

Abstract
Monoclonal antibodies that block inhibitory immune checkpoint molecules and enhance anti-tumor responses show clinical promise in advanced solid tumors. Most of the preliminary evidence on therapeutic efficacy of immune checkpoint blockers comes from studies in melanoma, lung and renal cancer. To test the in vivo potential of programmed death-ligand 1 (PD-L1) blockade in ovarian cancer, we recently generated a new transplantable tumor model using human mucin 1 (MUC1)-expressing 2F8 cells. The MUC1 transgenic (MUC1.Tg) mice develop large number of intraperitoneal (IP) tumors following IP injection of 8 × 105 syngeneic 2F8 cells. The tumors are aggressive and display little T cell infiltration. Anti-PD-L1 antibody was administered IP every 2 weeks (200 μg/dose) for a total of three doses. Treatment was started 21 days post-tumor challenge, a time point which corresponds to late tumor stage. The anti-PD-L1 treatment led to substantial T cell infiltration within the tumor and significantly increased survival (p = 0.001) compared to isotype control-treated mice. When the same therapy was administered to wild-type mice challenged with 2F8 tumors, no survival benefit was observed, despite the presence of high titer anti-MUC1 antibodies. However, earlier treatment (day 11) and higher frequency of IP injections restored the T cell responses and led to prolonged survival. Splenocyte profiling via Nanostring using probes for 511 immune genes revealed a treatment-induced immune gene signature consistent with increased T cell-mediated immunity. These findings strongly support further preclinical and clinical strategies exploring PD-L1 blockade in ovarian cancer.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-015-1712-6) contains supplementary material, which is available to authorized users.
Keywords: Ovarian cancer, PD-L1, MUC1, Antibodies, T cells
Introduction
The standard treatment for ovarian cancer combines surgery and platinum–taxane chemotherapy [1]. Despite a favorable initial response to treatment, patients in advanced stages relapse and eventually succumb to platinum-resistant disease [2]. Therapeutic strategies involving immune modulators have received increasing attention due to encouraging results from clinical trials [3, 4]. Tumor-infiltrating lymphocytes (TILs) with anti-tumor effector functions (mainly cytotoxic CD8 T cells) are prognostic indicators of increased survival [5, 6], and the ultimate goal of immune therapy is to increase the quantity and quality of anti-tumor TILs [7]. However, achieving this aim faces many layers of complexity, given that TILs are finely regulated by the balance between several co-stimulatory and inhibitory signals, collectively known as the immune checkpoint molecules [8, 9]. Majority of ongoing studies focus on therapeutic blockade of inhibitory signals, mainly through cytotoxic T lymphocyte antigen-4 (CTLA-4) and programmed death-1 (PD-1). CTLA-4 is a homologue of the co-stimulatory CD28 molecule, expressed on activated T cells. Because it binds to the same ligands, but with much higher affinity, CTLA-4 outcompetes the ability of CD28 to bind to CD80/CD86 on antigen-presenting cells and activates phosphatases that lead to downregulation of effector T cell activity [9]. PD-1 is also expressed by activated T cells, and engagement of its primary ligand, programmed death-ligand 1 (PD-L1 or B7-H1), triggers an inhibitory signal through Src homology 2 domain-containing phosphatase-2 (SHP-2) [9, 10]. PD-L1 is broadly expressed by many tissue types and plays a role in maintaining peripheral tolerance [10]. During carcinogenesis, tumor cells as well as other cells in the tumor microenvironment upregulate PD-L1 in response to inflammatory stimuli and use the PD-1/PD-L1 pathway to inhibit T cell-mediated anti-tumor responses [9, 11].
Several therapeutic approaches that hinder inhibitory immune checkpoint signaling through blocking antibodies such as anti-CTLA-4, anti-PD1 or anti-PD-L1 are currently in various stages of clinical trials, for different cancers [12, 13]. However, most of the emerging data stem from the FDA-approved ipilimumab (anti-CTLA4) and pembrolizumab (anti-PD-1) [14–18]. In ovarian cancer, PD-L1 expression correlates negatively with infiltrating CD8 T lymphocytes and is a negative prognostic factor [19, 20], providing the rationale for PD-L1 blockade as ovarian cancer treatment. Nevertheless, additional evidence for in vivo efficacy of immune checkpoint blockade in ovarian cancer requires further studies, in adequate preclinical models that support future clinical trials.
We describe here the preclinical in vivo efficacy of PD-L1 blockade in ovarian cancer, using a new transplantable tumor model based on the 2F8 cells. The 2F8 murine ovarian cancer cell line was derived from an orthotopic tumor isolated from our previously described triple transgenic MUC1KrasPten mice [21], which express human mucin 1 (MUC1) as a transgene. MUC1 is an epithelial cell membrane protein overexpressed by the vast majority of adenocarcinomas, including epithelial ovarian cancer, regardless of histology [22]. Intrabursal injection of Cre recombinase-encoding adenovirus (AdCre), which activates oncogenic KrasG12D and induces Pten loss in the ovarian surface epithelium, leads to orthotopic endometrioid ovarian tumors that overexpress MUC1 similarly to the human disease [21, 23].
Our results demonstrate that despite their non-immunogenic profile and aggressive behavior, 2F8 tumors respond well to anti-PD-L1 blockade, due to increased systemic T cell responses and intratumoral T cell accumulation.
Materials and methods
Transplantable mouse ovarian cancer model and treatment protocols
The generation of MUC1KrasPten mice and genotyping of littermates for identification of MUC1 transgene carriers and MUC1 negative littermates (wild type, WT) were previously described [21]. Upon injection of AdCre (Gene Transfer Vector Core Facility, University of Iowa) under the ovarian bursa, the MUC1KrasPten mice develop orthotopic, human MUC1-expressing ovarian tumors with endometrioid histology [21]. Ovarian tumor tissue was isolated at necropsy from a MUC1KrasPten mouse with an orthotopic tumor. The tumor tissue was subjected to enzymatic digestion in 0.1 % trypsin, 0.02 % ethylenediaminetetraacetic acid (EDTA) at 37 °C, 5 % CO2 for 1 h. Tumor cells were subsequently cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10 % fetal bovine serum (FBS, Gibco), 10,000 U/L penicillin (Sigma), 10,000 μg/L streptomycin (Sigma) 2 mmol/L l-glutamine, 1 % nonessential amino acids, 1 mM sodium pyruvate and 0.1 mM beta-mercaptoethanol (all from Gibco), until a cell line was established. Clone 2F8 was selected through limiting dilution [24]. Intraperitoneal (IP) injections employed 800,000 2F8 cells. Tumor penetrance is 100 %, and median survival is 29 days (range 19–39 days post-tumor challenge).
Anti-PD-L1 antibody (Clone 10F.9G2) and its rat IgG isotype control were purchased from BioXcell. In the late treatment protocol, therapy was initiated 21 days after the 2F8 tumor challenge. This protocol was applied to MUC1 expressing (MUC1.Tg) and its non-MUC1 transgenic, WT littermates. The antibodies were administered IP every 2 weeks, three doses in total.
In the early, dose-intense treatment protocol, the WT mice were given anti-PD-L1 treatment on day 11 post-tumor challenge and then weekly and for a total of seven doses.
Mice were euthanized either due to tumor burden for ethical reasons or at the completion of experiment (day 53–56 post-tumor challenge). Treatment protocols are depicted in schematic diagrams in Figs. 1f and 4c. For combination therapy, mouse IFNα (10,000 IU, Miltenyi Biotec) was added to the weekly IP dosing of anti-PD-L1 antibody. Serum, spleens and lymph nodes were collected at necropsy, processed into single-cell suspension through mechanical disruption using a 70-μm sieve and cryopreserved for future assays.
Fig. 1.
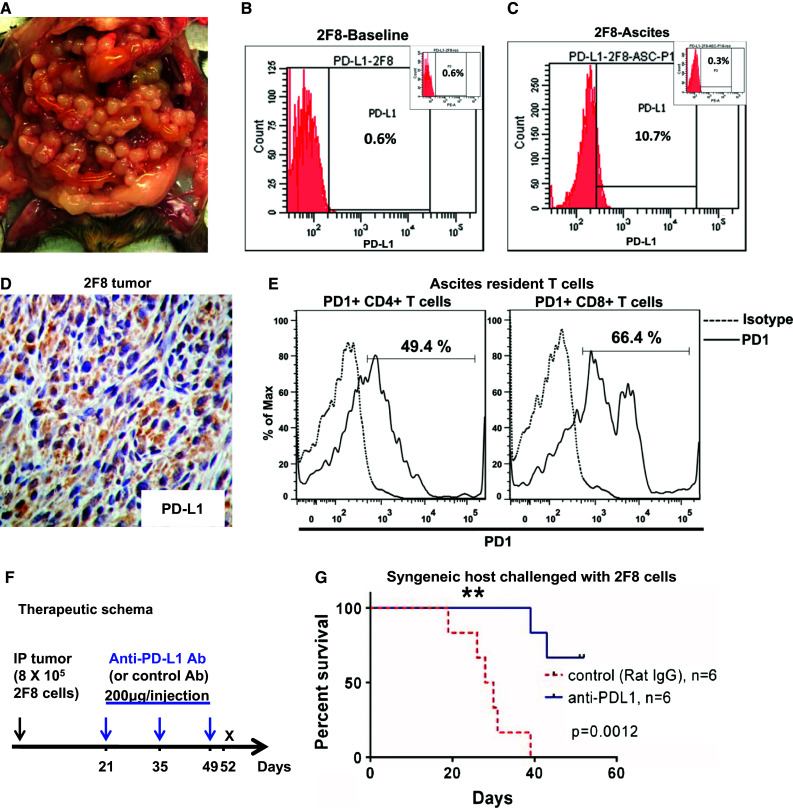
Late treatment with low-dose anti-PD-L1 antibody significantly improves survival. a MUC1.Tg mice challenged IP with 8 × 105 syngeneic 2F8 cells. Image representative of tumor burden at day 29. b, c Flow cytometry staining for cell surface PD-L1 protein expression on 2F8 cells in culture at baseline (b) and after isolation from ascites, post in vivo growth (c). Percentages show positive cells measured outside the isotype control, shown as insets. d Tumor PD-L1 by IHC. e Flow cytometry detection of PD-1+ CD4+ and PD-1+ CD8+ T cells isolated from ascites of 2F8 tumor-bearing mice. Dotted histograms represent staining with isotype control antibody; solid histograms are representative of cells stained with anti-PD-1 antibody. Percentages represent PD-1 positive cells, gated under the CD4 (left) and CD8 (right) populations, respectively. f Therapeutic schema (n = 12 mice): Protocol was started 21 days after IP tumor challenge with 8 × 105 2F8 cells. Half of the mice (n = 6) received IP 200 µg of anti-PD-L1 antibody. The remaining (n = 6) received control rat IgG. All mice received a total of three doses, 2 weeks apart. g Kaplan–Meier survival curve of survival of mice that received anti-PD-L1 antibody (blue) and control IgG (red, p = 0.001)
Fig. 4.
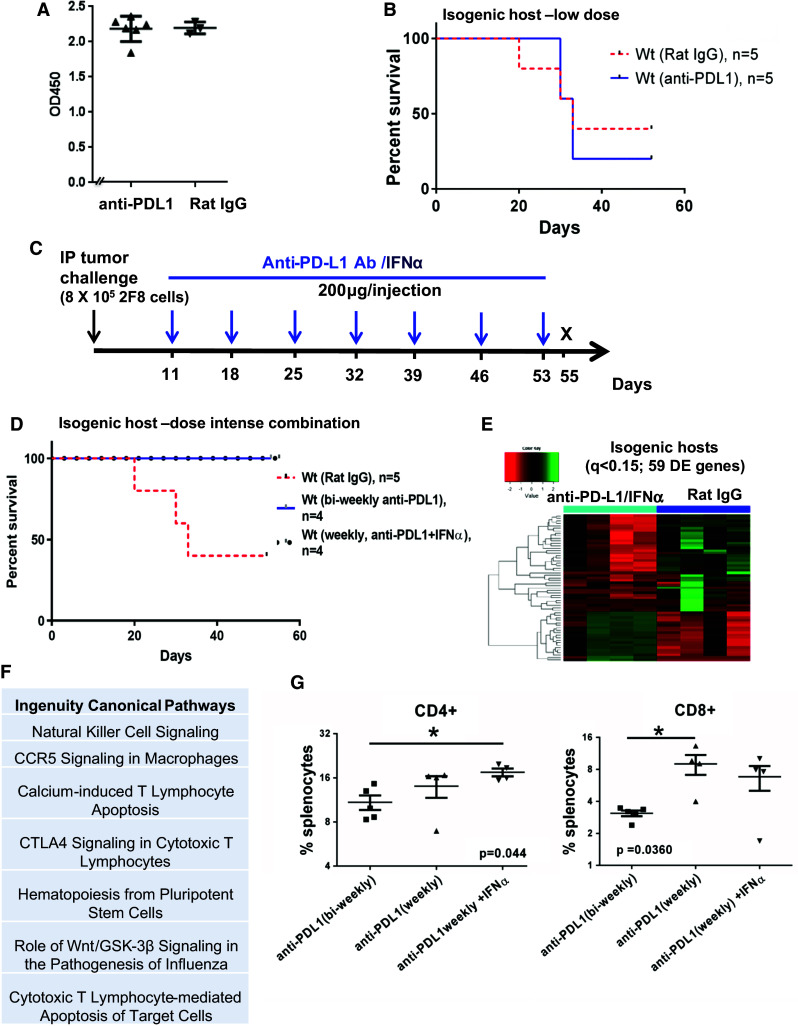
Weekly anti-PD-L1 intraperitoneal injections started early increase survival in WT mice with high levels of naturally occurring anti-tumor antibodies. a ELISA measurement of anti-MUC1 IgG antibodies in sera from non-MUC1 transgenic littermates (WT mice) challenged IP with 8 × 105 2F8 cells. The optical density (OD) at 450 nm is shown on the y axis. b Kaplan–Meier survival curve of survival in wild-type mice treated biweekly with isotype control (n = 5, dotted line) or anti-PD-L1 antibody (n = 5, solid line). c Modified protocol schema for dose-intense weekly treatment, starting at day 11 post-tumor challenge. Mice received weekly anti-PD-L1 alone or in combination with IFNα (10,000 IU). d Kaplan–Meier curve of survival for WT mice treated with IgG (n = 5, red intermittent line), anti-PD-L1 (black circles) or anti-PD-L1/IFNα (n = 4, blue solid line). e Heat map analysis of 59 DE genes by Nanostring (q < 0.15) of which 20 were upregulated and 39 were downregulated in anti-PD-L1/IFNα treated (n = 4) versus control animals (n = 4). f Top seven canonical pathways identified through IPA, using the 59 genes in e. g Percentages of splenic CD3+ T cells by flow cytometry. **p < 0.001, ANOVA
Immunohistochemistry (IHC)
Tumors were collected at necropsy, formalin-fixed and processed for paraffin embedding. Heat-induced epitope retrieval was performed by boiling in TRIS–EDTA buffer (pH 9.0) for CD3, FoxP3 and perforin, or in citrate buffer (pH 6.0) for PD-L1, for 20 min. Five micron sections were stained for CD3ε (M-20, sc-1127; Santa Cruz Biotechnology; 1:100), perforin (Clone CB5.4; LifeSpan Biosciences; 1:40) and PD-L1 (ab58810, Abcam, 1: 40). Secondary antibodies to rat IgG-horseradish peroxidase (HRP) (1:100, eBioscience), rabbit IgG-HRP (Dako EnVision System-HRP) or goat IgG-HRP (sc-2020; Santa Cruz Biotechnology, 1:200) were followed by 3,3′-diaminobenzidine (DAB, Abcam) plus counterstaining with hematoxylin (Sigma Aldrich).
Flow cytometry
Splenocyte suspensions were prepared by mechanical disruption and passing small fragments through a 70-µm cell sieve (BD Falcon, Franklin Lake, NJ, USA). The cells were cryopreserved in freezing medium (90 % FBS and 10 % DMSO) until ready to use. T cell stimulation was performed in 96 well plates coated with anti-CD3ε antibody (5.0 µg/mL, clone 145-2C11, BD Biosciences). PE-CD107a (1D4B, BD Biosciences) was added along with Golgi Plug and Golgi Stop (BD Biosciences) according to manufacturer’s protocol. Surface staining was performed using AlexaFluor 488-TCRβ (H57-597, BioLegend), V450-CD4 (RM4-5, #560468, BD Biosciences) and PerCP-Cy5.5-CD8a (53–6.7, BD Biosciences). Intracellular staining was carried out with Foxp3 staining buffer kit (eBioscience), as per manufacturer’s protocol, using allophycocyanin-FoxP3 (FJK-16 s, eBioscience) and phycoerythrin/Cy7-IFNγ (XMG 1.2, BD Biosciences). Data were acquired using LSR-II flow cytometer (BD Biosciences) and analyzed using FACSDIVATM (BD Biosciences) and FlowJo software (Tree Star). Gating was performed based on isotype controls. Statistical analysis was performed using Prism 6.0 (GraphPad).
ELISA
Serum anti-MUC1 IgG antibody levels were measured using an indirect enzyme-linked immunosorbent assay (ELISA) as previously described [25, 26]. Serum samples (1:50) were incubated for 1 h at room temperature, followed by goat anti-mouse IgG-HRP (1:2000, Sigma). Samples were run in duplicates, and results were calculated after extracting the values from control BSA-coated wells, using Ascent Software for Multiskan version 2.6 (Thermo Scientific).
Nanostring and nCounter data processing
RNA was extracted from whole splenocytes using RNeasy Mini kit (Qiagen) as per manufacturer’s protocol and subjected to Nanostring measurement using the nCounter Mouse Inflammation kit which contains probes for 547 immune-related genes and 14 control (housekeeping) genes.
Experiments were compliant with nCounter mRNA Expression Assay protocol (http://www.genetics.pitt.edu/forms/nCounter_Gene_Expression_Data_Analysis_Guidelines.pdf). To minimize the impact of lane-to-lane variation, we normalized the data to the sum of positive control count values. We used the mean plus two standard deviation values of the negative control probes to estimate the background threshold and confirm specificity. Further, to correct for the RNA content among different samples, we also normalized the data to the geometric means of 14 invariant housekeeping genes. Samples with either positive control normalization factor outside the recommended range of 0.3–3 or estimated background greater than 3 standard deviations from the mean were considered outliers and were removed from analyses. In addition, endogenous genes with zero counts in all samples were filtered out.
Statistical analyses
The edgeR package in Bioconductor was used to identify DE genes between two specified groups. After obtaining gene-wise dispersion, a negative binomial generalized linear model was fit to the count data and differential expression was determined using a likelihood ratio test. We used Benjamini–Hochberg procedure to correct for multiple comparisons. Differentially expressed (DE) genes were then loaded to Ingenuity Pathway Analyses (IPA) for functional analysis.
To analytically characterize samples across the groups, we carried out unsupervised clustering. The count data were first transformed into log2 counts per million as suggested in edgeR. Then, we performed feature filtering by removing genes with means or standard deviations under the median of all the genes. Based on the remaining 357 genes (with standard deviation/mean >0.2), hierarchical clustering with Ward linkage was applied. All statistical programming was implemented in R.
Results
New transplantable ovarian tumor model shows PD-L1 upregulation in vivo
We recently reported that triple transgenic MUC1KrasPten mice carrying conditional (Cre-loxP) oncogenic KrasG12D and Pten deletion mutations progress to human MUC1-expressing endometrioid ovarian tumors that mirror the human disease [21]. Here, we harvested orthotopic tumor tissue from one MUC1KrasPten mouse and used it to generate a new syngeneic ovarian cancer cell line. Tumor cells were expanded in vitro and were subsequently cloned via limiting dilution. Clone 2F8 was chosen due to its rapid doubling time in vitro (14.3 h) and aggressive growth in vivo. Intraperitoneal injection of 800,000 2F8 cells into syngeneic mice leads to widespread peritoneal carcinomatosis (Fig. 1a) and median survival of 29 days. PD-L1 expression is not detectable on the surface of 2F8 cells at baseline (Fig. 1b). However, PD-L1 can be detected on cancer cells isolated from ascites fluid (2F8-Asc) (Fig. 1c) and on cancer cells within the tumor implants (Fig. 1d), suggesting that PD-L1 is upregulated in vivo, in line with recent reports showing in vitro and in vivo plasticity of immune checkpoint molecules [27]. Importantly, the ascites resident CD4 and CD8 T cells from 2F8 tumor-bearing mice express PD-1 (Fig. 1e), suggesting that this in vivo model replicates the inflammatory environment typically found in human ovarian cancer [28] and is adequate for preclinical testing of immune checkpoint PD-1/PD-L1 blockade.
Anti-PD-L1 increases survival in late-stage disease and triggers increased expression of cytotoxic immune genes in splenocytes and increased intratumoral T cell infiltration
We postulated that despite their aggressive behavior in vitro and in vivo, 2F8 tumors may respond to IP administration of anti-PD-L1 treatment. Therapy was instituted according to the schema in Fig. 1f, starting at day 21, which corresponds to late-stage disease. Injections of 200 μg anti-PD-L1 antibody (clone 10F.9G2), previously shown to block PD-L1 in vivo [28], were administered every 2 weeks, for a total of three doses. Control mice received same IP dose of isotype control antibody. Primary endpoint was survival at day 52, 3 days after the completion of treatment protocol. Mice treated with anti-PD-L1 (n = 6) survived significantly longer than control mice (n = 6) (Fig. 1g, p = 0.0012), suggesting in vivo efficacy of IP anti-PD-L1.
To profile the treatment-induced immune gene expression changes in splenocytes, we used Nanostring measurements of 511 immune genes. Data analysis revealed a total of 79 genes that were DE between the anti-PD-L1 (n = 6) and control-treated animals (n = 5) (Supplementary Table 1, Benjamini-Hochberg multiple comparison adjusted q < 0.15). The heat map profile of the top 20 genes (q < 0.05, Fig. 2a) shows that several of the 10 genes that were upregulated in PD-L1-treated mice encode for proteins essential for CD8 T cell function, including T cell co-receptor function (CD8β), intracellular signaling (CD3e, CD3δ, Lck) and cytotoxicity (granzyme A).
Fig. 2.
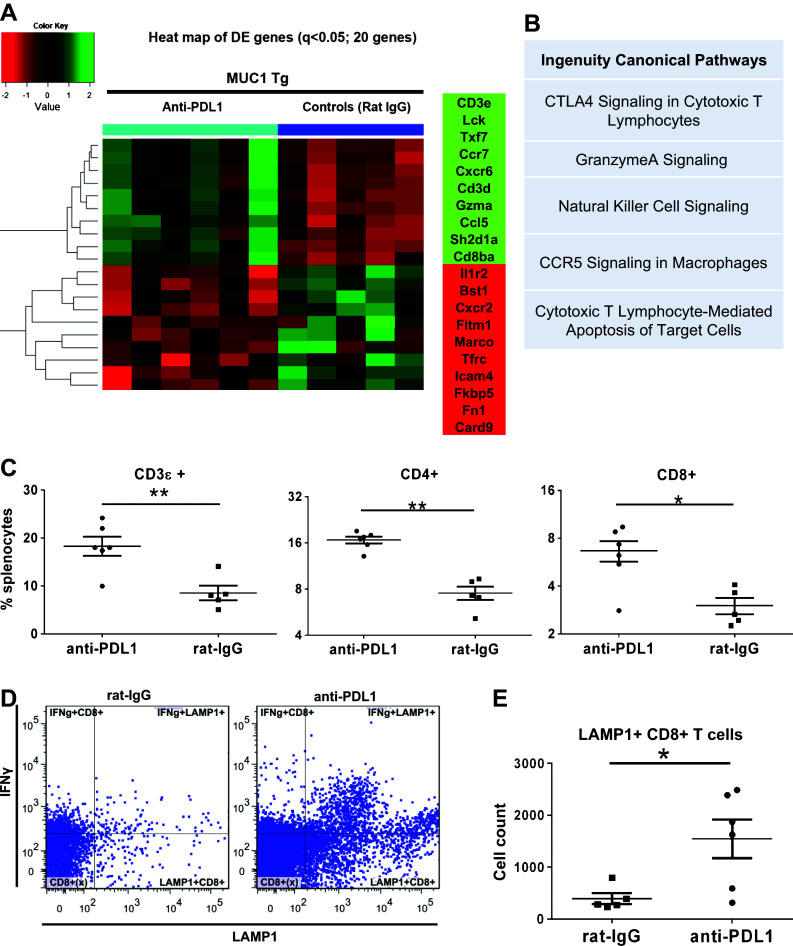
Treatment-induced changes in splenocyte T cell populations and immune gene expression profiles. a Heat map of top 20 DE genes (adjusted q < 0.05) in splenocytes of MUC1.Tg mice that received anti-PD-L1 or rat IgG (controls), detected via Nanostring. b Top five canonical pathways identified by IPA, using n = 79 DE genes. c Phenotypic analysis via flow multicolor cytometry of splenocytes from mice treated with either anti-PD-L1 or control rat IgG. Percent cells positive for CD3 (left), CD4 (middle) and CD8 (right) are shown. d Flow cytometry dot plots showing intracellular staining for IFNγ and LAMP1 following ex vivo stimulation of whole splenocytes in anti-CD3-coated 96 well plates. Data shown are from one representative mouse from either the isotype control (left) or anti-PD-L1 treatment group (right). e Total counts for CD8+ T cells expressing the degranulation marker LAMP1 (CD107a) from n = 5 control-treated and n = 6 anti-PD-L1-treated mice. *p < 0.05; **p < 0.001, Mann–Whitney t test
Among the top 10 downregulated genes in the PD-L1 group were the genes encoding for CXCR2 (neutrophil chemotactic factor), FK506 binding protein 5 (FKBP5, an immunophilin associated with immune suppression, epithelial to mesenchymal transition and cancer progression) [29] and interferon-induced transmembrane protein 1 (Ifitm1, associated with tumor cell proliferation and migration) [30]. Strikingly, IPA results for all 79 DE genes confirmed that four of the top five pathways modified by anti-PD-L1 treatment are related to immune cell-mediated cytotoxicity, with the top pathway being an immune checkpoint pathway (Fig. 2b). Although listed as the CTLA4 pathway, genes identified in this category include CD247 (Cd3ζ), Fyn, Lck, CD3ε, Fcer1 g, Cd8α, Cd3δ, Cd8β, which are also triggered downstream of PD-1.
In line with findings on gene expression, flow cytometry analysis of splenocytes showed increased splenic CD4 and CD8 T cells in the experimental group (Fig. 2c) confirming T cell responses in these mice.
Although many of the detected genes are often considered to be upregulated in response to IFNγ (such as granzyme, perforin, Stat1), we could not detect significant changes in IFNγ expression at either RNA level through Nanostring (Supplementary Table 1) or protein level by flow cytometry, although the latter showed a trend in increase in IFNγ+ T cells (Supplementary Figure 1, p = 0.05). Nevertheless, the number of CD8 T cells positive for the degranulation marker CD107a (LAMP-1) was significantly higher in mice treated with anti-PD-L1 (Fig. 2d, e), further suggesting a treatment-induced cytotoxic response.
We also investigated whether and how the composition of tumor-infiltrating lymphocytes changes with anti-PD-L1 treatment. Because only half of the anti-PD-L1-treated mice had any visible tumors at necropsy and access to tumor tissue was limited in the remaining mice, we could not perform comprehensive phenotyping of tumor-infiltrating TILs via flow cytometry. Consequently, the available tumors were processed via formalin fixation and paraffin embedding, followed by IHC with anti-CD3 antibodies (for entire T cell fraction) and anti-perforin antibodies (for cytotoxic T and NK cells) (Fig. 3a). Remarkably, tumors from control mice displayed very little T cell infiltration, while the tumors isolated from anti-PD-L1-treated mice showed significant T cell accumulation inside the tumor (Fig. 3a). A subset of the cells stained positive for perforin, a cytotoxic cell marker. Although Foxp3-positive regulatory T cells (Tregs) are present in tumors from both control- and antibody-treated mice (Supplementary Figure 2), tumor infiltration of perforin + cells is much higher in anti-PD-L1-treated tumors, in line with previous reports showing that high effector to Treg ratio is predictor of good prognosis in ovarian cancer [6]. Taken together, these results suggest that PD-L1 administration increases the effector arm of T cell-mediated immunity through increased systemic (splenic) cytotoxic T cell responses (Fig. 2c–e) and increased T cell infiltration of cytotoxic, perforin-positive cells inside the tumor mass (Fig. 3a).
Fig. 3.
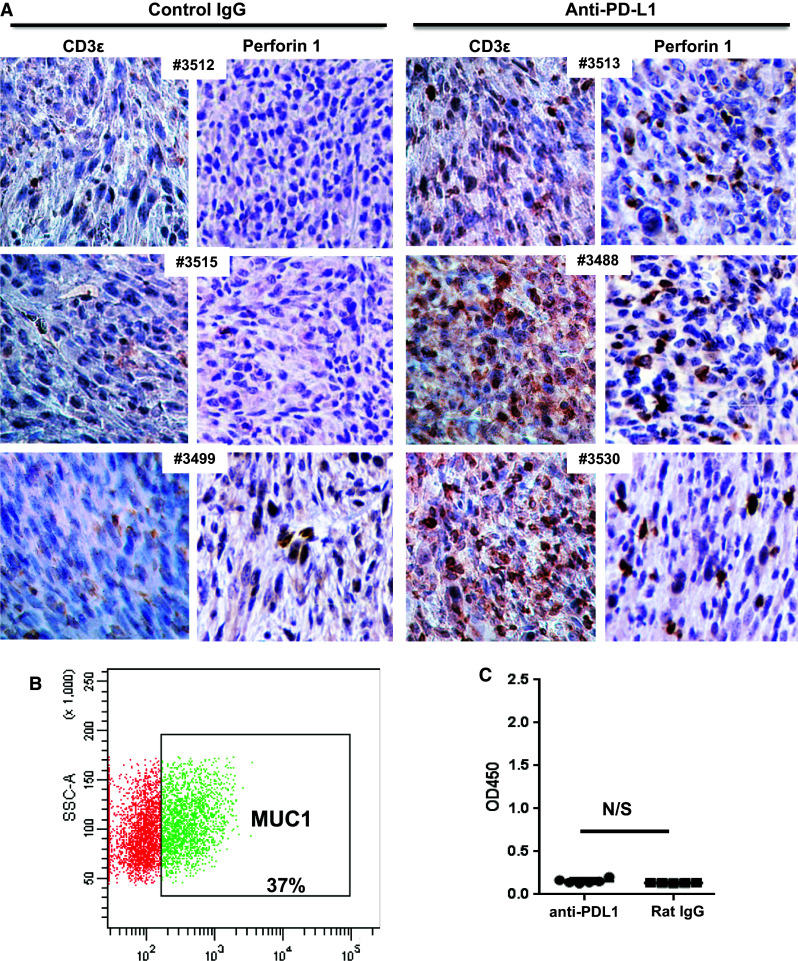
Anti-PD-L1 blockade enhances T cell infiltration but does not trigger anti-tumor antibody responses. a IHC analysis of tumors from six different mice, treated with either control rat IgG (left) or anti-PD-L1 antibody (right). Antibodies for CD3 perforin (clone CB5.4). All images were taken with a Nikon digital camera, coupled to an Olympus microscope, at ×20 magnification. b Expression of cell surface MUC1 on 2F8 via flow cytometry. Gate shows percent MUC1-positive cells, outside of isotype control area. c ELISA measurement of anti-MUC1 IgG antibodies. The optical density (OD) is shown on the y axis. Values shown represent average values from duplicate wells, calculated after background extraction (sera incubated on MUC1 peptide-coated plates minus same sera incubated on bovine serum albumin-coated plates). OD optical density, N/S not significant, Student’s t test
Similarly to the MUC1KrasPten mice with human MUC1-expressing orthotopic ovarian tumors, [21] the MUC1-positive 2F8 cells (Fig. 3b) trigger low but detectable anti-MUC1 circulating antibodies in control-treated tumor-bearing hosts (Fig. 3c). Notably, PD-L1 blockade did not change the anti-MUC1 antibody levels, suggesting that while this therapeutic approach can effectively induce T cells (Figs. 2, 3a), it does not trigger de novo anti-tumor humoral immunity (Fig. 3c).
Antibodies correlate inversely with anti-PD-L1 response
It is well established that a number of cancer patients can naturally develop anti-tumor antibodies, often found in high titers at the time of diagnosis [31]. However, the protective roles of baseline antibodies during immune therapeutic protocols that primarily target T cells remain unclear.
Because MUC1.Tg hosts do not seem to produce high antibody levels against 2F8 cells (Fig. 3c), we designed an isogenic in vivo model, in which the host and the 2F8 tumor cells differ by a single protein (MUC1). In this isogenic model, the host mice do not carry the MUC1 transgene, and therefore, they are not tolerant to MUC1 on 2F8 cells used as transplantable tumors. Nevertheless, the WT hosts are on an identical background to MUC1.Tg mice from which 2F8 cells were derived and are thus tolerant to all 2F8-expressed antigens, except for MUC1. We hypothesized that isogenic hosts will mount MUC1-specific anti-tumor antibodies and may respond even more efficiently to PD-L1 blockade (which as seen above triggers primarily T cell responses) due to the concomitant involvement of T cell and antibody-mediated immune responses. In line with this hypothesis, the human MUC1-expressing 2F8 cells injected into non-MUC1 transgenic hosts triggered very high antibody levels to MUC1 glycoprotein, seen as xenogeneic (Fig. 4a). However, despite a robust humoral immune response in tumor-challenged hosts, the 2F8 tumors were not rejected and developed at similar rates as the ones seen in the fully syngeneic system (Fig. 1a). Furthermore, although prevalence of splenic PD-1-expressing T cells and PD-L1 tumor expression were similar in MUC1.Tg and WT hosts (Supplementary Figure 3), treatment with anti-PD-L1, using a protocol identical to the one tested in completely syngeneic MUC1.Tg mice (Fig. 1f), led to no survival benefit in WT mice (Fig. 4b), suggesting that high humoral immunity at baseline counteracts, instead of acting alongside, the anti-PD-L1-induced T cell responses. To test whether a different treatment protocol would overcome this effect, we employed a dose-intense regimen that started 10 days earlier (at day 11, instead of day 21 post-tumor challenge) and applied weekly anti-PD-L1, for a total of seven doses (Fig. 4c). To ensure further support for cellular cytotoxicity, we added IFNα (10,000 IU) to each injection (Fig. 4c). Using this dose-intense combination regimen, all WT mice survived the protocol (56 days post-tumor challenge) compared to control animals (n = 4), of which only one survived (Fig. 4d). Gene expression profiling of splenocytes revealed a total of 59 DE genes of which 20 were upregulated and 39 were downregulated in the experimental versus control animals (Fig. 4e; Supplementary Table 2, q < 0.15). The top pathways triggered by the dose-intense combination (Fig. 4f) reveal the involvement of cytotoxic effector function. Furthermore, as in MUC1.Tg mice responding to the biweekly anti-PD-L1 protocol, splenic increases in T cells from WT mice were detected by flow cytometry, confirming T cell responses as the hallmark of PD-L1 blockade in vivo (Fig. 4g). Nanostring analyses revealed that although expression of certain DE genes was lowered in the presence of IFNα, several important immune effector genes (including those encoding for CD8, IFNG and MHC-II) were boosted in the combination versus single-agent therapy group (Supplementary Figure 4). The increased expression of Ia antigen-associated invariant chain (Ii) CD74 and H2-A genes (Supplementary Figure 4) also explains the greater involvement of CD4+ T cells along with CD8+ T cells in these mice. Notably, exposure of 2F8 tumors to IFNα (and similarly to IFNγ, as previously demonstrated) [32] may also trigger PD-L1 upregulation (Supplementary Figure 5), supporting the rationale for PD-L1 blockade in interferon-containing regimens. To monitor occurrence of autoimmunity, all mice, regardless of genotype or treatment protocol, were screened for mononuclear infiltration in lungs and kidneys, and no changes were observed.
Immune gene signature associated with increased survival points to enhanced T cell function
These results (Figs. 2, 4) demonstrate that PD-L1 blockade effectively increases survival in both MUC1.Tg and isogenic hosts, albeit through different dose regimens. To explore whether common pathways were triggered in responding mice, we analyzed the splenocyte gene expression data obtained using Nanostring from all groups (n = 24): MUC1.Tg mice (n = 11, of which six received anti-PD-L1 and five were controls) and wild-type mice (n = 13, of which five received low-dose anti-PD-L1 with no survival benefit, four received dose-dense anti-PD-L1/IFNα with increased survival and four were IgG controls). Unsupervised clustering showed two major groups, of 10 and 14 mice, respectively (Fig. 5a). In the group of 10 mice (left cluster), eight received PD-L1 blockade that led to increased survival (i.e., the low-dose PD-L1-treated MUC1.Tg mice and the dose-intense PD-L1/IFNα-treated isogenic mice). The second group of 14 mice (right cluster) contains eight of the nine control-treated mice and four of the five anti-PD-L1-treated wild-type mice with no survival benefit. Thus, unsupervised clustering correctly groups 8 of the 10 (80 %) animals with treatment-induced increased survival and 12 of the 14 (86 %) mice with no survival benefit (i.e., all control IgG mice plus low-dose, PD-L1-treated WT mice) (Fig. 5a).
Fig. 5.
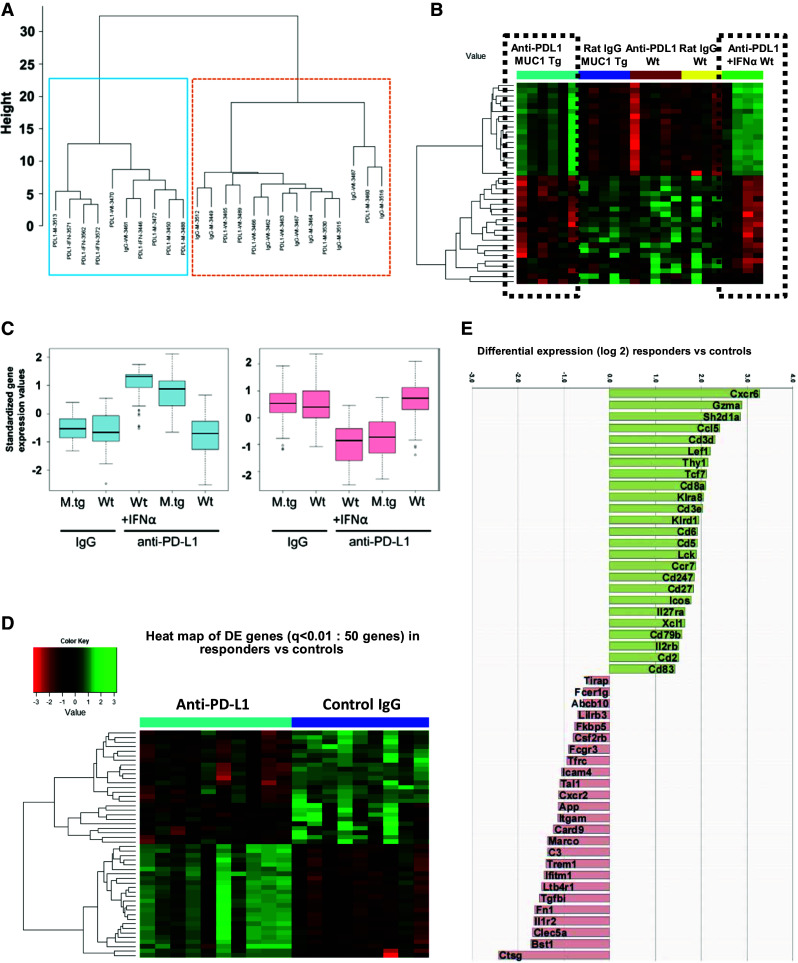
Identification of genes associated with anti-PD-L1-induced increase in survival. a Unsupervised cluster analysis using gene expression of n = 357 filtered genes, from five mouse groups (n = 24): MUC1.Tg mice (n = 11, of which six received anti-PD-L1 and 5 were controls) and wild-type mice (n = 13, of which five received low-dose anti-PD-L1 with no survival benefit, four received dose-dense anti-PD/L1/IFNα with increased survival and four were IgG controls). Two major clusters are observed of 10 (left, blue box) and 14 mice (right, orange box), respectively. b Heat map of the top 39 DE genes, from five mouse group comparisons (ANOVA, p < 0.025). Two clusters of genes (of 18 and 21 genes, respectively) show similar patterns in the anti-PD-L1 groups with increased survival (black dotted boxes), in contrast to the three other groups with no survival benefit, which also share a similar pattern. c Gene expressions of the two clusters, stratified by groups. Y axis is the standardized expression values for the genes in the cluster. Boxplots show that the anti-PD-L1 groups have clearly higher expression than the other groups in the first cluster but lower expression in the second cluster. d Heat map of top 50 DE genes (q < 0.01) from the comparison of anti-PD-L1-treated mice with increased survival (n = 10 mice, of which n = 6 MUC1.Tg mice treated with anti-PD-L1 and n = 4 WT mice treated with dose-intense anti-PD-L1/IFNα) versus isotype control-treated mice (non-responders, n = 9 mice). e Graph bars of the top 50 DE genes shown in panel D heat map (q < 0.01). Values plotted represent average differential gene expression (log2). Positive values represent upregulated and negative values represent downregulated genes in anti-PD-L1-treated mice compared to controls
To identify the treatment-induced differentiating genes among all five mouse groups, we used analysis of variance (ANOVA) model, in which the DE genes are defined as those genes with significantly different expression levels between any two groups. The heat map in Fig. 5b shows top 39 DE genes (listed in Supplementary Table 3). Two clusters of genes, upregulated (n = 18 genes) and downregulated (n = 21), respectively, show similar patterns in the anti-PD-L1 groups with increased survival (i.e., anti-PD-L1-treated MUC1.Tg mice and anti-PD-L1/IFNα-treated WT mice, respectively), in contrast to the three other groups with no survival benefit (Fig. 5b). The magnitude of gene expressions stratified by groups (Fig. 5c) further demonstrates similarity among mice with therapeutic responses, compared to groups with no survival benefit.
To focus our analysis strictly on DE genes modified in animals that show significant survival benefit, we eliminated the WT mice treated with low-dose, low-frequency anti-PD-L1. The heat map in Fig. 5d shows the top 50 DE genes (q < 0.01) when comparing the anti-PD-L1 responders (n = 10 mice, of which n = 6 MUC1.Tg mice treated with anti-PD-L1 and n = 4 WT mice treated with dose-intense anti-PD-L1/IFNα) with isotype control-treated mice (n = 9 mice). All DE genes (n = 136) are listed in Supplementary Table 4. Strikingly, among the most upregulated is the gene encoding for granzyme A, which mediates the cytotoxic function of CD8 T cells (Fig. 5e). In addition, 21 of the 25 upregulated genes are involved in (albeit not being restricted to) T cell biology: T cell receptor complex (CD3d, CD3e, CD8a), co-stimulation (CD2, Icos), intracellular TCR-induced cell signaling (CD3ζ, Sh2d1a, Lef1, Lck, Tcf7, CD5, CD6), chemotaxis (Xcl1, Ccl5), memory T cell function and migration (CD27, CCR7) and T cell function (Thy1, Il2rb) (Fig. 5e).
In contrast to the profile of upregulated genes homogeneously pointing to T cell functionality, the downregulated genes evoke involvement of several different immune cells, mostly of the myeloid lineage: macrophage receptor with collagenous structure (Marco), triggering receptor expressed on myeloid cells 1 (Trem1) and C-type lectin domain family 5 member A (Clec5a). The most downregulated is the cathepsin G gene (Ctsg), encoding for a neutrophil-derived protease (Fig. 5e).
Overall, these multiplex immune gene expression profiles reveal that anti-PD-L1-induced increase in survival is associated with enhanced T cell function and increased T cell-mediated cytotoxicity.
Discussion
We report here a new transplantable syngeneic ovarian cancer model that, despite being non-immunogenic and highly aggressive, responds well to IP administration of anti-PD-L1. The treatment-induced increase in survival is associated with increased T cell biology and increased tumor infiltration with T cells, while tumor-specific antibody levels remain unchanged.
Promising data from preclinical studies and early phase clinical trials show that antibody-mediated immune checkpoint inhibition holds promise for enhancing the overall survival of cancer patients [3, 9]. Blockade of the PD-1/PD-L1 pathway alone or in combination with anti-CTLA-4 has been tested in patients, although most of the emerging data come from melanoma [14–16]. Results from ovarian cancer are scarce, with only one report presenting activity and safety of PD-L1 blockade from 17 patients with advanced cancer treated intravenously with escalating doses (0.3–10 mg per kilogram of body weight) of BMS-936559, a high affinity, fully human PD-L1-specific IgG4 antibody [14]. Although adverse effects were present, they were lower grade and relatively easier to manage compared to those seen in anti-CTLA-4-treated patients. The clinical activity showed a partial response in one case (6 %) and stable disease lasting at least 24 weeks in three patients (18 %) [14]. Design and implementation of additional clinical trials in ovarian cancer that explore immune checkpoint blockers alone or in combination therapies are needed. Progress in this area relies in part on adequate models for preclinical testing.
Several immune-competent transgenic mice that develop orthotopic ovarian tumors have been developed in the last years [33]. We have recently described a triple transgenic orthotopic model that progresses to human MUC1-expressing endometrioid ovarian tumors and favorably responds to a MUC1 peptide-loaded dendritic cell vaccine [21].Furthermore, MUC1 vaccination of mice that carry MUC1 transgene does not trigger autoimmunity, in line with findings from numerous MUC1 vaccine clinical trials [36].
Unlike the healthy ovarian surface epithelium (OSE)-derived ID8 and IG10 cell lines, currently employed in the vast majority of transplantable ovarian cancer studies [34, 35], the 2F8 cells employed here originate from an orthotopic ovarian tumor with well-defined genetic traits (oncogenic KrasG12D mutation and Pten deletion) [21]. In addition, 2F8 cells also express MUC1, a widely studied tumor-associated antigen and immune therapy target [36, 37]. By using the 2F8 cells, we were able to monitor anti-tumor humoral (MUC1-specific) immunity in tumor-bearing hosts and assess the efficacy of PD-L1 blockade in mice with or without anti-MUC1 antibodies, using isogenic (WT, non-MUC1.Tg) and syngeneic (MUC1.Tg) hosts, respectively. These two groups of mice served here as surrogate representatives of patients who have either high or low anti-tumor (including anti-MUC1) antibody levels at the time of diagnosis. Given that the MUC1.Tg mice see human MUC1 as a self-antigen, all natural and immune checkpoint blockade-induced immune responses against MUC1-expressing 2F8 tumors are expected to be similar to those seen in wild-type animals challenged with syngeneic tumors [28], with no additional risks of autoimmunity.
Unlike T and NK cells whose roles in eliminating tumors are well established [38, 39], the role of B cells and anti-tumor antibody responses are still a matter of debate [40]. We have previously reported that increased anti-MUC1 antibody levels are prognostic for poor clinical response and reduced overall survival in platinum-resistant or platinum-refractory ovarian cancer patients who received IP interleukin 2 (IL-2) [25, 26]. In line with these findings, anti-PD-L1 treatment employed here (which like IL-2, is intended to support T cell immunity, albeit through different mechanisms) showed significantly diminished efficacy in tumor-bearing mice with high MUC1-specific antibodies, suggesting that a potential “bias” for humoral immunity may interfere with PD-L1 blockade, despite similar PD-1 and/or PD-L1 expression levels at baseline. However, the treatment efficacy and overall survival could be increased upon dose adjustment and addition of IFNα, which further supports cytotoxic immunity. We acknowledge that the requirement for additional immune modulators (like the highly potent IFNα employed here, which triggers IFNγ, IFNγ-induced genes and MHC upregulation) needs to be further clarified and translatability of this dose-intense regimen carefully considered. Our findings also raise the question whether screening for baseline anti-tumor antibodies could identify patients who may benefit from more personalized approaches, through dose adjustment or combination regimens.
In summary, our preclinical study shows that ovarian tumors that are aggressive and non-immunogenic may benefit from IP administration of anti-PD-L1 antibody-mediated blockade. In addition to significantly increasing the survival, treatment triggers the expansion of splenic T cells and LAMP1-positive CD8+ T cells, together with increased migration and infiltration of T cells, including perforin-positive cells into the tumor mass. Among the DE immune genes identified in splenocytes that were associated with survival, many are typically involved in T cell functionality and cytotoxic anti-tumor immune responses. The increased availability of immune checkpoint reagents and accelerated emergence of clinical data from ongoing trials will provide new opportunities to validate the gene signatures reported here as correlates of survival and for additional correlative studies on anti-tumor antibodies (including but not limited to MUC1-specific antibodies) in responding and non-responding patients.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This study was partly supported by the Department of Defense (DOD) Ovarian Cancer Academy Award W81XWH-10-1-0525 and National Cancer Institute (NCI) R01 CA163462 (to A. Vlad) and P50 CA159981 (to R. Edwards and A. Vlad). Xin Huang is Ovarian Cancer Research Fund Liz Tilberis Scholar (OCRF 258940) and American Cancer Society (ACS) Research Scholar (RSG-12-188-01-RMC). This project used the UPCI Peptide Synthesis Facility that is supported in part by NCI award P30 CA047904.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standard
All animal studies have been approved by University of Pittsburgh Institutional Animal Care and Use Committee (IACUC), according to the Guide for the Care and Use of Laboratory Animals from the National Research Center of the National Academies.
Abbreviations
- AdCre
Adenovirus encoding for Cre recombinase
- ANOVA
Analysis of variance
- BSA
Bovine serum albumin
- CD
Cluster of differentiation
- CTLA-4
Cytotoxic T lymphocyte antigen-4
- DAB
3,3′-Diaminobenzidine
- DE
Differentially expressed
- DMEM
Dulbecco’s modified Eagle’s medium
- EDTA
Ethylenediaminetetraacetic acid
- ELISA
Enzyme-linked immunosorbent assay
- HRP
Horseradish peroxidase
- IFN
Interferon
- IHC
Immunohistochemistry
- IL-2
Interleukin 2
- IP
Intraperitoneal
- IPA
Ingenuity Pathway Analysis
- LAMP-1
Lysosome-associated membrane protein-1
- MUC1
Mucin 1
- NK
Natural killer
- OSE
Ovarian surface epithelium
- PD-1
Programmed death-1
- PD-L1
Programmed death-ligand 1
- SHP-1
Src homology region 2 domain-containing phosphatase-1
- TILs
Tumor-infiltrating lymphocytes
- Tg
Transgenic
- Tregs
Regulatory T cells
- TMB
Tetramethylbenzidine
- WT
Wild type
References
- 1.Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014;384:1376–1388. doi: 10.1016/S0140-6736(13)62146-7. [DOI] [PubMed] [Google Scholar]
- 2.Davis A, Tinker AV, Friedlander M. “Platinum resistant” ovarian cancer: what is it, who to treat and how to measure benefit? Gynecol Oncol. 2014;133:624–631. doi: 10.1016/j.ygyno.2014.02.038. [DOI] [PubMed] [Google Scholar]
- 3.Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med. 2014;65:185–202. doi: 10.1146/annurev-med-092012-112807. [DOI] [PubMed] [Google Scholar]
- 4.Finn OJ (2012) Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol. 23(Suppl 8):viii6–viii9. doi:10.1093/annonc/mds256 [DOI] [PMC free article] [PubMed]
- 5.Hwang WT, Adams SF, Tahirovic E, Hagemann IS, Coukos G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol. 2012;124:192–198. doi: 10.1016/j.ygyno.2011.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA. 2005;102:18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257:56–71. doi: 10.1111/imr.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ostrand-Rosenberg S, Horn LA, Haile ST. The programmed death-1 immune-suppressive pathway: barrier to antitumor immunity. J Immunol. 2014;193:3835–3841. doi: 10.4049/jimmunol.1401572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haile ST, Horn LA, Ostrand-Rosenberg S. A soluble form of CD80 enhances antitumor immunity by neutralizing programmed death ligand-1 and simultaneously providing costimulation. Cancer Immunol Res. 2014;2:610–615. doi: 10.1158/2326-6066.CIR-13-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harvey RD. Immunologic and clinical effects of targeting PD-1 in lung cancer. Clin Pharmacol Ther. 2014;96:214–223. doi: 10.1038/clpt.2014.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naidoo J, Page DB, Wolchok JD. Immune modulation for cancer therapy. Br J Cancer. 2014;111:2214–2219. doi: 10.1038/bjc.2014.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–562. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- 19.Abiko K, Mandai M, Hamanishi J, et al. PD-L1 on tumor cells is induced in ascites and promotes peritoneal dissemination of ovarian cancer through CTL dysfunction. Clin Cancer Res. 2013;19:1363–1374. doi: 10.1158/1078-0432.CCR-12-2199. [DOI] [PubMed] [Google Scholar]
- 20.Hamanishi J, Mandai M, Iwasaki M, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci USA. 2007;104:3360–3365. doi: 10.1073/pnas.0611533104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Budiu RA, Elishaev E, Brozick J, Lee M, Edwards RP, Kalinski P, Vlad AM. Immunobiology of human mucin 1 in a preclinical ovarian tumor model. Oncogene. 2013;32:3664–3675. doi: 10.1038/onc.2012.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lau SK, Weiss LM, Chu PG. Differential expression of MUC1, MUC2, and MUC5AC in carcinomas of various sites: an immunohistochemical study. Am J Clin Pathol. 2004;122:61–69. doi: 10.1309/9R6673QEC06D86Y4. [DOI] [PubMed] [Google Scholar]
- 23.Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11:63–70. doi: 10.1038/nm1173. [DOI] [PubMed] [Google Scholar]
- 24.Liu H, Zhang W, Jia Y, et al. Single-cell clones of liver cancer stem cells have the potential of differentiating into different types of tumor cells. Cell Death Dis. 2013;4:e857. doi: 10.1038/cddis.2013.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Budiu RA, Mantia-Smaldone G, Elishaev E, Chu T, Thaller J, McCabe K, Lenzner D, Edwards RP, Vlad AM. Soluble MUC1 and serum MUC1-specific antibodies are potential prognostic biomarkers for platinum-resistant ovarian cancer. Cancer Immunol Immunother. 2011;60:975–984. doi: 10.1007/s00262-011-1010-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vlad AM, Budiu RA, Lenzner DE, et al. A phase II trial of intraperitoneal interleukin-2 in patients with platinum-resistant or platinum-refractory ovarian cancer. Cancer Immunol Immunother. 2010;59:293–301. doi: 10.1007/s00262-009-0750-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith JB, Stashwick C, Powell DJ., Jr B7-H4 as a potential target for immunotherapy for gynecologic cancers: a closer look. Gynecol Oncol. 2014;134:181–189. doi: 10.1016/j.ygyno.2014.03.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013;73:6900–6912. doi: 10.1158/0008-5472.CAN-13-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romano S, Nappo G, Sorrentino A, Romano MF. The large immunophilin FKBP51 in apoptosis and cancer. Curr Med Chem. 2011;18(35):5424–5429. doi: 10.2174/092986711798194333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu F, Ng SS, Chow BK, Sze J, Lu G, Poon WS, Kung HF, Lin MC. Knockdown of interferon-induced transmembrane protein 1 (IFITM1) inhibits proliferation, migration, and invasion of glioma cells. J Neurooncol. 2011;103:187–195. doi: 10.1007/s11060-010-0377-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luborsky JL, Barua A, Shatavi SV, Kebede T, Abramowicz J, Rotmensch J. Anti-tumor antibodies in ovarian cancer. Am J Reprod Immunol. 2005;54:55–62. doi: 10.1111/j.1600-0897.2005.00287.x. [DOI] [PubMed] [Google Scholar]
- 32.Muhlbauer M, Fleck M, Schutz C, Weiss T, Froh M, Blank C, Scholmerich J, Hellerbrand C. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J Hepatol. 2006;45:520–528. doi: 10.1016/j.jhep.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 33.Lengyel E, Burdette JE, Kenny HA, Matei D, Pilrose J, Haluska P, Nephew KP, Hales DB, Stack MS. Epithelial ovarian cancer experimental models. Oncogene. 2014;33:3619–3633. doi: 10.1038/onc.2013.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenaway J, Moorehead R, Shaw P, Petrik J. Epithelial-stromal interaction increases cell proliferation, survival and tumorigenicity in a mouse model of human epithelial ovarian cancer. Gynecol Oncol. 2008;108:385–394. doi: 10.1016/j.ygyno.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 35.Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, Persons DL, Smith PG, Terranova PF. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 2000;21:585–591. doi: 10.1093/carcin/21.4.585. [DOI] [PubMed] [Google Scholar]
- 36.Kimura T, Finn OJ. MUC1 immunotherapy is here to stay. Expert Opin Biol Ther. 2013;13:35–49. doi: 10.1517/14712598.2012.725719. [DOI] [PubMed] [Google Scholar]
- 37.Cheever MA, Allison JP, Ferris AS, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12:239–252. doi: 10.1038/nri3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fremd C, Schuetz F, Sohn C, Beckhove P, Domschke C. B cell-regulated immune responses in tumor models and cancer patients. Oncoimmunology. 2013;2:e25443. doi: 10.4161/onci.25443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.