, much more work, including in vivo manipulation of specific pro- and anti-autophagy molecules and pathways, will be necessary before its mechanisms and consequences can be more fully understood.
The most commonly invoked mechanisms for upregulation of autophagy flux after TBI include increased BECN1 protein levels and decreased BECN1/BCL2 complexes (20, 28, 64), which should increase the type III PI3 kinase activity and autophagosome biogenesis. The requirement for the activity of this kinase in injury models leading to increase in autophagy flux is supported by the finding that accumulation of autophagosomes and degradation of SQSTM1 are attenuated after focal cerebellar lesion in Becn1+/− mice (64).
It remains unknown how levels of BECN1 may be upregulated after CNS trauma. One potential mechanism was proposed by Keane and colleagues (9), who noted that in the brains of sham rats some BECN1 protein was localized to membrane rafts, where it formed complexes with the N-methyl-d-aspartate receptor 2B (NR2B). TBI caused dissociation of BECN1/NR2B complexes and release of BECN1, which may have contributed to the induction of autophagy. Another potential mechanism could involve the gap junction protein connexin 43/CX43 (GJA1), which was recently shown to directly interact with the BECN1 complex and regulate autophagosome biogenesis (7). Levels and activity of CX43 are affected after TBI and may contribute to regulation of autophagy (59). Other proposed pathways include mTOR, GSK3β, JNK, and BNIP3 (9, 35, 75); however, their involvement will need to be confirmed by further studies.
Decreased levels and activity of the lysosomal protein cathepsin D (CTSD) and in numbers of CTSD-positive lysosomes was recently reported to accompany inhibition of autophagy flux in both TBI and SCI (38, 53). This suggests the possibility that SCI trauma may lead to lysosomal damage and dysfunction, which could cause defects in autophagy flux. Both studies also noted a decrease in proportion of LC3+/CTSD+ autolysosomes as compared with total autophagosomes, consistent with deficiencies in autophagosome processing (Fig. 5). It remains to be determined whether this may be due to a specific defect in autophagosome-lysosome fusion or to a general decline in lysosomal function, leading to accumulation of unprocessed autophagosomes.
FIG. 5.

Potential mechanisms and consequences of inhibition of autophagy flux after CNS trauma. SCI or severe TBI lead to accumulation of ROS, increased levels of pro-apoptotic BCL2 family proteins, and a decrease in HSP70 chaperone. This can cause lysosomal damage and consequent inhibition of autophagy flux, leading to neuronal and oligodendrocyte cell death and increasing functional deficits after injury. ROS, reactive oxygen species. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
It is also not clear how lysosomes may be damaged after CNS trauma. The simplest explanation would be mechanical trauma leading to loss of lysosomal membrane integrity. However, no gross leakage of soluble lysosomal enzymes into the cytosol was observed, at least after TBI (53). This does not exclude the possibility of more subtle lysosomal membrane damage. Such limited lysosomal membrane permeabilization (LMP) has been linked to neuronal damage in ischemia and neurodegenerative diseases (10, 73), but its mechanisms are only now being elucidated. Potential mediators include channel formation by pro-apoptotic Bcl-2 family proteins such as BAX (10), and calpain-mediated cleavage and inhibition of the chaperone heat shock protein HSP70, leading to lysosomal membrane destabilization (73). Both BAX and HSP70 have been shown to be involved after CNS trauma (50, 79), but their potential contribution to LMP in this context has yet to be investigated. Importantly, lysosomal defects can lead to cytotoxicity in both an autophagy-dependent and -independent manner, further underscoring the need to understand their mechanisms and effects after CNS trauma.
Another factor that could contribute to altered autophagy flux after CNS trauma is reactive oxygen species (ROS). ROS have been shown to both positively and negatively regulate autophagy, depending on the levels and context (24, 36). The positive effects of ROS on autophagy flux can be exerted directly via redox-sensitive mediators such as ATG4, or indirectly via upstream regulatory pathways such as the adenosine 5′-monophosphate-activated protein kinase (AMPK) (80). On the other hand, excessive ROS as well as reactive nitrogen species (RNS) can inhibit autophagy, for example, by means of S-nitrosylation of the components of JNK and mTOR pathways (52). HSP70 can also be carbonylated under oxidative conditions, making it a better substrate for calpain-mediated cleavage. This has been shown to contribute to LMP during brain ischemia (73) and could play a similar function after TBI and SCI. Alternatively, lysosomal membranes could be directly subject to oxidative damage. Therefore, depending on the level and mechanism of injury, ROS and RNS could contribute to both activation and inhibition of autophagy flux.
Function of Autophagy in Neurons After CNS Trauma
The function of autophagy in the secondary neurodegeneration after TBI and SCI has long been a source of controversy, with both beneficial (23, 26, 55, 61) and detrimental roles proposed (31, 41). This may reflect the fact that either induction or inhibition of autophagy flux may occur after CNS injury. The function of autophagy can change drastically depending on flux, with unobstructed flux usually contributing to cytoprotection and flux inhibition promoting cell death. In addition, since autophagy is often induced as a response to cell stress (12, 72), care must be taken to distinguish between association and causation of cell death.
The early TBI and SCI studies noted co-localization of autophagy markers in brain and spinal cord regions showing high levels of neuronal cell death, leading to conclusions that autophagy may be contributing to neurodegeneration (20, 28). This view was supported by reports demonstrating decreased levels of autophagy markers after neuroprotective drug treatments (19, 31). However, most of the treatments used were not specific inhibitors of autophagy and without knowing whether autophagy flux was increased or decreased after injury, it is difficult to interpret these results. For example, signs of autophagy could be decreased because neuroprotective treatment increased autophagy flux and thus clearance of accumulated autophagosomes. Alternatively, the treatment could act independently of autophagy by making cells healthier. Thus, if autophagy flux was increased as a protective response by injured cells, healthier cells may no longer upregulate it.
More recent data support the idea that in models where autophagy flux is increased after injury, it likely serves a protective function. The best evidence comes from the focal cerebellar lesion model, where induction of autophagy flux was decreased and functional outcomes were worsened in autophagy-deficient Becn1+/− mice as compared with wild-type controls (64). This was associated with decreased neuronal survival, indicating that increased autophagy flux was neuroprotective. This must, however, be confirmed in other injury models, including SCI and contusive TBI.
The mechanisms by which increased autophagy flux contributes to neuroprotection after injury have yet to be investigated. CNS trauma can lead to generation of damaged cellular components such as mitochondria, lysosomes, and peroxisomes, which are both vulnerable to and a source of oxidative stress. Increased autophagy flux could help eliminate these injured organelles to protect the cells from further damage. Consistent with this possibility, accumulation of autophagosomes, including damaged mitochondria and increased BNIP3, both of which suggest induction of mitophagy, have been noted after SCI (75). However, its function and mechanisms remain to be further investigated. Autophagy may also serve to remove toxic ubiquitin-positive protein aggregates generated after injury (40), and it provides necessary building blocks as well as energy required for the initiation of restorative processes after trauma.
In models where CNS injury leads to inhibition of autophagy flux, it likely contributes to neuronal cell death. This is supported by recent reports indicating specific accumulation of SQSTM1 in dying neuronal cells after both TBI and SCI (38, 53). The cell death markers included those specific for both apoptotic and non-apoptotic mechanisms, suggesting that inhibition of autophagy flux after CNS injury may contribute to the induction and/or amplification of multiple cell death pathways. However, a causative role for inhibited autophagy flux has yet to be directly confirmed in those models.
In both TBI and SCI, a specific connection was noted between inhibition of autophagy flux and ER stress in neurons (38, 53). ER stress has been implicated as a part of the secondary injury after CNS trauma (32, 47), but its mechanisms remain unknown. As autophagy is commonly increased and can serve as a protective mechanism in response to ER stress (12), its inhibition after injury could exacerbate ER stress, contributing to the induction of apoptosis.
Inhibition of autophagy flux could also contribute to non-apoptotic neuronal cell death after CNS trauma. For example, TBI neurons with signs of inhibited autophagy also accumulated AIF (AIFM1) (53). During caspase-independent cell death, AIF translocates from the mitochondrial inner membrane to the cytosol. As damaged mitochondria can be targeted by autophagy/mitophagy, this suggests the possibility that impaired autophagic clearance could contribute to accumulation of damaged mitochondria after TBI. Another cell death pathway that could be affected by autophagy is necroptosis, an RIPK1/RIPK3-dependent regulated necrosis (62). Inhibition of necroptosis can improve functional outcomes after both TBI and SCI, demonstrating involvement of this pathway in the secondary injury (67, 74). Recent data suggest that autophagosomes can serve as platforms for the assembly of necroptosis-inducing complexes (necrosomes), including RIPK1, RIPK3, and ATG5 (4). Therefore, accumulation of autophagosomes after CNS trauma could also potentially contribute to necroptosis.
Defects in autophagy flux have been noted in multiple neurodegenerative and lysosomal diseases, where they contribute to neuronal cell death. In lysosomal storage diseases, defects in specific lysosomal hydrolases lead to lysosomal dysfunction and, as a consequence, inhibition of autophagy flux (56). Lysosomal function abnormalities have been also reported in some neurodegenerative diseases and proposed to contribute to pathological accumulation of autophagosomes and to neuronal dysfunction and death (63). It appears that a similar situation may occur after CNS trauma, with impairment of lysosomal function contributing to defects in autophagic clearance and neuronal cell death. This suggests lysosomal and autophagy flux defects as a potential common mechanism contributing to neuronal cell death due to chronic (neurodegenerative and lysosomal storage diseases) and acute (CNS trauma) insults.
It is also important to note that in models where autophagy flux is inhibited after trauma, this effect may be only transient (38, 53). Therefore, early after SCI or severe TBI, the accumulation of autophagosomes may contribute to neuronal cell death, whereas at later time points when autophagic flux is restored, autophagy may play a neuroprotective role (Fig. 4). While it is not clear how autophagy flux may be restored after CNS trauma, the timing correlates with increased lysosomal biogenesis (53, 78), which could compensate for the initial damage to these organelles. Alternatively, increased flux could reflect death of the affected cells.
Function of Autophagy in Other Cell Types
In addition to neurons, markers of autophagy are increased in other cell types after CNS trauma, including all major glial sub-types (astrocytes, oligodendrocytes, and microglia). However, much less is known about either the mechanisms or the function of autophagy in these cells. Oligodendrocyte cell death is prevalent after CNS injury and is a major component of secondary injury (5). Similar to the situation observed in neurons, depending on whether autophagy flux is activated or inhibited, it could contribute to either survival or death of oligodendrocytes. In addition, after TBI, increased markers of autophagy have been observed not only in mature oligodendrocytes but also in neural/glial antigen 2 (NG2/CSPG4)-positive oligodendrocyte progenitors (53). Autophagy can promote oligodendrocyte precursor survival and myelin development in a myelin mutant rat model (57), and it is possible that it may play a similar function after CNS trauma.
Recent data indicate that autophagy can regulate inflammatory responses via the NFκB pathway (70). For example, in cancer-associated macrophages, autophagy can selectively degrade NFκB RELA/p65, thereby reducing production of pro-inflammatory cytokines (15). SQSTM1 can also stimulate the NFκB pathway through its interaction with TRAF6. It is possible that autophagy may similarly contribute to regulation of inflammatory responses in microglia and/or infiltrating macrophages after CNS injury. However, further work is needed to determine how autophagy may affect glial cell survival and function after CNS trauma.
Autophagy-Lysosomal Pathway as a New Therapeutic Target Against CNS Trauma
Treatments that increase autophagy flux have been shown to offer neuroprotection in both TBI and SCI. The most commonly used agent is the mTOR inhibitor Rapamycin. Even a single administration of Rapamycin early (till 4 h) after injury decreased neuronal cell death and inflammation and improved functional recovery till a month later in multiple models of CNS trauma (16, 23, 55, 61, 64, 68). Several of these studies demonstrated increased autophagy flux after Rapamycin treatment, suggesting the neuroprotective potential of increasing autophagy after CNS injury. However, Rapamycin is known to affect other cellular pathways and functions in addition to autophagy. This includes a well-documented anti-inflammatory function, which may help attenuate neuroinflammation after SCI (16).
The best evidence for the involvement of autophagy in Rapamycin-induced neuroprotection comes from the acute focal cerebellar lesion model (64), where both induction of autophagy flux and the beneficial effects of Rapamycin on neuronal cell survival and functional recovery were attenuated in autophagy hypomorph Becn1+/− mice. Therefore, the neuroprotective role of Rapamycin appears to depend on its ability to induce autophagy in this system. Similar results were obtained in a neonatal hypoxia-ischemia injury model, where protective effects of Rapamycin were attenuated in animals treated with the type III PI3 kinase inhibitor 3-methyladenine (3MA), which inhibits autophagosome formation (13). In the latter model, Rapamycin-induced neuroprotection was also attenuated by AKT1 inhibition, suggesting that both autophagy and AKT1 signaling may be involved downstream of mTOR. However, since mechanisms of hypoxia-ischemia-induced neuronal damage differ in many ways from those after CNS trauma, involvement of this pathway remains to be confirmed in TBI and SCI.
It will also be important to confirm that Rapamycin or other pro-autophagy treatments are able to restore flux in CNS trauma models where it is inhibited. This is an important consideration, as under conditions where autophagic degradation cannot proceed to completion, increasing input into the system may be deleterious rather than beneficial (36). In fact, at least one report has noted the beneficial effects of upstream inhibition of autophagy flux with 3MA after TBI (41), although this has not been confirmed in other models (13, 61).
Based on our preliminary data (Lipinski and Wu, unpublished), treatment with Rapamycin leads to restoration of flux after SCI. This could be mediated through enhanced activity of the master regulator of lysosomal biosynthesis, the transcription factor EB, which is negatively regulated by mTOR (48) (Fig. 6). Increasing lysosomal biogenesis can augment autophagy flux and improve outcomes in animal models of neurodegenerative diseases (11). This supports the idea that enhancing lysosomal biogenesis and autophagy-lysosomal function may also represent a potential treatment strategy after CNS trauma. Such interventions may directly decrease the extent of neuronal and oligodendrocyte cell death as well as attenuate neuroinflammation and promote re-myelination that is necessary for long-term recovery. However, mTOR activity has been recently shown to play a vital function in processes that are necessary for recovery after neurotrauma, such as oligodendrocyte differentiation and myelination (66) and axonal sprouting (34). Therefore, drugs that are capable of promoting lysosomal biogenesis and autophagy flux in an mTOR-independent manner may be necessary for achieving the best therapeutic benefit.
FIG. 6.
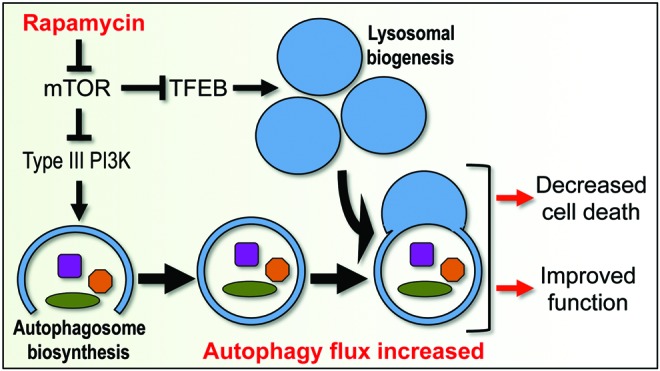
Potential mechanisms and consequences of restoring or increasing autophagy flux after CNS trauma. Inhibition of mTOR can increase initiation of autophagy by stimulating activity of the type III PI3 kinase that is necessary for initiation of autophagosome biogenesis. Inhibition of mTOR can also lead to activation of transcription of lysosomal genes by TFEB and increased lysosomal biogenesis. This could restore and/or increase autophagy flux after CNS trauma, leading to neuroprotection and functional improvements. TFEB, transcription factor EB. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Conclusions
Recently, there has been considerable progress in characterizing autophagy and its potential involvement in CNS trauma. However, much work remains to be done. More detailed delineation of changes in autophagy flux over time after TBI and SCI is needed. Such temporal resolution is essential, as recent studies indicate that the rate of autophagy flux may change over time after injury (38, 53), suggesting potentially important changes in function and mechanisms. Functional studies in transgenic animals with defects in autophagy, such as the Becn1+/− mice, should also be performed. In addition, investigation of selective types of autophagy such as mitophagy (75) after CNS trauma is only now beginning. Finally, the major focus thus far has been on the function and mechanisms of autophagy in neurons. As markers of autophagy may be also altered in astrocytes, microglia, and oligodendrocytes, it will be important to determine its function and mechanisms in these cell types.
Emerging data suggest that depending on the location and severity of the injury, autophagy flux may be either increased or decreased after CNS trauma. Therefore, autophagy may play either beneficial or detrimental functional roles after injury. However, it appears that in all cases restoration and/or augmentation of autophagy flux can increase cell survival and improve functional recovery after injury, suggesting the autophagy pathway as a potential therapeutic target for TBI and SCI. Because of the unique importance of oligodendrocyte differentiation and axonal sprouting for recovery after injury, this may require the development of novel drugs that are able to increase lysosomal biogenesis and autophagy flux without mTOR inhibition.
Abbreviations Used
- 3MA
3-methyladenine
- AIF
apoptosis inducing factor
- CNS
central nervous system
- CQ
chloroquine
- CTSD
cathepsin D
- ER
endoplasmic reticulum
- LMP
lysosomal membrane permeabilization
- NR2B
N-methyl-d-aspartate receptor 2B
- PE
phosphatidylethanolamine
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SCI
spinal cord injury
- TBI
traumatic brain injury
- TFEB
transcription factor EB
Acknowledgments
The authors are supported by 2014-MSCRFE-0587 from Maryland Stem Cell Research Fund, NIH R03NS087338 and R01NS091128 to M.M.L.; NIH R21NR014053 and P30NR014129 to J.W.; and NIH R01NS052568 and R01NR013601 to A.I.F.
References
- 1.Injury Prevention and Control: Traumatic Brain Injury Center for Disease Control and Prevention. http://cdc.gov/traumaticbraininjury/2014 (Accessed date November/26/2014)
- 2.Spinal Cord Injury (SCI): Fact Sheet Center for Disease Control and Prevention. http://cdc.gov/TraumaticBrainInjury/scifacts.html 2014. (Accessed date November/26/2014)
- 3.Bains M. and Hall ED. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta 1822: 675–684, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basit F, Cristofanon S, and Fulda S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ 20: 1161–1173, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beattie MS, Hermann GE, Rogers RC, and Bresnahan JC. Cell death in models of spinal cord injury. Prog Brain Res 137: 37–47, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Beck KD, Nguyen HX, Galvan MD, Salazar DL, Woodruff TM, and Anderson AJ. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 133: 433–447, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bejarano E, Yuste A, Patel B, Stout RF, Jr., Spray DC, and Cuervo AM. Connexins modulate autophagosome biogenesis. Nat Cell Biol 16: 401–414, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berliocchi L, Russo R, Maiaru M, Levato A, Bagetta G, and Corasaniti MT. Autophagy impairment in a mouse model of neuropathic pain. Mol Pain 7: 83, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bigford GE, Alonso OF, Dietrich D, and Keane RW. A novel protein complex in membrane rafts linking the NR2B glutamate receptor and autophagy is disrupted following traumatic brain injury. J Neurotrauma 26: 703–720, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bove J, Martinez-Vicente M, Dehay B, Perier C, Recasens A, Bombrun A, Antonsson B, and Vila M. BAX channel activity mediates lysosomal disruption linked to Parkinson disease. Autophagy 10: 889–900, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bove J, Martinez-Vicente M, and Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci 12: 437–452, 2011 [DOI] [PubMed] [Google Scholar]
- 12.Boyce M, Lipinski MM, Py BF, and Yuan J. Endoplasmic reticulum stress response in cell death and cell survival. In:Apoptosis: Physiology and Pathology, edited by Reed JC. and Green DR. Cambridge, United Kingdom: Cambridge University Press, 2011, pp. 51–62 [Google Scholar]
- 13.Carloni S, Girelli S, Scopa C, Buonocore G, Longini M, and Balduini W. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy 6: 366–377, 2010 [DOI] [PubMed] [Google Scholar]
- 14.Cernak I. Animal models of head trauma. NeuroRx 2: 410–422, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang CP, Su YC, Hu CW, and Lei HY. TLR2-dependent selective autophagy regulates NF-kappaB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ 20: 515–523, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen HC, Fong TH, Hsu PW, and Chiu WT. Multifaceted effects of rapamycin on functional recovery after spinal cord injury in rats through autophagy promotion, anti-inflammation, and neuroprotection. J Surg Res 179: e203–e210, 2013 [DOI] [PubMed] [Google Scholar]
- 17.Chen Z, Fu Q, Shen B, Huang X, Wang K, He P, Li F, Zhang F, and Shen H. Enhanced p62 expression triggers concomitant autophagy and apoptosis in a rat chronic spinal cord compression model. Mol Med Rep 9: 2091–2096, 2014 [DOI] [PubMed] [Google Scholar]
- 18.Cheriyan T, Ryan DJ, Weinreb JH, Cheriyan J, Paul JC, Lafage V, Kirsch T, and Errico TJ. Spinal cord injury models: a review. Spinal Cord 52: 588–595, 2014 [DOI] [PubMed] [Google Scholar]
- 19.Clark RS, Bayir H, Chu CT, Alber SM, Kochanek PM, and Watkins SC. Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy 4: 88–90, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Diskin T, Tal-Or P, Erlich S, Mizrachy L, Alexandrovich A, Shohami E, and Pinkas-Kramarski R. Closed head injury induces upregulation of Beclin 1 at the cortical site of injury. J Neurotrauma 22: 750–762, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Dowler RN, Harrington DL, Haaland KY, Swanda RM, Fee F, and Fiedler K. Profiles of cognitive functioning in chronic spinal cord injury and the role of moderating variables. J Int Neuropsychol Soc 3: 464–472, 1997 [PubMed] [Google Scholar]
- 22.Dumont RJ, Okonkwo DO, Verma S, Hurlbert RJ, Boulos PT, Ellegala DB, and Dumont AS. Acute spinal cord injury, part I: pathophysiologic mechanisms. Clin Neuropharmacol 24: 254–264, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Erlich S, Alexandrovich A, Shohami E, and Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis 26: 86–93, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Filomeni G, De Zio D, and Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 22: 377–388, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fox GB, Fan L, Levasseur RA, and Faden AI. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma 15: 599–614, 1998 [DOI] [PubMed] [Google Scholar]
- 26.Guo D, Zeng L, Brody DL, and Wong M. Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PloS One 8: e64078, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou H, Zhang L, Liu D, Xiong Q, Du H, and Tang P. Acute spinal cord injury could cause activation of autophagy in dorsal root ganglia. Spinal Cord 51: 679–682, 2013 [DOI] [PubMed] [Google Scholar]
- 28.Kanno H, Ozawa H, Sekiguchi A, Yamaya S, and Itoi E. Induction of autophagy and autophagic cell death in damaged neural tissue after acute spinal cord injury in mice. Spine 36: E1427–E1434, 2011 [DOI] [PubMed] [Google Scholar]
- 29.Kanno H, Ozawa H, Sekiguchi A, Yamaya S, Tateda S, Yahata K, and Itoi E. The role of mTOR signaling pathway in spinal cord injury. Cell Cycle 11: 3175–3179, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, Aoki H, Apostolova N, Arancia G, Aris JP, Asanuma K, Asare NY, Ashida H, Askanas V, Askew DS, Auberger P, Baba M, Backues SK, Baehrecke EH, Bahr BA, Bai XY, Bailly Y, Baiocchi R, Baldini G, Balduini W, Ballabio A, Bamber BA, Bampton ET, Banhegyi G, Bartholomew CR, Bassham DC, Bast RC, Jr., Batoko H, Bay BH, Beau I, Bechet DM, Begley TJ, Behl C, Behrends C, Bekri S, Bellaire B, Bendall LJ, Benetti L, Berliocchi L, Bernardi H, Bernassola F, Besteiro S, Bhatia-Kissova I, Bi X, Biard-Piechaczyk M, Blum JS, Boise LH, Bonaldo P, Boone DL, Bornhauser BC, Bortoluci KR, Bossis I, Bost F, Bourquin JP, Boya P, Boyer-Guittaut M, Bozhkov PV, Brady NR, Brancolini C, Brech A, Brenman JE, Brennand A, Bresnick EH, Brest P, Bridges D, Bristol ML, Brookes PS, Brown EJ, Brumell JH, Brunetti-Pierri N, Brunk UT, Bulman DE, Bultman SJ, Bultynck G, Burbulla LF, Bursch W, Butchar JP, Buzgariu W, Bydlowski SP, Cadwell K, Cahova M, Cai D, Cai J, Cai Q, Calabretta B, Calvo-Garrido J, Camougrand N, Campanella M, Campos-Salinas J, Candi E, Cao L, Caplan AB, Carding SR, Cardoso SM, Carew JS, Carlin CR, Carmignac V, Carneiro LA, Carra S, Caruso RA, Casari G, Casas C, Castino R, Cebollero E, Cecconi F, Celli J, Chaachouay H, Chae HJ, Chai CY, Chan DC, Chan EY, Chang RC, Che CM, Chen CC, Chen GC, Chen GQ, Chen M, Chen Q, Chen SS, Chen W, Chen X, Chen YG, Chen Y, Chen YJ, Chen Z, Cheng A, Cheng CH, Cheng Y, Cheong H, Cheong JH, Cherry S, Chess-Williams R, Cheung ZH, Chevet E, Chiang HL, Chiarelli R, Chiba T, Chin LS, Chiou SH, Chisari FV, Cho CH, Cho DH, Choi AM, Choi D, Choi KS, Choi ME, Chouaib S, Choubey D, Choubey V, Chu CT, Chuang TH, Chueh SH, Chun T, Chwae YJ, Chye ML, Ciarcia R, Ciriolo MR, Clague MJ, Clark RS, Clarke PG, Clarke R, Codogno P, Coller HA, Colombo MI, Comincini S, Condello M, Condorelli F, Cookson MR, Coombs GH, Coppens I, Corbalan R, Cossart P, Costelli P, Costes S, Coto-Montes A, Couve E, Coxon FP, Cregg JM, Crespo JL, Cronje MJ, Cuervo AM, Cullen JJ, Czaja MJ, D'Amelio M, Darfeuille-Michaud A, Davids LM, Davies FE, De Felici M, de Groot JF, de Haan CA, De Martino L, De Milito A, De Tata V, Debnath J, Degterev A, Dehay B, Delbridge LM, Demarchi F, Deng YZ, Dengjel J, Dent P, Denton D, Deretic V, Desai SD, Devenish RJ, Di Gioacchino M, Di Paolo G, Di Pietro C, Diaz-Araya G, Diaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dikic I, Dinesh-Kumar SP, Ding WX, Distelhorst CW, Diwan A, Djavaheri-Mergny M, Dokudovskaya S, Dong Z, Dorsey FC, Dosenko V, Dowling JJ, Doxsey S, Dreux M, Drew ME, Duan Q, Duchosal MA, Duff K, Dugail I, Durbeej M, Duszenko M, Edelstein CL, Edinger AL, Egea G, Eichinger L, Eissa NT, Ekmekcioglu S, El-Deiry WS, Elazar Z, Elgendy M, Ellerby LM, Eng KE, Engelbrecht AM, Engelender S, Erenpreisa J, Escalante R, Esclatine A, Eskelinen EL, Espert L, Espina V, Fan H, Fan J, Fan QW, Fan Z, Fang S, Fang Y, Fanto M, Fanzani A, Farkas T, Farre JC, Faure M, Fechheimer M, Feng CG, Feng J, Feng Q, Feng Y, Fesus L, Feuer R, Figueiredo-Pereira ME, Fimia GM, Fingar DC, Finkbeiner S, Finkel T, Finley KD, Fiorito F, Fisher EA, Fisher PB, Flajolet M, Florez-McClure ML, Florio S, Fon EA, Fornai F, Fortunato F, Fotedar R, Fowler DH, Fox HS, Franco R, Frankel LB, Fransen M, Fuentes JM, Fueyo J, Fujii J, Fujisaki K, Fujita E, Fukuda M, Furukawa RH, Gaestel M, Gailly P, Gajewska M, Galliot B, Galy V, Ganesh S, Ganetzky B, Ganley IG, Gao FB, Gao GF, Gao J, Garcia L, Garcia-Manero G, Garcia-Marcos M, Garmyn M, Gartel AL, Gatti E, Gautel M, Gawriluk TR, Gegg ME, Geng J, Germain M, Gestwicki JE, Gewirtz DA, Ghavami S, Ghosh P, Giammarioli AM, Giatromanolaki AN, Gibson SB, Gilkerson RW, Ginger ML, Ginsberg HN, Golab J, Goligorsky MS, Golstein P, Gomez-Manzano C, Goncu E, Gongora C, Gonzalez CD, Gonzalez R, Gonzalez-Estevez C, Gonzalez-Polo RA, Gonzalez-Rey E, Gorbunov NV, Gorski S, Goruppi S, Gottlieb RA, Gozuacik D, Granato GE, Grant GD, Green KN, Gregorc A, Gros F, Grose C, Grunt TW, Gual P, Guan JL, Guan KL, Guichard SM, Gukovskaya AS, Gukovsky I, Gunst J, Gustafsson AB, Halayko AJ, Hale AN, Halonen SK, Hamasaki M, Han F, Han T, Hancock MK, Hansen M, Harada H, Harada M, Hardt SE, Harper JW, Harris AL, Harris J, Harris SD, Hashimoto M, Haspel JA, Hayashi S, Hazelhurst LA, He C, He YW, Hebert MJ, Heidenreich KA, Helfrich MH, Helgason GV, Henske EP, Herman B, Herman PK, Hetz C, Hilfiker S, Hill JA, Hocking LJ, Hofman P, Hofmann TG, Hohfeld J, Holyoake TL, Hong MH, Hood DA, Hotamisligil GS, Houwerzijl EJ, Hoyer-Hansen M, Hu B, Hu CA, Hu HM, Hua Y, Huang C, Huang J, Huang S, Huang WP, Huber TB, Huh WK, Hung TH, Hupp TR, Hur GM, Hurley JB, Hussain SN, Hussey PJ, Hwang JJ, Hwang S, Ichihara A, Ilkhanizadeh S, Inoki K, Into T, Iovane V, Iovanna JL, Ip NY, Isaka Y, Ishida H, Isidoro C, Isobe K, Iwasaki A, Izquierdo M, Izumi Y, Jaakkola PM, Jaattela M, Jackson GR, Jackson WT, Janji B, Jendrach M, Jeon JH, Jeung EB, Jiang H, Jiang JX, Jiang M, Jiang Q, Jiang X, Jimenez A, Jin M, Jin S, Joe CO, Johansen T, Johnson DE, Johnson GV, Jones NL, Joseph B, Joseph SK, Joubert AM, Juhasz G, Juillerat-Jeanneret L, Jung CH, Jung YK, Kaarniranta K, Kaasik A, Kabuta T, Kadowaki M, Kagedal K, Kamada Y, Kaminskyy VO, Kampinga HH, Kanamori H, Kang C, Kang KB, Kang KI, Kang R, Kang YA, Kanki T, Kanneganti TD, Kanno H, Kanthasamy AG, Kanthasamy A, Karantza V, Kaushal GP, Kaushik S, Kawazoe Y, Ke PY, Kehrl JH, Kelekar A, Kerkhoff C, Kessel DH, Khalil H, Kiel JA, Kiger AA, Kihara A, Kim DR, Kim DH, Kim EK, Kim HR, Kim JS, Kim JH, Kim JC, Kim JK, Kim PK, Kim SW, Kim YS, Kim Y, Kimchi A, Kimmelman AC, King JS, Kinsella TJ, Kirkin V, Kirshenbaum LA, Kitamoto K, Kitazato K, Klein L, Klimecki WT, Klucken J, Knecht E, Ko BC, Koch JC, Koga H, Koh JY, Koh YH, Koike M, Komatsu M, Kominami E, Kong HJ, Kong WJ, Korolchuk VI, Kotake Y, Koukourakis MI, Kouri Flores JB, Kovacs AL, Kraft C, Krainc D, Kramer H, Kretz-Remy C, Krichevsky AM, Kroemer G, Kruger R, Krut O, Ktistakis NT, Kuan CY, Kucharczyk R, Kumar A, Kumar R, Kumar S, Kundu M, Kung HJ, Kurz T, Kwon HJ, La Spada AR, Lafont F, Lamark T, Landry J, Lane JD, Lapaquette P, Laporte JF, Laszlo L, Lavandero S, Lavoie JN, Layfield R, Lazo PA, Le W, Le Cam L, Ledbetter DJ, Lee AJ, Lee BW, Lee GM, Lee J, Lee JH, Lee M, Lee MS, Lee SH, Leeuwenburgh C, Legembre P, Legouis R, Lehmann M, Lei HY, Lei QY, Leib DA, Leiro J, Lemasters JJ, Lemoine A, Lesniak MS, Lev D, Levenson VV, Levine B, Levy E, Li F, Li JL, Li L, Li S, Li W, Li XJ, Li YB, Li YP, Liang C, Liang Q, Liao YF, Liberski PP, Lieberman A, Lim HJ, Lim KL, Lim K, Lin CF, Lin FC, Lin J, Lin JD, Lin K, Lin WW, Lin WC, Lin YL, Linden R, Lingor P, Lippincott-Schwartz J, Lisanti MP, Liton PB, Liu B, Liu CF, Liu K, Liu L, Liu QA, Liu W, Liu YC, Liu Y, Lockshin RA, Lok CN, Lonial S, Loos B, Lopez-Berestein G, Lopez-Otin C, Lossi L, Lotze MT, Low P, Lu B, Lu Z, Luciano F, Lukacs NW, Lund AH, Lynch-Day MA, Ma Y, Macian F, MacKeigan JP, Macleod KF, Madeo F, Maiuri L, Maiuri MC, Malagoli D, Malicdan MC, Malorni W, Man N, Mandelkow EM, Manon S, Manov I, Mao K, Mao X, Mao Z, Marambaud P, Marazziti D, Marcel YL, Marchbank K, Marchetti P, Marciniak SJ, Marcondes M, Mardi M, Marfe G, Marino G, Markaki M, Marten MR, Martin SJ, Martinand-Mari C, Martinet W, Martinez-Vicente M, Masini M, Matarrese P, Matsuo S, Matteoni R, Mayer A, Mazure NM, McConkey DJ, McConnell MJ, McDermott C, McDonald C, McInerney GM, McKenna SL, McLaughlin B, McLean PJ, McMaster CR, McQuibban GA, Meijer AJ, Meisler MH, Melendez A, Melia TJ, Melino G, Mena MA, Menendez JA, Menna-Barreto RF, Menon MB, Menzies FM, Mercer CA, Merighi A, Merry DE, Meschini S, Meyer CG, Meyer TF, Miao CY, Miao JY, Michels PA, Michiels C, Mijaljica D, Milojkovic A, Minucci S, Miracco C, Miranti CK, Mitroulis I, Miyazawa K, Mizushima N, Mograbi B, Mohseni S, Molero X, Mollereau B, Mollinedo F, Momoi T, Monastyrska I, Monick MM, Monteiro MJ, Moore MN, Mora R, Moreau K, Moreira PI, Moriyasu Y, Moscat J, Mostowy S, Mottram JC, Motyl T, Moussa CE, Muller S, Munger K, Munz C, Murphy LO, Murphy ME, Musaro A, Mysorekar I, Nagata E, Nagata K, Nahimana A, Nair U, Nakagawa T, Nakahira K, Nakano H, Nakatogawa H, Nanjundan M, Naqvi NI, Narendra DP, Narita M, Navarro M, Nawrocki ST, Nazarko TY, Nemchenko A, Netea MG, Neufeld TP, Ney PA, Nezis IP, Nguyen HP, Nie D, Nishino I, Nislow C, Nixon RA, Noda T, Noegel AA, Nogalska A, Noguchi S, Notterpek L, Novak I, Nozaki T, Nukina N, Nurnberger T, Nyfeler B, Obara K, Oberley TD, Oddo S, Ogawa M, Ohashi T, Okamoto K, Oleinick NL, Oliver FJ, Olsen LJ, Olsson S, Opota O, Osborne TF, Ostrander GK, Otsu K, Ou JH, Ouimet M, Overholtzer M, Ozpolat B, Paganetti P, Pagnini U, Pallet N, Palmer GE, Palumbo C, Pan T, Panaretakis T, Pandey UB, Papackova Z, Papassideri I, Paris I, Park J, Park OK, Parys JB, Parzych KR, Patschan S, Patterson C, Pattingre S, Pawelek JM, Peng J, Perlmutter DH, Perrotta I, Perry G, Pervaiz S, Peter M, Peters GJ, Petersen M, Petrovski G, Phang JM, Piacentini M, Pierre P, Pierrefite-Carle V, Pierron G, Pinkas-Kramarski R, Piras A, Piri N, Platanias LC, Poggeler S, Poirot M, Poletti A, Pous C, Pozuelo-Rubio M, Praetorius-Ibba M, Prasad A, Prescott M, Priault M, Produit-Zengaffinen N, Progulske-Fox A, Proikas-Cezanne T, Przedborski S, Przyklenk K, Puertollano R, Puyal J, Qian SB, Qin L, Qin ZH, Quaggin SE, Raben N, Rabinowich H, Rabkin SW, Rahman I, Rami A, Ramm G, Randall G, Randow F, Rao VA, Rathmell JC, Ravikumar B, Ray SK, Reed BH, Reed JC, Reggiori F, Regnier-Vigouroux A, Reichert AS, Reiners JJ, Jr., Reiter RJ, Ren J, Revuelta JL, Rhodes CJ, Ritis K, Rizzo E, Robbins J, Roberge M, Roca H, Roccheri MC, Rocchi S, Rodemann HP, Rodriguez de Cordoba S, Rohrer B, Roninson IB, Rosen K, Rost-Roszkowska MM, Rouis M, Rouschop KM, Rovetta F, Rubin BP, Rubinsztein DC, Ruckdeschel K, Rucker EB, 3rd, Rudich A, Rudolf E, Ruiz-Opazo N, Russo R, Rusten TE, Ryan KM, Ryter SW, Sabatini DM, Sadoshima J, Saha T, Saitoh T, Sakagami H, Sakai Y, Salekdeh GH, Salomoni P, Salvaterra PM, Salvesen G, Salvioli R, Sanchez AM, Sanchez-Alcazar JA, Sanchez-Prieto R, Sandri M, Sankar U, Sansanwal P, Santambrogio L, Saran S, Sarkar S, Sarwal M, Sasakawa C, Sasnauskiene A, Sass M, Sato K, Sato M, Schapira AH, Scharl M, Schatzl HM, Scheper W, Schiaffino S, Schneider C, Schneider ME, Schneider-Stock R, Schoenlein PV, Schorderet DF, Schuller C, Schwartz GK, Scorrano L, Sealy L, Seglen PO, Segura-Aguilar J, Seiliez I, Seleverstov O, Sell C, Seo JB, Separovic D, Setaluri V, Setoguchi T, Settembre C, Shacka JJ, Shanmugam M, Shapiro IM, Shaulian E, Shaw RJ, Shelhamer JH, Shen HM, Shen WC, Sheng ZH, Shi Y, Shibuya K, Shidoji Y, Shieh JJ, Shih CM, Shimada Y, Shimizu S, Shintani T, Shirihai OS, Shore GC, Sibirny AA, Sidhu SB, Sikorska B, Silva-Zacarin EC, Simmons A, Simon AK, Simon HU, Simone C, Simonsen A, Sinclair DA, Singh R, Sinha D, Sinicrope FA, Sirko A, Siu PM, Sivridis E, Skop V, Skulachev VP, Slack RS, Smaili SS, Smith DR, Soengas MS, Soldati T, Song X, Sood AK, Soong TW, Sotgia F, Spector SA, Spies CD, Springer W, Srinivasula SM, Stefanis L, Steffan JS, Stendel R, Stenmark H, Stephanou A, Stern ST, Sternberg C, Stork B, Stralfors P, Subauste CS, Sui X, Sulzer D, Sun J, Sun SY, Sun ZJ, Sung JJ, Suzuki K, Suzuki T, Swanson MS, Swanton C, Sweeney ST, Sy LK, Szabadkai G, Tabas I, Taegtmeyer H, Tafani M, Takacs-Vellai K, Takano Y, Takegawa K, Takemura G, Takeshita F, Talbot NJ, Tan KS, Tanaka K, Tang D, Tanida I, Tannous BA, Tavernarakis N, Taylor GS, Taylor GA, Taylor JP, Terada LS, Terman A, Tettamanti G, Thevissen K, Thompson CB, Thorburn A, Thumm M, Tian F, Tian Y, Tocchini-Valentini G, Tolkovsky AM, Tomino Y, Tonges L, Tooze SA, Tournier C, Tower J, Towns R, Trajkovic V, Travassos LH, Tsai TF, Tschan MP, Tsubata T, Tsung A, Turk B, Turner LS, Tyagi SC, Uchiyama Y, Ueno T, Umekawa M, Umemiya-Shirafuji R, Unni VK, Vaccaro MI, Valente EM, Van den Berghe G, van der Klei IJ, van Doorn W, van Dyk LF, van Egmond M, van Grunsven LA, Vandenabeele P, Vandenberghe WP, Vanhorebeek I, Vaquero EC, Velasco G, Vellai T, Vicencio JM, Vierstra RD, Vila M, Vindis C, Viola G, Viscomi MT, Voitsekhovskaja OV, von Haefen C, Votruba M, Wada K, Wade-Martins R, Walker CL, Walsh CM, Walter J, Wan XB, Wang A, Wang C, Wang D, Wang F, Wang G, Wang H, Wang HG, Wang HD, Wang J, Wang K, Wang M, Wang RC, Wang X, Wang YJ, Wang Y, Wang Z, Wang ZC, Wansink DG, Ward DM, Watada H, Waters SL, Webster P, Wei L, Weihl CC, Weiss WA, Welford SM, Wen LP, Whitehouse CA, Whitton JL, Whitworth AJ, Wileman T, Wiley JW, Wilkinson S, Willbold D, Williams RL, Williamson PR, Wouters BG, Wu C, Wu DC, Wu WK, Wyttenbach A, Xavier RJ, Xi Z, Xia P, Xiao G, Xie Z, Xu DZ, Xu J, Xu L, Xu X, Yamamoto A, Yamashina S, Yamashita M, Yan X, Yanagida M, Yang DS, Yang E, Yang JM, Yang SY, Yang W, Yang WY, Yang Z, Yao MC, Yao TP, Yeganeh B, Yen WL, Yin JJ, Yin XM, Yoo OJ, Yoon G, Yoon SY, Yorimitsu T, Yoshikawa Y, Yoshimori T, Yoshimoto K, You HJ, Youle RJ, Younes A, Yu L, Yu SW, Yu WH, Yuan ZM, Yue Z, Yun CH, Yuzaki M, Zabirnyk O, Silva-Zacarin E, Zacks D, Zacksenhaus E, Zaffaroni N, Zakeri Z, Zeh HJ, 3rd, Zeitlin SO, Zhang H, Zhang HL, Zhang J, Zhang JP, Zhang L, Zhang MY, Zhang XD, Zhao M, Zhao YF, Zhao Y, Zhao ZJ, Zheng X, Zhivotovsky B, Zhong Q, Zhou CZ, Zhu C, Zhu WG, Zhu XF, Zhu X, Zhu Y, Zoladek T, Zong WX, Zorzano A, Zschocke J, and Zuckerbraun B. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8: 445–544, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai Y, Hickey RW, Chen Y, Bayir H, Sullivan ML, Chu CT, Kochanek PM, Dixon CE, Jenkins LW, Graham SH, Watkins SC, and Clark RS. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab 28: 540–550, 2008 [DOI] [PubMed] [Google Scholar]
- 32.Larner SF, Hayes RL, McKinsey DM, Pike BR, and Wang KK. Increased expression and processing of caspase-12 after traumatic brain injury in rats. J Neurochem 88: 78–90, 2004 [DOI] [PubMed] [Google Scholar]
- 33.Lazzaro I, Tran Y, Wijesuriya N, and Craig A. Central correlates of impaired information processing in people with spinal cord injury. J Clin Neurophysiol 30: 59–65, 2013 [DOI] [PubMed] [Google Scholar]
- 34.Lee DH, Luo X, Yungher BJ, Bray E, Lee JK, and Park KK. Mammalian target of rapamycin's distinct roles and effectiveness in promoting compensatory axonal sprouting in the injured CNS. J Neurosci 34: 15347–15355, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin CJ, Chen TH, Yang LY, and Shih CM. Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis 5: e1147, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR, and Yuan J. A genome-wide analysis reveals differential regulation of autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci U S A 107: 14164–14169, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu CL, Chen S, Dietrich D, and Hu BR. Changes in autophagy after traumatic brain injury. J Cereb Blood Flow Metab 28: 674–683, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu S, Sarkar S, Dinizo M, Faden AI, Koh EY, Lipinski MM, and Wu J. Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell Death Dis 6: e1582, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loane DJ. and Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci 31: 596–604, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loane DJ, Pocivavsek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI, Rebeck GW, and Burns MP. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med 15: 377–379, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo CL, Li BX, Li QQ, Chen XP, Sun YX, Bao HJ, Dai DK, Shen YW, Xu HF, Ni H, Wan L, Qin ZH, Tao LY, and Zhao ZQ. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience 184: 54–63, 2011 [DOI] [PubMed] [Google Scholar]
- 42.Mizushima N, Levine B, Cuervo AM, and Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 451: 1069–1075, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morrison B, 3rd, Elkin BS, Dolle JP, and Yarmush ML. In vitro models of traumatic brain injury. Annu Rev Biomed Eng 13: 91–126, 2011 [DOI] [PubMed] [Google Scholar]
- 44.Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci 29: 528–535, 2006 [DOI] [PubMed] [Google Scholar]
- 45.Norenberg MD, Smith J, and Marcillo A. The pathology of human spinal cord injury: defining the problems. J Neurotrauma 21: 429–440, 2004 [DOI] [PubMed] [Google Scholar]
- 46.O'Connor WT, Smyth A, and Gilchrist MD. Animal models of traumatic brain injury: a critical evaluation. Pharmacol Ther 130: 106–113, 2011 [DOI] [PubMed] [Google Scholar]
- 47.Ohri SS, Hetman M, and Whittemore SR. Restoring endoplasmic reticulum homeostasis improves functional recovery after spinal cord injury. Neurobiol Dis 58: 29–37, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TA, Zou L, Xie XJ, Corey DR, and Brugarolas J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J 30: 3242–3258, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ribas VT, Schnepf B, Challagundla M, Koch JC, Bahr M, and Lingor P. Early and sustained activation of autophagy in degenerating axons after spinal cord injury. Brain Pathol 25: 157–170, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sabirzhanov B, Zhao Z, Stoica BA, Loane DJ, Wu J, Borroto C, Dorsey SG, and Faden AI. Downregulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. J Neurosci 34: 10055–10071, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakai K, Fukuda T, and Iwadate K. Immunohistochemical analysis of the ubiquitin proteasome system and autophagy lysosome system induced after traumatic intracranial injury: association with time between the injury and death. Am J Forensic Med Pathol 35: 38–44, 2014 [DOI] [PubMed] [Google Scholar]
- 52.Sarkar S, Korolchuk VI, Renna M, Imarisio S, Fleming A, Williams A, Garcia-Arencibia M, Rose C, Luo S, Underwood BR, Kroemer G, O'Kane CJ, and Rubinsztein DC. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell 43: 19–32, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarkar S, Zhao Z, Aungst S, Sabirzhanov B, Faden AI, and Lipinski MM. Impaired autophagy flux is associated with neuronal cell death after TBI. Autophagy 10: 2208–2222, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schoch KM, Madathil SK, and Saatman KE. Genetic manipulation of cell death and neuroplasticity pathways in traumatic brain injury. Neurotherapeutics 9: 323–337, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sekiguchi A, Kanno H, Ozawa H, Yamaya S, and Itoi E. Rapamycin promotes autophagy and reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. J Neurotrauma 29: 946–956, 2012 [DOI] [PubMed] [Google Scholar]
- 56.Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D, de Pablo R, Tacchetti C, Rubinsztein DC, and Ballabio A. A block of autophagy in lysosomal storage disorders. Hum Mol Genet 17: 119–129, 2008 [DOI] [PubMed] [Google Scholar]
- 57.Smith CM, Mayer JA, and Duncan ID. Autophagy promotes oligodendrocyte survival and function following dysmyelination in a long-lived myelin mutant. J Neurosci 33: 8088–8100, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stoica BA. and Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 7: 3–12, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun LQ, Gao JL, Cui CM, Cui Y, Jing XB, Zhao MM, Wang YC, Tian YX, Wang KJ, and Cui JZ. Astrocytic p-connexin 43 regulates neuronal autophagy in the hippocampus following traumatic brain injury in rats. Mol Med Rep 9: 77–82, 2014 [DOI] [PubMed] [Google Scholar]
- 60.Tanabe F, Yone K, Kawabata N, Sakakima H, Matsuda F, Ishidou Y, Maeda S, Abematsu M, Komiya S, and Setoguchi T. Accumulation of p62 in degenerated spinal cord under chronic mechanical compression: functional analysis of p62 and autophagy in hypoxic neuronal cells. Autophagy 7: 1462–1471, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang P, Hou H, Zhang L, Lan X, Mao Z, Liu D, He C, and Du H. Autophagy reduces neuronal damage and promotes locomotor recovery via inhibition of apoptosis after spinal cord injury in rats. Mol Neurobiol 49: 276–287, 2014 [DOI] [PubMed] [Google Scholar]
- 62.Vandenabeele P, Galluzzi L, Vanden Berghe T, and Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11: 700–714, 2010 [DOI] [PubMed] [Google Scholar]
- 63.Vila M, Bove J, Dehay B, Rodriguez-Muela N, and Boya P. Lysosomal membrane permeabilization in Parkinson disease. Autophagy 7: 98–100, 2011 [DOI] [PubMed] [Google Scholar]
- 64.Viscomi MT, D'Amelio M, Cavallucci V, Latini L, Bisicchia E, Nazio F, Fanelli F, Maccarrone M, Moreno S, Cecconi F, and Molinari M. Stimulation of autophagy by rapamycin protects neurons from remote degeneration after acute focal brain damage. Autophagy 8: 222–235, 2012 [DOI] [PubMed] [Google Scholar]
- 65.Viscomi MT. and Molinari M. Remote neurodegeneration: multiple actors for one play. Mol Neurobiol 50: 368–389, 2014 [DOI] [PubMed] [Google Scholar]
- 66.Wahl SE, McLane LE, Bercury KK, Macklin WB, and Wood TL. Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. J Neurosci 34: 4453–4465, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Y, Wang H, Tao Y, Zhang S, Wang J, and Feng X. Necroptosis inhibitor necrostatin-1 promotes cell protection and physiological function in traumatic spinal cord injury. Neuroscience 266: 91–101, 2014 [DOI] [PubMed] [Google Scholar]
- 68.Wang Z, Shi XY, Yin J, Zuo G, Zhang J, and Chen G. Role of autophagy in early brain injury after experimental subarachnoid hemorrhage. J Mol Neurosci 46: 192–202, 2012 [DOI] [PubMed] [Google Scholar]
- 69.Werner C. and Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth 99: 4–9, 2007 [DOI] [PubMed] [Google Scholar]
- 70.White E, Karp C, Strohecker AM, Guo Y, and Mathew R. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol 22: 212–217, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu J, Pajoohesh-Ganji A, Stoica BA, Dinizo M, Guanciale K, and Faden AI. Delayed expression of cell cycle proteins contributes to astroglial scar formation and chronic inflammation after rat spinal cord contusion. J Neuroinflammation 9: 169, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu Y, Yuan J, and Lipinski MM. Live imaging and single-cell analysis reveal differential dynamics of autophagy and apoptosis. Autophagy 9: 1418–1430, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamashima T. and Oikawa S. The role of lysosomal rupture in neuronal death. Prog Neurobiol 89: 343–358, 2009 [DOI] [PubMed] [Google Scholar]
- 74.You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, and Whalen MJ. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab 28: 1564–1573, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu D, Li M, Ni B, Kong J, and Zhang Z. Induction of neuronal mitophagy in acute spinal cord injury in rats. Neurotox Res 24: 512–522, 2013 [DOI] [PubMed] [Google Scholar]
- 76.Zhang B. and Gensel JC. Is neuroinflammation in the injured spinal cord different than in the brain? Examining intrinsic differences between the brain and spinal cord. Exp Neurol 258: 112–120, 2014 [DOI] [PubMed] [Google Scholar]
- 77.Zhang E, Yi MH, Ko Y, Kim HW, Seo JH, Lee YH, Lee W, and Kim DW. Expression of LC3 and Beclin 1 in the spinal dorsal horn following spinal nerve ligation-induced neuropathic pain. Brain Res 1519: 31–39, 2013 [DOI] [PubMed] [Google Scholar]
- 78.Zhang Q, Huang C, Meng B, Tang TS, and Yang HL. Changes in autophagy proteins in a rat model of spinal cord injury. Chin J Traumatol 17: 193–197, 2014 [PubMed] [Google Scholar]
- 79.Zhao Z, Faden AI, Loane DJ, Lipinski MM, Sabirzhanov B, and Stoica BA. Neuroprotective effects of geranylgeranylacetone in experimental traumatic brain injury. J Cereb Blood Flow Metab 33: 1897–1908, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, and Abraham E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem 285: 33154–33164, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]