

Abstract
The development of a cation clock method based on the intramolecular Sakurai reaction for probing the concentration dependence of the nucleophile in glycosylation reactions is described. The method is developed for the sulfoxide and trichloroacetimidate glycosylation protocols. The method reveals that O-glycosylation reactions have stronger concentration dependencies than C-glycosylation reactions consistent with a more associative, SN2-like character. For the 4,6-O-benzylidene-directed mannosylation reaction a significant difference in concentration dependence is found for the formation of the β- and α-anomers suggesting a difference in mechanism and a rationale for the optimization of selectivity regardless of the type of donor employed. In the mannose series the cyclization reaction employed as clock results in the formation of cis and trans-fused oxabicyclo[4,4,0]decanes as products with the latter being strongly indicative of the involvement of a conformationally mobile transient glycosyl oxocarbenium ion. With identical protecting group arrays cyclization in the glucopyranose series is more rapid than in the mannopyranose manifold. The potential application of related clock reactions in other carbenium ion-based branches of organic synthesis is considered.
Introduction
In the realm of glycoscience the formation of glycosidic bonds reigns over all other covalent bond forming processes, being central to the preparation of homogeneous glycoconjugates and (oligo)saccharides of all shades. Dating back more than one hundred and thirty years to the Michael synthesis of aryl glycosides,1 the Fischer glycosidation,2 and the Koenigs-Knorr reactions,3 the chemical synthesis of glycosidic bonds has been extensively researched through the investigation of innumerable combinations of glycosyl donors, glycosyl acceptors, promoters, solvents, temperature and additives. Indeed, if the year 1994 is taken as representative of the annual level of activity in the last several decades,4 it could be suggested that the entire history of glycosidic bond forming reactions can be classified as a century-long, world-wide exercise in the combinatorial exploration of reaction conditions. This situation arises in part because of the enormous variety of glycosidic linkages possible, both natural and artificial, but also because of uncertainty in the location of any one particular reaction on the SN1-SN2 continuum of mechanisms for nucleophilic substitution reactions. It follows that any given glycosylation reaction might be more rationally optimized if its mechanism could be pinpointed, or at least localized, without undue expenditure of effort.
According to Ingold and Hughes,5 the mechanism of a substitution reaction is characterized by its stereochemistry and its kinetics, yet the enormous majority of glycosylation reactions reported in the literature are discussed only in terms of their stereoselectivity. There is no doubt that this situation pertains because of the preparative significance of the selectivity, but it is also to some extent due to the difficulty in conducting kinetic studies on many glycosylation reactions. The role of ion pairs in nucleophilic substitution reactions in general has been appreciated6–7 since Winstein’s seminal contribution,8 and in glycosylation in particular since the work of the Vernon,9 Schuerch,10 and Lemieux groups,11 yet the glycosylation literature is replete with depictions of naked glycosyl oxocarbenium ions lacking their essential counterions, a fact which is even more surprising when the sparse physical evidence for existence of such glycosyl oxocarbenium ions is taken into account.12–20 Even admitting the transient existence of glycosyl oxocarbenium ions, the widespread (and disputed21–22) application of basic stereoelectronic principles23–24 to the rationalization of their face selectivity is of a relatively recent vintage.25–26
In our laboratory, driven by the desire to rationalize the benzylidene-directed β-mannopyranosylation,27–28 and conscious of the major contribution of kinetic isotope effect (KIE) measurements to the understanding of the mechanisms of chemical and enzymatic glycosidic bond hydrolysis and transfer,29–33 we have introduced34–35 variants on the Singleton NMR method36 for the determination of kinetic isotope effects in glycosylation reactions. Nevertheless, however informative these experiments, they are highly instrument intensive and consequently not routinely applicable. Therefore, we sought simple alternative methods for the determination of the relative kinetics and molecularity of glycosylation reactions such as might be applied in synthetic glycochemistry laboratories and so inform the rational optimization of glycosylation reactions. To this end we turned to the development of competition experiments for assessing the relative kinetics of two reactions, a concept that is deeply ingrained in the field of organic (and inorganic) chemistry.37–54
In carbohydrate chemistry the Jencks azide clock reaction played an important role in estimating the lifetimes of transient glycosyl oxocarbenium ions in aqueous media,49–50 but it is not adaptable to the study of glycosylation reactions in organic solution at low temperature. Seeking an operational simple method we focused on cyclization reactions as unimolecular clocks for the investigation of intermolecular glycosylation reactions. In this Article we report in full on our initial design55 of such glycosylation clock reactions, present further examples, discuss the evidence for the transient formation of certain glycosyl oxocarbenium ion intermediates, and apply the results to rationalize the variation of glycosylation stereoselectivity according to conditions, most notably the passage from solution to solid phase. Although this Article focuses on glycosylation and oxocarbenium ions, we note that the concept of cyclization reactions as simple cation clocks for estimating the molecularity of nucleophilic attack on transient carbenium ions should, with suitable adaptation, find application in cognate fields.
Results and Discussion
Design
Our design of cyclization reactions for use of intramolecular glycosylation clocks was informed by the general concept of intramolecular aglycone delivery56–60 and, more particularly, by two instances in our own laboratories of the cyclization of protecting groups at O2 of glycosyl donors onto the anomeric center in the course of previous studies (Scheme 1). Thus it was observed61 that on warming above 5 °C in CD2Cl2 the α-mannosyl triflate 1 underwent decomposition with cyclization onto a benzyl group to give the tricyclic product 2, which was isolated in 56% yield. On the other hand, the corresponding α-mannosyl triflate derived by in situ activation of the sulfoxide 3 underwent cyclization at −78 °C in CH2Cl2 onto the naphthylpropargyl system in competition with reaction with an external alcohol,62 with yields varying as an inverse function of the reactivity of the external alcohol. In contrast, simple allyl ethers,63–65 propargyl ethers66–67 and [3-(4-trifluoromethylphenyl)propargyl] ethers68–69 may be employed as protecting groups for the O2-postion in mannosylation reactions without complications arising from cyclization. Thus, while the precedent certainly gave rise to the potential for the use of carbon-carbon bond forming cyclization reaction onto O2 protecting groups as an intramolecular clock reaction for glycosylation reactions, it also highlighted the sensitivity of such cyclizations to the structure and reactivity of the nucleophilic function.
Scheme 1.

Cyclization of Protecting Groups at the 2-Position onto the Anomeric Center.
Recalling Denmark’s use of allylsilanes as nucleophiles in a series of cyclization-based probes designed to interrogate the mechanism of the Lewis acid promoted reaction of allylsilanes with acetals (Scheme 2)70 and other instances of the intramolecular Sakurai reaction,71 we designed an initial system 5 (Scheme 3) employing a 2-O-(2-trimethylsilylmethyl)allyl group as intramolecular nucleophile. In addition to excluding the formation of an analysis-complicating additional stereogenic center, as observed in the model cyclization of 3 to 4 (Scheme 1), this system takes advantage of the much increased nucleophilicity of allylsilanes over simple alkenes in their reaction with carbocations,72 thereby increasing the likelihood that cyclization will compete with trapping by an external alcohol nucleophile. Finally, the system envisaged finds precedent in the well-known C-glycoside-forming intermolecular reaction of allylsilanes with putative anomeric oxocarbenium ions.25–26,73–90 The use of more potent tethered nucleophiles, such as alcohols, for the clock cyclization was not explored as it was considered that the high effective molarity of an intramolecular alcohol would lead to an excessively fast cyclization that would outcompete the intermolecular process.
Scheme 2.
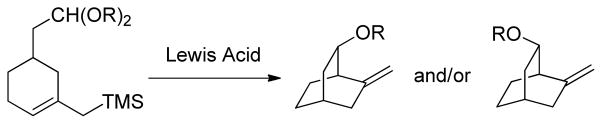
Use of an Intramolecular Allylsilane-Acetal Reaction as a Probe of Mechanism.
Scheme 3.
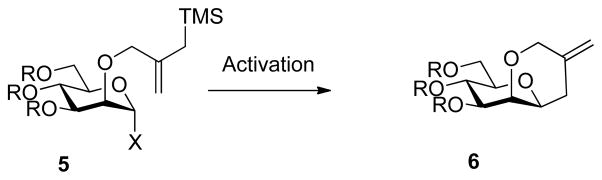
Initial Clock Cyclization Design.
Because of the important role that the 4,6-O-benzylidene protected α-mannopyranosyl triflates91 have played in current debate on glycosylation mechanisms,12,15–20,27–28,92–98 the initial work was focused on the use of the mannosyl sulfoxides as precursors of the mannosyl triflates. Conscious of the ever-widening range of donor types capable of providing β-mannopyranosides when used in conjunction with the 4,6-O-benzylidene or a related acetal,93–94,99–110 in this study we extend the glycosyl clock method to include the use of the very popular glycosyl trichloroacetimidates.111 The method is also extended to the 3,4,6-tri-O-benzyl mannopyranoside and the 3-O-benzyl-4,6-O-benzylidene-glucopyranoside series, although it must be recognized that this does not permit direct comparisons of relative glycosylation rates between the differing series owing to structural differences in the clock cyclizations.
Synthesis
The synthesis of the 4,6-O-benzylidene mannopyranosyl sulfoxide clock began with phenyl 4,6-O-benzylidene-α-D-thiomannopyranoside 7112–114 while that of the corresponding trichloroacetimidate followed a parallel path from 2-phenylthioethyl 4,6-O-benzylidene-α-D-mannopyranoside 8 (Scheme 4), which was prepared by standard means from pentaacetyl mannopyranose and phenylthioethanol (Supporting Information). Standard115 regioselective monobenzylation of 7 and 8 with dibutyltin oxide, cesium fluoride and benzyl bromide gave 9116 and 10, respectively, ready for the installation of the allylsilane moiety. In the phenyl thioglycoside series studied initially this was achieved with sodium hydride and commercial 2-(chloromethyl)allyl trimethylsilane in hot THF and gave the anticipated product 11 in 47% yield after 7 days. Subsequently, working with the phenylthioethyl glycosides, we have preferred initial conversion of the chloromethylallysilane to the corresponding iodomethylallylsilane117 with sodium iodide in acetone followed by reaction with substrate and sodium hydride in THF at 0 °C in the presence of 15-crown-5 when the product 12 was obtained in 81% with greatly reduced reaction times. Controlled oxidation of the thioglycoside 11 with mCPBA in dichloromethane at −72 °C gave the sulfoxide 13 as a 16:1 mixture of diastereomers in which the major isomer is assigned the R configuration at sulfur consistent with the precedent.118–120 Treatment of the phenylthioethyl glycoside with lithium naphthenalide121 in THF at −78 °C gave the mannopyranose 14 in 78% yield, which was converted to the α-trichloroacetimidate 15 in 78% yield on reaction with trichloroacetonitrile in the presence of DBU122 (Scheme 4).
Scheme 4.

Synthesis of the 4,6-O-Benzylidene-protected Mannopyranosyl Clocks
The synthesis of the corresponding 3,4,6-tri-O-benzyl mannopyranosyl sulfoxide clock 18 was achieved analogously from phenyl 3,4,6-tri-O-benzyl-α-D-thiomannopyranoside 16123 (Scheme 5).
Scheme 5.

Synthesis of the Perbenzyl Mannopyranosyl Clock
In the 4,6-O-benzylidene glucopyranose series (Scheme 6) synthesis of the sulfoxide clock 21 set out from the 3-O-benzyl derivative 19,124 of which alkylation with sodium hydride and iodomethylallyl trimethylsilane in hot THF gave 20 albeit only in poor yield. Oxidation with mCPBA then gave a 3:2 mixture of the two isomers of the sulfoxide 21 in 76% yield (Scheme 6), with the major isomer assigned as the R configuration at sulfur consistent with the precedent.119–120 The two diastereomers of 21 were separable chromatographically but were typically used as mixtures in the kinetic runs that follow. The synthesis of the trichloroacetimidate clock 27 began with 2-phenylthioethyl 1,2,4,6-tetra-O-acetyl-β-D-glucopyranoside125 22, itself obtained by standard means from 3-O-benzyl-1,2;5,6-di-O-isopropylidene-β-D-glucofuranose (Supporting Information). Thus, saponification of 22 gave the triol 23 onto which the benzylidene group was installed in the usual manner to afford 2-phenylthioethyl 3-O-benzyl-4,6-O-benzilidene-β-D-glucopyranose 24. Trimethylsilylmethallylation under the improved conditions then gave 25 in 89% yield. Finally, cleavage of the phenylthioethyl group was achieved with lithium naphthenalide to give 26, and the trichloroacetimidate was installed with the aid of sodium hydride122 affording the trichloroacetimidate clock 27 (Scheme 6).
Scheme 6.

Synthesis of the 4,6-O-Benzylidene-protected Glucopyranosyl Clocks
Clock Cyclization Reactions
Treatment of the 4,6-O-benzylidene-protected mannosyl sulfoxide 13 with triflic anhydride in dichloromethane at −72 °C in the presence of the hindered non-nucleophilic base 2,4,6-tri-tert-butylpyrimidine (TTBP) with quenching at −72 °C after stirring for 2.5 h resulted in the isolation of two tricyclic products 28 and 29 in 45 and 25% isolated yield, respectively (Chart 1, Table 1, entry 1). Attempted activation of the corresponding trichloroacetimidate 15 with catalytic TMSOTf at the same temperature resulted in a very slow reaction. However, raising the temperature to −20 °C enabled a rapid smooth reaction resulting in the isolation of 28 and 29 in 70 and 25 % yields, respectively (Chart 1, Table 1, entry 2). We note that the reported use of 3-O-allyl-2-O-benzyl-4,6-O-benzylidene-α-D-mannopyranosyl trichloroacetimidate as a β-selective mannosyl donor was conducted at −50 °C,99 presumably also for reasons of reduced reactivity in standard dry ice/acetone cooling baths. The 3,4,6-tri-O-benzyl protected mannopyranosyl sulfoxide 18 cyclized smoothly on activation with triflic anhydride in the presence of TTBP at −72 °C giving the two bicyclic products 30 and 31 in 52 and 20% isolated yield, respectively (Chart 1, Table 1, entry 3). In the glucopyranose series, however, working with the sulfoxide clock 21 complications arising from sulfenyl transfer to the allylsilane were dominant and resulted in the formation of the anticipated cyclization product 32 in only low yield. Sulfenyl group transfer to substrate-based nucleophiles has been observed previously and can typically be suppressed by working in the presence of a sacrificial alkene.62,126 However, even working in the presence of multiple equivalents of 1-octene or the highly reactive β-pinene127–128 we were unable to obtain a satisfactory yield of 32 (Chart 1, Table 1, entry 4). This observation, coupled with the relative paucity of such side reactions in the mannose series (Table 1, entries 1–3), suggests an intramolecular process and caused us to abandon the use of the gluco-configured sulfoxide 21 in clock reactions. Fortunately, the glucosyl trichloroacetimidate 27 behaved in the anticipated manner resulting in a 92% isolated yield of the cyclization product 32 following activation with TMSOTf in the presence of TTBP at −20 °C (Chart 1, Table 1, entry 5).
Chart 1.

Clock Cyclization Products
Table 1.
Cyclization Reactions in the Absence of Added External Nucleophile
| Entry | Substrate | Conditions | Products, % yield, (pyranose conformation, method) |
|---|---|---|---|
| 1 | 13 | Tf2O, TTBP, −72 °C | 28, 45% (4C1, X-ray, NMR) + 29, 25% (1S5, X-ray, NMR) |
| 2 | 15 | TMSOTf, TTBP, −20 °C | 28, 70% + 29, 25% |
| 3 | 18 | Tf2O, TTBP, −72 °C, 1-octene | 30, 52% (4C1, X-ray, NMR), + 31, 20% (1S5, NMR) |
| 4 | 21 | Tf2O, TTBP, −72 °C, 1-octene or β-pinene | 32, 20% (1S5, X-ray, NMR), |
| 5 | 27 | TMSOTf, TTBP, −20 °C | 32, 92% |
The structure of the manno-configured trans,trans-tricyclic system 29, with the pyranose ring in the 1S5 twist boat conformation and the benzylidene and newly formed rings in chair conformations, was confirmed by X-ray crystallography (CSD refcode YEYDUD55). That the overall conformation of 29 does not change on going from the crystal to the solution phase is apparent from the 10.5 Hz coupling of the vicinal trans-pseudo-diaxial hydrogen atoms at the bridgehead positions of the newly formed ring. Crystals of the trans-fused bicyclic analog 31 suitable for X-ray crystallographic analysis could not be obtained, however, the 1S5 twist boat conformation of the pyranose ring is again revealed by the large coupling constant between the two bridgehead hydrogens. The major cis-fused products 28 and 30 in the mannose series were both examined crystallographically (CSD Refcode YEYFAL55 and CCDC 1403928) and were both found to contain only chair conformers of the various six-membered rings. The single cyclization product 32 formed in the glucopyranose series (Table 1, entries 4 and 5) also adopts the 1S5 twist boat conformation of the pyranose ring in the crystal (CCDC 878445), while the benzylidene and newly-formed rings take up chair conformations. This conformation is also retained in solution as indicated by the 3J1,2 and 3J2,3 (glucose numbering) coupling constants of 3.8 and 5.2 Hz, respectively.
The formation of the trans-fused products 29 and 31 as minor isomers from the cyclization of the mannopyranosyl clocks 13, 15, and 18 (Table 1, entries 1–3) is instructive and points to the formation of a mannopyranosyl oxocarbenium ion as transient intermediate that populates the B2,5 or related conformation. Thus, the mannosyl triflate91 33 formed on activation of the donor can be considered to be in equilibrium with the mannosyl oxocarbenium/triflate ion pair 34, which can adopt several conformations. The 4H3 and B2,5 conformations of 34 are accessible for both the 4,6-O-benzylidene and tri-O-benzyl systems, while the latter, being less conformationally constrained, also can access the 3H4 half-chair (Scheme 7). In the B2,5 conformation the pendant allylsilane adopts a pseudoequatorial position and is poised to react with either face of the oxocarbenium ion, thereby enabling the formation of the cis and trans-fused products (Scheme 7). Formation of the trans-fused product is also possible from the 3H4 conformer in the case of the tri-O-benzyl protected system. The major cis-fused product can be formed from any of the three accessible conformations (Scheme 7).
Scheme 7.

Conformation and Selectivity of the Mannopyranosyl Oxocarbenium Ion
In the glucopyranose system there are only two conformations, 4H3 and B2,5, of the oxocarbenium ion/triflate ion pair 36 in equilibrium with the initially formed covalent triflate 35, and both lead to the cis-fused product 32 (Scheme 8). The excellent cis-selectivity observed in this kinetic ring closure contrasts with the thermodynamic trans-selectivity seen on ring closure of 2-O-(2-thioethyl)glucopyranosyl cations and related systems to hetero-bicyclo[4.4.0]decane-like systems, which are governed by the steric factors in the product.129–131 The preference of the tricyclic glucose derivative 32 for the 1S5 conformation of the pyranose ring as opposed to the 4C1 chair must arise because of a combination two unfavorable steric interactions in the chair: the 1,3-diaxial interaction between the axial anomeric CC bond and the axial C3-H3 bond and the gauche butane interaction emphasized in red in Scheme 8. This is because simple 4,6-O-benzylidene protected α-C-glucopyranosides, with an axial substituent at C1 but lacking the third ring, exist predominantly as 4C1 conformers (vide infra).80–81
Scheme 8.

Conformation and Selectivity of the Glucopyranosyl Oxocarbenium Ion
Schemes 7 and 8 employ triflate as the counterion given that activations were conducted with either triflic anhydride or TMSOTf for the sulfoxide and trichloroacetimidate donors, respectively. This does not restrict the general concept of the cation clock method to the use of triflate-based activating systems and triflates as counterions. The concept is equally valid for glycosylation reactions conducted with other activating systems and leaving groups.
Competition Kinetics
For the sulfoxide donors competition kinetics were conducted at −72 °C in CH2Cl2 by addition of triflic anhydride to a mixture of the donor and TTBP followed by addition of incremental amounts of isopropanol or methallyltrimethylsilane as glycosyl acceptor. After stirring for 5 min at −72 °C the reactions were quenched at that temperature, worked up, and the product ratios analyzed by UHPLC giving the data presented in Tables 2 and 4. The crude reaction mixtures from multiple runs were combined and subjected to purification over silica gel yielding pure samples of the glycosides 37 – 41 (Chart 2). For the trichloroacetimidates 15 and 27, TMSOTf was simply added to preformed mixtures of the donor and acceptors at −20 °C followed by continued stirring at that temperature. After quenching at −20 °C reaction mixtures were again analyzed by UHPLC (Tables 3 and 5), and pooling of crude reaction mixtures afforded sufficient material for the chromatographic purification and full characterization of the glycosides 42–44 (Chart 2). The data from Tables 2–5 are presented graphically in the form of plots of glycoside/cyclized product ratios against acceptor concentration in Figures 1a–d.
Table 2.
O - and C-Mannosylation with Donor 13.
| Entry | Nucleophile | Equiv (M conc) | β-gly/cycl | α-gly/cycl |
|---|---|---|---|---|
|
| ||||
| 37/(28+29)c | 38/(28+29)c | |||
| 1a | iPrOH | 0.8 (0.014) | 2.17 | 0.15 |
| 2a | iPrOH | 1.2 (0.020) | 3.66 | 0.28 |
| 3a | iPrOH | 1.5 (0.026) | 5.36 | 0.44 |
| 4a | iPrOH | 2.5 (0.043) | 10.99 | 0.99 |
| 5a | iPrOH | 3 (0.051) | 13.09 | 1.28 |
| 6a | iPrOH | 4 (0.068) | 15.75 | 1.14 |
| 7a | iPrOH | 5 (0.085) | 19.38 | 1.53 |
| 8a | iPrOH | 8 (0.136) | 24.34 | 1.60 |
| 39/(28+29)c | - | |||
| 9b | TMSCH2C(Me)=CH2 | 2 (0.034) | 0.06 | - |
| 10b | TMSCH2C(Me)=CH2 | 4 (0.068) | 0.18 | - |
| 11b | TMSCH2C(Me)=CH2 | 8 (0.136) | 0.40 | - |
| 12b | TMSCH2C(Me)=CH2 | 12 (0.204) | 0.55 | - |
| 13b | TMSCH2C(Me)=CH2 | 15 (0.255) | 0.69 | - |
| 14b | TMSCH2C(Me)=CH2 | 20 (0.34) | 0.87 | - |
| 15b | TMSCH2C(Me)=CH2 | 30 (0.51) | 1.40 | - |
Experimental conditions: TTBP (4 equiv), 1-octene (10 equiv), molecular sieves 4 Å, Tf2O (1.2 equiv) at −72 °C;
Experimental conditions: TTBP (4 equiv), molecular sieves 4 Å, Tf2O (1.2 equiv) at − 72 °C;
Molar ratios were determined by UHPLC/UV/MS
Table 4.
O-Mannosylation with Donor 18.
| Entry | Nucleophile | Equiv (M conc) | β-gly/cycl | α-gly/cycl |
|---|---|---|---|---|
|
| ||||
| 40/(30+31)c | 41/(30+31)c | |||
| 1a | iPrOH | 1 (0.019) | 0.27 | 0.27 |
| 2a | iPrOH | 2 (0.038) | 0.95 | 0.85 |
| 3a | iPrOH | 2.5 (0.048) | 1.11 | 1.08 |
| 4a | iPrOH | 3 (0.057) | 1.27 | 1.20 |
| 5a | iPrOH | 4 (0.076) | 1.60 | 1.39 |
| 6a | iPrOH | 5 (0.095) | 1.75 | 1.67 |
| 7a | iPrOH | 7.5 (0.143) | 1.84 | 1.73 |
| 8a | iPrOH | 10 (0.190) | 1.87 | 1.78 |
Experimental conditions: TTBP (10 equiv), β-pinene (30 equiv), molecular sieves 4 Å, Tf2O (1.2 equiv) at − 60 °C;
Molar ratios were determined by UHPLC/UV/MS
Chart 2.

Glycosides From Competition Kinetic Experiments
Table 3.
O- and C-Mannosylation with Donor 15.
| Entry | Nucleophile | Equiv (M conc) | β-gly/cycl | α-gly/cycl |
|---|---|---|---|---|
|
| ||||
| 37/(28+29)c | 38/(28+29)c | |||
| 1a | iPrOH | 0.81 (0.013) | 2.5702 | 0.30827 |
| 2a | iPrOH | 1.63 (0.026) | 5.0541 | 0.54899 |
| 3a | iPrOH | 2.4 (0.052) | 10.1031 | 1.33043 |
| 4a | iPrOH | 3.25 (0.078) | 13.3632 | 1.83040 |
| 5a | iPrOH | 4.88 (0.104) | 19.7443 | 3.20719 |
| 6a | iPrOH | 8.12 (0.130) | 24.2748 | 4.83401 |
| 39/(28+29)c | - | |||
| 7b | TMSCH2C(Me)=CH2 | 2.1 (0.032) | 0.0729 | - |
| 8b | TMSCH2C(Me)=CH2 | 5.3 (0.085) | 0.1046 | - |
| 9b | TMSCH2C(Me)=CH2 | 10.7 (0.171) | 0.2996 | - |
| 10b | TMSCH2C(Me)=CH2 | 16.0 (0.256) | 0.4712 | - |
| 11b | TMSCH2C(Me)=CH2 | 21.3 (0.341) | 0.6673 | - |
| 12b | TMSCH2C(Me)=CH2 | 26.7(0.427) | 0.9509 | - |
| 13b | TMSCH2C(Me)=CH2 | 32.0 (0.512) | 1.1419 | - |
Experimental conditions: TTBP (0.3 equiv), TMSOTf (0.3 equiv.) at −20 °C; molecular sieves 4 Å;
Experimental conditions: TTBP (0.3 equiv), TMSOTf(0.3 equiv.) at −20 °C; molecular sieves 4 Å;
Molar ratios were determined by UHPLC/UV/MS
Table 5.
O- and C-Glucosylation with Donor 27.
| Entry | Nucleophile | Equiv (M conc) | β-gly/cycl | α-gly/cycl |
|---|---|---|---|---|
|
| ||||
| 42/(32)c | 43/(32)c | |||
| 1a | iPrOH | 1 (0.048) | 0.0067 | 0.0227 |
| 2a | iPrOH | 1.5 (0.072) | 0.0243 | 0.0708 |
| 3a | iPrOH | 2 (0.096) | 0.0550 | 0.1297 |
| 4a | iPrOH | 3 (0.144 | 0.1143 | 0.2498 |
| 5a | iPrOH | 5 (0.239) | 0.4043 | 0.5135 |
| 6a | iPrOH | 8 (0.382) | 0.9845 | 1.0176 |
| 44/(32)c | ||||
| 7b | TMSCH2C(Me)=CH2 | 23.8 (1.14) | - | 0.0899 |
| 8b | TMSCH2C(Me)=CH2 | 35.7 (1.71) | - | 0.1078 |
| 9b | TMSCH2C(Me)=CH2 | 47.6 (2.28) | - | 0.1273 |
| 10b | TMSCH2C(Me)=CH2 | 59.5 (2.85) | - | 0.1957 |
| 11b | TMSCH2C(Me)=CH2 | 71.4 (3.41) | - | 0.2331 |
Experimental conditions: TTBP (0.3 equiv), TMSOTf (0.3 equiv.) at −20 °C; molecular sieves 4 Å;
Experimental conditions: TTBP (0.3 equiv), TMSOTf (0.3 equiv.) at −20 °C; molecular sieves 4 Å;
Molar ratios were determined by UHPLC/UV/MS
Figure 1.

Graphic Representation of Clock Reactions
Comparison of Figures 1a and b reveals that the 4,6-O-benzylidene mannosyl donors 13 and 15 display largely parallel behavior toward both isopropanol and methylallyltrimethylsilane in spite of the different reaction temperatures and leaving groups. Thus, with both 13 and 15 the formation of the β-mannoside 37 shows a much stronger concentration dependence than that of the α-anomer 38, with the latter showing a slightly greater concentration dependence than the formation of the β-C-mannoside 39. Consistent with earlier observations only the β-anomer 39 is formed in the 4,6-O-benzylidene-directed C-mannosylation.80 These observations are most consistent with an SN2-like associative displacement of an axial leaving group for the formation of the β-O-mannoside 37 and of a much more dissociative SN1-like reaction for the formation of the α-O-mannoside 38, as determined previously using 13C-primary kinetic isotope effect measurements for the case of triflate as leaving group in 4,6-O-benzylidene protected mannosyl donors.35 The very low concentration dependence observed for the formation of the C-mannoside 39 is consistent with a highly dissociative mechanism proceeding via an oxocarbenium ion 34 (Scheme 7, R-R = PhCH) that is only loosely associated with the counterion.132 This in turn is consistent with methallyltrimethylsilane being a much weaker nucleophile than isopropanol and requiring the potent electrophile at the dissociative end of the mechanistic spectrum.133
Comparison of Figures 1a and c reveals a very different pattern of behavior between the 4,6-O-benzylidene and perbenzyl protected mannosyl donors 13 and 18. Thus, in contrast to the case of 13, both the β- and α-perbenzyl mannosides 40 and 41 formed from 18 on coupling to isopropanol display essentially the same concentration dependence. Superposition of Figures 1a and c, as in Figure 1e, reveals the very shallow concentration dependence for the formation of 40 and 41 to approximate that for the formation of the 4,6-O-benzylidene protected α-mannoside 38 and strongly suggests that 40 and 41 are formed by dissociative mechanisms that involve the oxocarbenium ion 34 (Scheme 7, R = Bn) only loosely associated with the counterion. This observation is consistent with the strongly disarming nature of the 4,6-O-benzylidene acetal,52,134–136 as compared to two benzyl ethers, which in turn is a function of the imposition of the trans-gauche conformation137 of the C5-C6 bond in which the electron-withdrawing effect of the C5-O5 bond is maximized.138–140 A caveat to this argument concerns the use of different clock reactions in Figures 1a and c (vide infra), however, we believe that the comparison is justified here in view of the similarity of the two oxocarbenium ions (Scheme 7, 34, R = Bn and R-R = PhCH).
Comparison of Figures 1b and d reveals the very different influence of the 4,6-O-benzylidene acetal protecting group in the manno- and glucopyranose series. Thus, in contrast to the benzylidene-protected O-mannosylation, both anomers 42 and 43 of the benzylidene-protected glucosides are formed with the same concentration dependence. The formation of both O-glucosides 42 and 43 shows a much stronger concentration dependence than that of the corresponding C-glucoside 44 (Figure 1d), which was formed as a single α-anomer consistent with previous reports.80–81 If C-glucoside formation is interpreted as representative of the concentration dependence of a dissociative SN1-like mechanism with a weak nucleophile as in the mannose case, these results are again consistent with primary 13C kinetic isotope effect studies on benzylidene-protected O-glucosylation with triflate as leaving group which point to both anomers being formed by associative SN2-like mechanisms from a pair of rapidly equilibrating anomeric glucosyl triflates.35 The inherent difference in cyclization rates between the mannosyl and glucosyl clocks are most readily appreciated from a direct comparison of C-glycoside formation in the two series (Figure 1f). Thus, in order for C-glycosylation to compete with the cyclization of 27 to 32 significantly more methallylsilane is required than for C-glycosylation to compete with the cyclization of 15 to 28 and 29 under the same reaction conditions. It follows that C-glycosylation in the glucose series is less concentration dependent than in the mannose series suggesting that it is closer to a pure SN1 mechanism.
Conclusions
A cation clock method based on the intramolecular Sakurai reaction has been developed and used to probe the concentration dependence of representative O- and C-glycosylation reactions. The method is applicable to the use of glycosyl sulfoxides with activation by triflic anhydride and for the use of glycosyl trichloroacetimidates with activation by trimethylsilyl triflate. 4,6-O-Benzylidene-directed β-mannosylation is demonstrated to proceed with a strong dependence on the concentration of the acceptor alcohol, whereas the α-anomer is much less concentration dependent. Concentration therefore plays a critical role in the 4,6-O-benzylidene-directed β-mannosylation process and can be used to optimize selectivity. The reduced selectivity observed on trapping of 4,6-O-benzylidene-protected mannosyl donors by polymer-supported alcohols141 can be understood in terms of the reduced concentration of the acceptor. Analogous results are observed in 4,6-O-benzylidene-directed β-mannosylation conducted by the sulfoxide and trichloroacetimidate methods suggesting a commonality of mechanism if not necessarily of leaving group. In the 3,4,6-tri-O-benzyl protected mannopyranosyl and 3-O-benzyl-4,6-O-benzylidene glucopyranosyl systems the formation of both anomers of the isopropyl glycosides display very similar concentration dependencies which, are nevertheless, greater than that of the formation of C-glycosides. The formation of trans-fused products from the clock reaction in the mannopyranose series is interpreted in terms of transient glycosyl oxocarbenium ions that are capable of accessing the B2,5 and/or 3H4 conformers. Further development and application of the method is in progress and will be reported in due course.
Beyond the immediate context of carbohydrate chemistry and the glycosylation reaction, the concept of competition kinetics using a simple cyclization as clock reaction should find broader application in estimating the molecularity of other cation-based processes in organic synthesis. Such reactions might include but are not limited to oxocarbenium-like ions in the nucleophilic ring opening of chiral acetals in simple ether-forming reactions and in Mukaiyama-type aldol reactions, acylium ion-based processes, iminium and N-acyl iminium ion chemistry, thiacarbenium ions in Pummerer-type chemistry, and the many reactions of carbenium ions themselves.
Supplementary Material
Acknowledgments
We thank the NIH (GM62160) and the ICSN for financial support, the NSF for funds in support of the 600 MHz spectrometer in the Lumigen Instrument Center at WSU (MRI-084043), and Dr Philip D Martin in the Lumigen Core X-ray Facility at Wayne State University for determining the structure of compound 31. MH is grateful to the Ministère de l’Education Nationale, de la Recherche, et de la Technologie, France for fellowship support, DM thanks the Ministry of Science and Technology, India, for a Boyscast Fellowship.
Footnotes
Supporting Information Available. Full experimental details and 1H and 13C NMR spectra for all new compounds. The Supporting Information is available free of charge on the ACS Publication website.
References
- 1.Michael A. Comptes Rendu. 1879;LXXXIX [Google Scholar]
- 2.Fischer E. Ber Deut Chem Ges. 1893:2400–2412. [Google Scholar]
- 3.Koenigs W, Knorr E. Sitzungsber Bayr Akad Wiss. 1900:103–105. [Google Scholar]
- 4.Barresi F, Hindsgaul O. J Carbohydr Chem. 1995;14:1043–1087. [Google Scholar]
- 5.Ingold CK. Structure and Mechanism in Organic Chemistry. 2. Cornell Univ Press; Ithaca: 1969. [Google Scholar]
- 6.Richard JP, Amyes Tl, Toteva MM, Tsuji Y. Adv Phys Org Chem. 2004;39:1–26. doi: 10.1016/B978-0-12-386047-7.00002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters KS. Chem Rev. 2007;107:859–873. doi: 10.1021/cr068021k. [DOI] [PubMed] [Google Scholar]
- 8.Winstein S, Clippinger E, Fainberg AH, Heck R, Robinson GC. J Am Chem Soc. 1956;78:328–335. [Google Scholar]
- 9.Rhind-Tutt AJ, Vernon CA. J Chem Soc. 1960:4637–4644. [Google Scholar]
- 10.Fréchet JM, Schuerch C. J Am Chem Soc. 1972;94:604–609. [Google Scholar]
- 11.Lemieux RU, Hendriks KB, Stick RV, James K. J Am Chem Soc. 1975;97:4056–4062. [Google Scholar]
- 12.Bohé L, Crich D. CR Chimie. 2011;14:3–16. [Google Scholar]
- 13.Satoh H, Manabe S. Chem Soc Rev. 2013;42:4297–4309. doi: 10.1039/c3cs35457a. [DOI] [PubMed] [Google Scholar]
- 14.Walvoort MTC, van der Marel GA, Overkleeft HS, Codée JDC. Chem Sci. 2013;4:897–906. [Google Scholar]
- 15.Whitfield DM. Adv Carbohydr Chem Biochem. 2009;62:83–159. doi: 10.1016/S0065-2318(09)00004-3. [DOI] [PubMed] [Google Scholar]
- 16.Bohé L, Crich D. Carbohydr Res. 2015;403:48–59. doi: 10.1016/j.carres.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frihed TG, Bols M, Pedersen CM. Chem Rev. 2015;115:4963–5013. doi: 10.1021/cr500434x. [DOI] [PubMed] [Google Scholar]
- 18.Hosoya T, Takano T, Kosma P, Rosenau T. J Org Chem. 2014;79:7889–7894. doi: 10.1021/jo501012s. [DOI] [PubMed] [Google Scholar]
- 19.Hosoya T, Kosma P, Rosenau T. Carbohydr Res. 2015;401:127–131. doi: 10.1016/j.carres.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 20.Hosoya T, Kosma P, Rosenau T. Carbohydr Res. 2015;411:64–69. doi: 10.1016/j.carres.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 21.Sinnott ML. In: The Anomeric Effect and Associated Stereoelectronic Effects. Thatcher GRJ, editor. Vol. 539. ACS; Washington: 1993. pp. 97–113. [Google Scholar]
- 22.Sinnott ML. Adv Phys Org Chem. 1988;24:113–204. [Google Scholar]
- 23.Deslongchamps P. Stereoelectronic Effects in Organic Chemistry. Pergamon; Oxford: 1983. [Google Scholar]
- 24.Kirby AJ. The Anomeric Effect and Related Stereoelectronic Effects at Oxygen. Springer-Verlag; Berlin: 1983. [Google Scholar]
- 25.Lewis MD, Cha JK, Kishi Y. J Am Chem Soc. 1982;104:4976–4978. [Google Scholar]
- 26.Smith DM, Woerpel KA. Org Biomol Chem. 2006;4:1195–1201. doi: 10.1039/b600056h. [DOI] [PubMed] [Google Scholar]
- 27.Crich D. Acc Chem Res. 2010;43:1144–1153. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- 28.Aubry S, Sasaki K, Sharma I, Crich D. Top Curr Chem. 2011;301:141–188. doi: 10.1007/128_2010_102. [DOI] [PubMed] [Google Scholar]
- 29.Sinnott ML. Chem Rev. 1990;90:1171–1202. [Google Scholar]
- 30.Davies GJ, Sinnott ML, Withers SG. In: Comp Biol Catalysis. Sinnott ML, editor. Vol. 1. Academic Press; London: 1998. pp. 119–209. [Google Scholar]
- 31.Horenstein NA. Adv Phys Org Chem. 2006;41:275–314. [Google Scholar]
- 32.Schramm VL. ACS Chem Biol. 2013;8:71–81. doi: 10.1021/cb300631k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan J, Lewis AR, Gilbert M, Karwaski MF, Bennet AJ. Nature Chem Biol. 2010;6:405–407. doi: 10.1038/nchembio.352. [DOI] [PubMed] [Google Scholar]
- 34.Crich D, Chandrasekera NS. Angew Chem Int Ed. 2004;43:5386–5389. doi: 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]
- 35.Huang M, Garrett GE, Birlirakis N, Bohé L, Pratt DA, Crich D. Nat Chem. 2012;4:663–667. doi: 10.1038/nchem.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singleton DA, Thomas AA. J Am Chem Soc. 1995;117:9357–9358. [Google Scholar]
- 37.Francis AW, Hill J, Johnston J. J Am Chem Soc. 1925;47:2211–2232. [Google Scholar]
- 38.Ingold CK, Shaw FR. J Chem Soc. 1927:2918–2926. [Google Scholar]
- 39.Dewar MJS, Mole T, Warford EWT. J Chem Soc. 1956:3576–3580. [Google Scholar]
- 40.Tolgyesi WS. Can J Chem. 1965;43:343–355. [Google Scholar]
- 41.Hammett LP. Physical Organic Chemistry. McGraw-hill Kogakusha; Tokyo: 1970. [Google Scholar]
- 42.Felkin H, Frajerman C. Tetrahedron Lett. 1970;11:1045–1048. [Google Scholar]
- 43.Felkin H, Frajerman C, Roussi G. Ann Chimie. 1971;6:17–26. [Google Scholar]
- 44.Holm T. J Org Chem. 2000;65:1188–1192. doi: 10.1021/jo991777h. [DOI] [PubMed] [Google Scholar]
- 45.Olah GA. Acc Chem Res. 1971;4:240–248. [Google Scholar]
- 46.Ridd JH. Acc Chem Res. 1971;4:248–253. [Google Scholar]
- 47.Young PR, Jencks WP. J Am Chem Soc. 1977;99:8238–8248. [Google Scholar]
- 48.Ingold KU, Griller D. Acc Chem Res. 1980;13:317–323. [Google Scholar]
- 49.Amyes TL, Jencks WP. J Am Chem Soc. 1989;111:7888–7900. [Google Scholar]
- 50.Horenstein BA, Bruner M. J Am Chem Soc. 1998;120:1357–1362. [Google Scholar]
- 51.Newcomb M. Tetrahedron. 1993;49:1151–1176. [Google Scholar]
- 52.Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 53.Douglas NL, Ley SV, Lucking U, Warriner SL. J Chem Soc, Perkin Trans 1. 1998:51–65. [Google Scholar]
- 54.Blackmond DG, Schultz T, Mathew JS, Loew C, Rosner T, Pfaltz A. Synlett. 2006:3135–3139. [Google Scholar]
- 55.Huang M, Retailleau P, Bohé L, Crich D. J Am Chem Soc. 2012;134:14746–14749. doi: 10.1021/ja307266n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barresi F, Hindsgaul O. J Am Chem Soc. 1991;113:9376–9377. [Google Scholar]
- 57.Stork G, Kim G. J Am Chem Soc. 1992;114:1087–1088. [Google Scholar]
- 58.Ito Y, Ogawa T. Angew Chem Int Ed. 1994;33:1765–1767. [Google Scholar]
- 59.Jung KH, Muller M, Schmidt RR. Chem Rev. 2000;100:4423–4442. doi: 10.1021/cr990307k. [DOI] [PubMed] [Google Scholar]
- 60.Ishiwata A, Lee YJ, Ito Y. Org Biomol Chem. 2010;8:3596–3608. doi: 10.1039/c004281a. [DOI] [PubMed] [Google Scholar]
- 61.Crich D, Cai W, Dai Z. J Org Chem. 2000;65:1291–1297. doi: 10.1021/jo9910482. [DOI] [PubMed] [Google Scholar]
- 62.Crich D, Wu B. Org Lett. 2006;8:4879–4882. doi: 10.1021/ol061938l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crich D, Barba GR. Tetrahedron Lett. 1998;39:9339–9342. [Google Scholar]
- 64.Crich D, Dai Z. Tetrahedron. 1999;55:1569–1580. [Google Scholar]
- 65.Crich D, de la Mora MA, Cruz R. Tetrahedron. 2002;58:35–44. [Google Scholar]
- 66.Crich D, Jayalath P. Org Lett. 2005;7:2277–2280. doi: 10.1021/ol050680g. [DOI] [PubMed] [Google Scholar]
- 67.Crich D, Jayalath P, Hutton TK. J Org Chem. 2006;71:3064–3070. doi: 10.1021/jo0526789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Crich D, Karatholuvhu MS. J Org Chem. 2008;73:5173–5176. doi: 10.1021/jo7023398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsui R, Seto K, Sato Y, Suzuki T, Nakazaki A, Kobayashi S. Angew Chem Int Ed. 2011;50:680–683. doi: 10.1002/anie.201006230. [DOI] [PubMed] [Google Scholar]
- 70.Denmark SE, Willson TM. J Am Chem Soc. 1989;111:3475–3476. [Google Scholar]
- 71.Schinzer D. Synthesis. 1988:263–273. [Google Scholar]
- 72.Mayr H, Kempf B, Ofial AR. Acc Chem Res. 2003;36:66–77. doi: 10.1021/ar020094c. [DOI] [PubMed] [Google Scholar]
- 73.Danishefsky SJ, Kerwin JF. J Org Chem. 1982;47:3803–3805. [Google Scholar]
- 74.Kozikowski AP. Tetrahedron Lett. 1982;23:2281–2284. [Google Scholar]
- 75.Larsen CH, Ridgway BH, Shaw JT, Woerpel KA. J Am Chem Soc. 1999;121:11208–12209. doi: 10.1021/ja0524043. [DOI] [PubMed] [Google Scholar]
- 76.Larsen CH, Ridgway BH, Shaw JT, Smith DM, Woerpel KA. J Am Chem Soc. 2005;127:10879–10884. doi: 10.1021/ja0524043. [DOI] [PubMed] [Google Scholar]
- 77.Ayala L, Lucero CG, Romero JAC, Tabacco SA, Woerpel KA. J Am Chem Soc. 2003;125:15521–15528. doi: 10.1021/ja037935a. [DOI] [PubMed] [Google Scholar]
- 78.Beaver MG, Woerpel KA. J Org Chem. 2010;75:1107–1118. doi: 10.1021/jo902222a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lucero CG, Woerpel KA. J Org Chem. 2006;71:2641–2647. doi: 10.1021/jo0522963. [DOI] [PubMed] [Google Scholar]
- 80.Crich D, Sharma I. Org Lett. 2008;10:4731–4734. doi: 10.1021/ol8017038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McGarvey GJ, LeClair CA, Schmidtmann BA. Org Lett. 2008;10:4727–4730. doi: 10.1021/ol801710s. [DOI] [PubMed] [Google Scholar]
- 82.Miller GJ, Gardiner JM. Org Lett. 2010;12:5262–5265. doi: 10.1021/ol102310x. [DOI] [PubMed] [Google Scholar]
- 83.BeMiller JN, Yadav MP, Kalabokis VN, Myers RW. Carbohydr Res. 1990;200:111–126. [Google Scholar]
- 84.Giannis A, Sandhoff K. Tetrahedron Lett. 1985;26:1479–1482. [Google Scholar]
- 85.Levy DE, Tang C. The Chemistry of C-Glycosides. Pergamon; Oxford: 1995. [Google Scholar]
- 86.Postema MHD, Calimente D. In: Glycochemistry: Principles, Synthesis, and Applications. Wang PG, Bertozzi CR, editors. Dekker; New York: 2001. pp. 77–131. [Google Scholar]
- 87.Postema MHD, editor. C-Glycoside Synthesis. CRC; Boca Raton: 1995. [Google Scholar]
- 88.Gyorgydeak Z, Pelyvas IF. In: Glycoscience. Fraser-Reid B, Tatsuta K, Thiem J, editors. Vol. 1. Springer; Berlin: 2001. pp. 691–747. [Google Scholar]
- 89.Skrydstrup T, Vauzeilles B, Beau J-M. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinay P, editors. Vol. 1. Wiley-VCH; Weinheim: 2000. pp. 495–530. [Google Scholar]
- 90.Yuan XJ, Linhardt RJ. Current Topics in Medicinal Chemistry. 2005;5:1393–1430. doi: 10.2174/156802605774643033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Crich D, Sun S. J Am Chem Soc. 1997;119:11217–11223. [Google Scholar]
- 92.Kim KS, Suk DH. Top Curr Chem. 2011;301:109–140. doi: 10.1007/128_2010_107. [DOI] [PubMed] [Google Scholar]
- 93.Zhu Y, Yu B. Chem Eur J. 2015;21:8771–8780. doi: 10.1002/chem.201500648. [DOI] [PubMed] [Google Scholar]
- 94.Heuckendorff M, Bols PS, Barry C, Frihed TG, Pedersen CM, Bols M. Chem Commun. 2015 doi: 10.1039/C5CC04716A. [DOI] [PubMed] [Google Scholar]
- 95.Ranade SC, Demchenko AV. J Carbohydr Chem. 2013;32:1–43. [Google Scholar]
- 96.Satoh H, Nukada T. Trends in Glycosci Glycobiol. 2014;26:11–27. [Google Scholar]
- 97.Whitfield DM. Carbohydr Res. 2012;356:180–190. doi: 10.1016/j.carres.2012.03.040. [DOI] [PubMed] [Google Scholar]
- 98.Kononov LO. RSC Adv. 2015;5:46718–46734. [Google Scholar]
- 99.Weingart R, Schmidt RR. Tetrahedron Lett. 2000;41:8753–8758. [Google Scholar]
- 100.Yun M, Shin Y, Chun KH, Jen S. Bull Chem Soc Kor. 2000;21:562–566. [Google Scholar]
- 101.Kim KS, Kim JH, Lee YJ, Lee YJ, Park J. J Am Chem Soc. 2001;123:8477–8481. doi: 10.1021/ja015842s. [DOI] [PubMed] [Google Scholar]
- 102.Codée JDC, Hossain LH, Seeberger PH. Org Lett. 2005;7:3251–3254. doi: 10.1021/ol051038p. [DOI] [PubMed] [Google Scholar]
- 103.Tsuda T, Sato S, Nakamura S, Hashimoto S. Heterocycles. 2003;59:509–515. [Google Scholar]
- 104.Tanaka S-i, Takashima M, Tokimoto H, Fujimoto Y, Tanaka K, Fukase K. Synlett. 2005:2325–2328. [Google Scholar]
- 105.Baek JY, Choi TJ, Jeon HB, Kim KS. Angew Chem Int Ed. 2006;45:7436–7440. doi: 10.1002/anie.200602642. [DOI] [PubMed] [Google Scholar]
- 106.Kim KS, Fulse DB, Baek JY, Lee BY, Jeon HB. J Am Chem Soc. 2008;130:8537–8547. doi: 10.1021/ja710935z. [DOI] [PubMed] [Google Scholar]
- 107.Tanaka K, Mori Y, Fukase K. J Carbohydr Chem. 2009;28:1–11. [Google Scholar]
- 108.Chang S-SS, Che-Hao, Lai Kwun-Cheng, Mong Kwok-Kong Tony. Chem Asian J. 2010;5:1152–1162. doi: 10.1002/asia.200900765. [DOI] [PubMed] [Google Scholar]
- 109.Heuckendorff M, Bendix J, Pedersen CM, Bols M. Org Lett. 2014;16:1116–1119. doi: 10.1021/ol403722f. [DOI] [PubMed] [Google Scholar]
- 110.Sun P, Wang P, Zhang Y, Zhang X, Wang C, Liu S, Lu J, Li M. J Org Chem. 2015;80:4164–4175. doi: 10.1021/acs.joc.5b00140. [DOI] [PubMed] [Google Scholar]
- 111.Schmidt RR. In: Preparative Carbohydrate Chemistry. Hanessian S, editor. Dekker; New York: 1997. pp. 283–312. [Google Scholar]
- 112.Huang M, Tran H-A, Bohé L, Crich D. In: Carbohydrate Chemistry: Proven Synthetic Methods. van der Marel GA, Codée JDC, editors. Vol. 2. CRC Press; Boca Raton: 2014. pp. 175–181. [Google Scholar]
- 113.Chevalier R, Esnault J, Vandewalle P, Sendid B, Colombel JF, Poulain D, Mallet JM. Tetrahedron. 2005;61:7669–7677. [Google Scholar]
- 114.Tetsuta O, Masahiro T, Susuma K. Tetrahedron Lett. 1994;35:6493–6496. [Google Scholar]
- 115.David S, Hanessian S. Tetrahedron. 1985;41:643–663. [Google Scholar]
- 116.Szurmai Z, LB, Lipták A. Carbohydr Res. 1994;254:301–309. doi: 10.1016/0008-6215(94)84264-7. [DOI] [PubMed] [Google Scholar]
- 117.Lu L, Zhang W, Nam S, Horne DA, Jove R, Carter RG. J Org Chem. 2013;78:2213–2247. doi: 10.1021/jo3026077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Crich D, Mataka J, Zakharov LN, Rheingold AL, Wink DJ. J Am Chem Soc. 2002;124:6028–6036. doi: 10.1021/ja0122694. [DOI] [PubMed] [Google Scholar]
- 119.Crich D, Mataka J, Sun S, Lam K-C, Rheingold AR, Wink DJ. Chem Commun. 1998:2763–2764. [Google Scholar]
- 120.Liang H, MacKay M, Grindley TB, Robertson KN, Cameron TS. Can J Chem. 2010;88:1154–1174. [Google Scholar]
- 121.Screttas CG, Micha-Screttas M. J Org Chem. 1978;43:1064–1071. [Google Scholar]
- 122.Schmidt RR, Michel J, Roos M. Liebigs Ann Chem. 1984:1343–1357. [Google Scholar]
- 123.Zhang YM, Mallet JM, Sinaÿ P. Carbohydr Res. 1992;236:73–88. doi: 10.1016/0008-6215(92)85007-m. [DOI] [PubMed] [Google Scholar]
- 124.Pougny JR, Sinaÿ P. Tetrahedron Lett. 1976;17:4073–4076. [Google Scholar]
- 125.Koeppen BH. Carbohydr Res. 1972;24:154–158. [Google Scholar]
- 126.Gildersleeve J, Smith A, Sakurai D, Raghavan S, Kahne D. J Am Chem Soc. 1999;121:6176–6182. [Google Scholar]
- 127.Gu X, Chen L, Wang X, Liu X, You Q, Xi W, Gao L, Chen G, Chen YL, Xiong B, Shen J. J Org Chem. 2014;79:1100–1110. doi: 10.1021/jo402551x. [DOI] [PubMed] [Google Scholar]
- 128.Chen G, Yin Q, Yin J, Gu X, Liu X, You Q, Chen YL, Xiong B, Shen J. Org Biomol Chem. 2014;12:9781–9785. doi: 10.1039/c4ob01807a. [DOI] [PubMed] [Google Scholar]
- 129.Kim JH, Yang H, Park J, Boons GJ. J Am Chem Soc. 2005;127:12090–12097. doi: 10.1021/ja052548h. [DOI] [PubMed] [Google Scholar]
- 130.Fascione MA, Kilner CA, Leach AG, Turnbull WB. Chem Eur J. 2012;18:321–333. doi: 10.1002/chem.201101889. [DOI] [PubMed] [Google Scholar]
- 131.Cox DJ, Singh GP, Watson AJA, Fairbanks AJ. Eur J Org Chem. 2014:4624–4642. [Google Scholar]
- 132.Sammakia T, Smith RS. J Am Chem Soc. 1994;116:7915–7916. [Google Scholar]
- 133.Krumper JR, Salamant WA, Woerpel KA. J Org Chem. 2009;74:8039–8050. doi: 10.1021/jo901639b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fraser-Reid B, Wu ZC, Andrews W, Skowronski E. J Am Chem Soc. 1991;113:1434–1435. [Google Scholar]
- 135.Andrews CW, Rodebaugh R, Fraser-Reid B. J Org Chem. 1996;61:5280–5289. doi: 10.1021/jo961115h. [DOI] [PubMed] [Google Scholar]
- 136.Bülow A, Meyer T, Olszewski TK, Bols M. Eur J Org Chem. 2004:323–329. [Google Scholar]
- 137.Bock K, Duus JO. J Carbohydr Chem. 1994;13:513–543. [Google Scholar]
- 138.Jensen HH, Nordstrom M, Bols M. J Am Chem Soc. 2004;126:9205–9213. doi: 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]
- 139.Jensen HH, Bols M. Acc Chem Res. 2006;39:259–265. doi: 10.1021/ar050189p. [DOI] [PubMed] [Google Scholar]
- 140.Moumé-Pymbock M, Furukawa T, Mondal S, Crich D. J Am Chem Soc. 2013;135:14249–14255. doi: 10.1021/ja405588x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Codée JDC, Kröck L, Castagner B, Seeberger PH. Chem Eur J. 2008;14:3987–3994. doi: 10.1002/chem.200701864. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.