

SUMMARY
For some neurological disorders, disease is primarily RNA-mediated due to expression of non-coding microsatellite expansion RNAs (RNAexp). Toxicity is thought to result from enhanced binding of proteins to these expansions and depletion from their normal cellular targets. However, experimental evidence for this sequestration model is lacking. Here, we use HITS-CLIP and pre-mRNA processing analysis of human control versus myotonic dystrophy (DM) brains to provide compelling evidence for this RNA toxicity model. MBNL2 binds directly to DM repeat expansions in the brain resulting in depletion from its normal RNA targets with downstream effects on alternative splicing and polyadenylation. Similar RNA processing defects were detected in Mbnl compound knockout mice, highlighted by dysregulation of Mapt splicing and fetal tau isoform expression in adults. These results demonstrate that MBNL proteins are directly sequestered by RNAexp in the DM brain and introduce a powerful experimental tool to evaluate RNA-mediated toxicity in other expansion diseases.
Keywords: MBNL, microsatellite, myotonic dystrophy, non-coding RNA, RNA-mediated disease, RNA processing
Graphical Abstract
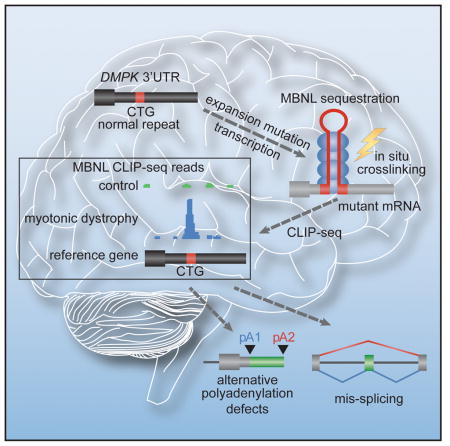
INTRODUCTION
Microsatellites, or simple sequence repeats of ≤ 10 bp, comprise ~3% of the human genome but are generally regarded as non-functional and neutrally evolving (Gemayel et al., 2010; Goodwin and Swanson, 2014). However, these repeats are highly polymorphic in size and expansions cause >40 hereditary neurological and neuromuscular disorders (Nelson et al., 2013). Current pathogenesis models propose that these diseases are most commonly caused by either mutant expansion proteins or RNAs depending on the location of the mutation within each affected gene. For example, coding region expansions generate mutant proteins containing elongated homopolymeric tracts while expansions in non-coding regions result in the synthesis of toxic RNAs that either sequester, or trigger the activation of, RNA processing factors (Echeverria and Cooper, 2012). However, repeat-associated non-ATG (RAN) translation also occurs in several expansion diseases, including spinocerebellar ataxia type 8 (SCA8) and C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia (C9 ALS/FTD), so toxic proteins may also be produced from classically defined non-coding regions (Kwon et al., 2014; Mizielinska et al., 2014; Mori et al., 2013b; Wen et al., 2014; Zu et al., 2011; Zu et al., 2013).
Studies on the molecular etiology of myotonic dystrophy (DM) have served as a model to investigate RNA-mediated toxicity mechanisms. Indeed, the sequestration and activation of RNA processing factors as well as RAN translation have been documented in this disease (Cleary and Ranum, 2014; Echeverria and Cooper, 2012; Mohan et al., 2014). DM types 1 and 2 (DM1, DM2) are progressive and multi-systemic neuromuscular disorders with cardinal manifestations including myotonia, muscle wasting, cardiomyopathy, excessive daytime sleepiness, cerebral atrophy, white matter lesions, cognitive impairments and neurofibrillary tangles (NFTs). DM1 is caused by CTGexp mutations in the 3′ untranslated region (3′ UTR) of the DMPK gene while DM2 is associated with a CCTGexp in intron 1 of CNBP/ZNF9. Transcription of these repeats results in the synthesis of C(C)UGexp RNAs that alter the RNA processing activities of several RNA-binding factors, including CELF and MBNL proteins (Echeverria and Cooper, 2012). For MBNL, C(C)UGexp RNAs co-localize with MBNL1-3 in nuclear RNA foci and this protein redistribution is thought to inhibit the normal functions of this protein family in alternative pre-mRNA splicing (AS), alternative polyadenylation (APA), pre-miR processing and RNA localization (Batra et al., 2014; Fardaei et al., 2002; Ho et al., 2004; Jiang et al., 2004; Miller et al., 2000; Rau et al., 2011; Wang et al., 2012). The MBNL loss-of-function model is also supported by studies on Mbnl single and compound knockout mice, which recapitulate many DM postnatal phenotypes (Charizanis et al., 2012; Kanadia et al., 2003; Lee et al., 2013a; Poulos et al., 2013). Nevertheless, the interaction of MBNL proteins with C(C)UGexp RNAs may be indirect and mediated by other factors in affected human tissues. Indeed, additional proteins bind to C(C)UGexp RNAs (Kim et al., 2005; Pettersson et al., 2014; Ravel-Chapuis et al., 2012) or co-localize with nuclear foci (Laurent et al., 2012).
Here, we use HITS-CLIP combined with pre-mRNA processing analysis to demonstrate that MBNL proteins regulate AS and APA in the human brain and these functions are dysregulated in DM due to direct MBNL binding to C(C)UGexp RNAs and depletion of these proteins from their normal RNA targets. Based on these results, we propose this in situ strategy to validate candidate sequestered factors in other microsatellite expansion diseases, including C9 ALS/FTD.
RESULTS
Direct Titration of MBNL2 by C(C)UGexp RNAs in DM1 and DM2 Brains
Since MBNL2 is the major MBNL family member responsible for DM-associated splicing abnormalities in the brain (Charizanis et al., 2012), we reasoned that if MBNL2 binds directly to, and is sequestered by, C(C)UGexp RNA in vivo, then MBNL2 HITS-CLIP performed on DM1 brain should produce CUG-rich reads that cluster over the CTG repeat region in the 3′ UTR of the DMPK reference gene (Figure 1A). To test this possibility, MBNL2 HITS-CLIP was performed for DM1 and neurological disease controls using autopsy tissue from two brain regions affected in DM, the hippocampus and frontal cortex (Table S1, available online). Sequencing reads were aligned back to the human reference genome and wiggle plots were used to visualize MBNL2 binding distribution. As predicted by the direct binding and sequestration model, a large peak of MBNL2 CLIP tags was observed over the DMPK 3′ UTR CTG repeat region in DM1 hippocampus compared to the few CLIP tags that mapped to this region for non-DM disease controls and DM2 (Figure 1B). The average read depth over the repeat region was significantly enriched in DM1 (>36-fold over controls). Similar binding to CUG repeats was observed in the frontal cortex (Figure S1A) demonstrating that MBNL2 was sequestered in both regions of the brain.
Figure 1. HITS-CLIP Identifies MBNL2-RNAexp Interactions in DM Brain.

(A) Strategy for identifying RBP-RNAexp binding interactions using HITS-CLIP. For DM1, a CTG repeat (red box) in the DMPK 3′UTR (coding exons, thick black boxes; UTRs, thin black boxes; introns, thin lines) expands in disease (CUGexp, red triangle). Upon transcription, the mRNA (grey) forms a stem-loop that sequesters MBNL2 (blue ovals). HITS-CLIP of MBNL proteins using DM1 (right), but not control (left), tissue generates a large increase in reads clustered over the repeat region (bottom right).
(B) MBNL2 binding profile reveals enriched binding to the DMPK CTGexp in DM1 brain. UCSC browser view showing wiggle plots of MBNL2 HITS-CLIP binding in the DMPK reference gene for control (orange), DM1 (green), and DM2 (blue) human hippocampus. Zoomed-in view of the terminal exon (bottom right) showing a clustered read peak over the CTG repeat region for DM1 only. Quantification (bottom left) of MBNL2 CLIP peak read depth (RPKM) over the DMPK CTG repeat region shows a significant enrichment (36-fold) over controls (n = 3 per group, data are reported ± SEM, *p < 0.05).
(C) MBNL2 HITS-CLIP binding profile for CNBP. Intron 1 (bottom right) containing the CCTG repeat region (red box) is shown. Quantification (bottom left) of average peak read depth over CCTG repeats showing a 79-fold enrichment in DM2 over controls (n = 3 per group, data are reported ± SEM, ***p < 0.001).
A parallel analysis was performed to assess MBNL2 binding to the CCUG repeat RNA in DM2 (Figure 1C). In contrast to DM1, a peak of MBNL2 binding occurred in the region corresponding to CNBP intron 1 in the CCUG repeat region in DM2 but not in control or DM1. Quantification of average read depth showed a 79-fold enrichment (1/10 standard deviation added over zero control reads) of MBNL2 binding to the CCUG repeat region in DM2 versus controls. Enrichment of CTG and CCTG repeat reads in the raw datasets was also assessed to address signal loss due to microsatellite misalignment. The excess of pre-alignment repeat reads confirmed the abundance of MBNL binding sites in DM1 and DM2 (Table S1 and Figure S1B). This strong enhancement of MBNL2 binding to the C(C)UGexp regions in DM1 and DM2 brains not only supported the protein sequestration model but also demonstrated the potential utility of HITS-CLIP for authenticating factors implicated in RNA toxicity in other microsatellite expansion disorders.
MBNL2 Sequestration and RNA Splicing Defects in the DM CNS
Since HITS-CLIP demonstrated enhanced MBNL2 binding to C(C)UG repeat regions in DM1 and DM2 brains, we next addressed the hypothesis that this toxic RNA binding results in depletion of MBNL2 binding to its normal pre-mRNA targets. Analysis of MBNL2 binding to normal RNA targets identified by HITS-CLIP of control brain revealed considerable overlap of target genes between the hippocampus and frontal cortex (Figure 2A). In addition, a significant portion of MBNL2 gene targets in the human hippocampus overlapped with hippocampal targets of murine Mbnl2 (Charizanis et al., 2012). Comparison of MBNL2 HITS-CLIP tags from human and mouse hippocampus indicated that tag distribution was similar with 67% of human MBNL2 tags mapping to the 3′ UTR (Figure 2B) versus 51% in mouse (Charizanis et al., 2012), and the preferred binding motif (YGCY, Y=T/U or C) was conserved between species (Figure S2A). Gene ontology (GO) analysis also indicated that MBNL2 targets were involved in similar pathways in human (Figure S2B) compared to mouse (Charizanis et al., 2012).
Figure 2. Depletion of MBNL2 Binding for Mis-regulated Exons in DM1.

(A) Venn diagram of overlapping MBNL2 target genes in human hippocampus and frontal cortex and mouse hippocampus.
(B) Pie chart of MBNL2 binding site distribution in human hippocampus.
(C) Venn diagram of common genes in DM1 and DM2 with MBNL2 depletion events identified by dCLIP analysis.
(D) UCSC browser view of MBNL2 dCLIP near CSNK1D exon 9 (left) (alternative exon, red box; flanking exons, thick black boxes; introns, gray lines) and APP exon 7 (right) showing loss of binding in DM1 (green) compared to controls (orange) (n = 3 each). RT-PCR splicing analyses are also shown for CSNK1D and APP in control versus DM1 brain with corresponding percent spliced in (ψ) values (n = 3 per group, data are reported ± SEM, ***p < 0.001, **p < 0.01).
To determine whether MBNL2 is titrated away from its normal pre-mRNA targets in DM, we performed a comparative analysis of MBNL binding in control and DM1 patient brains using differential CLIP (dCLIP) (Wang et al., 2014). The dCLIP computational pipeline normalizes HITS-CLIP read data across different datasets for comparison and identifies statistically significant changes in the amount of RBP binding between two different conditions using a hidden Markov model (HMM). The dCLIP comparative analysis of human MBNL2 binding interaction in control versus DM brain defined 2781 transcripts in DM1 and 2062 transcripts in DM2 that showed reduced binding of MBNL2 with considerable overlap (1765) between the two disease types (Figures 2C, Table S2). MBNL2 depletion occurred predominantly in 3′ UTRs followed by introns (Figures S2C and S2D) reflecting the typical binding distribution of MBNL2.
To determine if MBNL2 depletion near target alternative exons resulted in mis-regulated splicing in the DM brain, MBNL2 dCLIP profiles were analyzed for alternative cassette exons mis-spliced in DM1 (Figure S2E) (Charizanis et al., 2012; Jiang et al., 2004). Splicing patterns were analyzed by RT-PCR splicing assays using primers that annealed to exons flanking mis-regulated cassette exons and the percentage of exon inclusion was quantified as percent spliced in (ψ). For example, enhanced skipping of CSNK1D exon 9 and APP exon 7 occurs in the DM1 brain and reduced binding of MBNL2 near these alternative exons was observed by dCLIP (Figure 2D). These results indicate that expression of mutant DMPK and CNBP alleles leads to MBNL sequestration on C(C)UGexp RNAs resulting in dysregulation of RNA splicing in the brain.
Although dCLIP analysis supported the MBNL sequestration model, an alternative strategy was performed to confirm that depletion of MBNL activity in the brain recapitulated DM-associated RNA mis-splicing. Since we have recently demonstrated that MBNL1 and MBNL2 compensate for each other to regulate alternative splicing (Lee et al., 2013a), depletion of MBNL activity was achieved in a mouse Mbnl compound knockout model because prior work has shown that loss of both Mbnl1 and Mbnl2 expression in mouse skeletal muscle is required to recapitulate the RNA mis-processing events characteristic of DM (Batra et al., 2014; Lee et al., 2013a).
Mbnl1; Mbnl2 Knockout Mice Model DM1-Associated Mis-Splicing
Mbnl1; Mbnl2 conditional double knockout (Mbnl1−/−; Mbnl2c/c; Nestin-Cre+/− or Nestin-Cre DKO) mice were generated that were Mbnl1 constitutive nulls with Mbnl2 expression selectively ablated in the nervous system. Nestin-Cre DKOs were small, similar to Mbnl2−/− single knockouts (Figure S3A). In contrast to Mbnl2−/− mice (Charizanis et al., 2012), the DKOs only survived until ~23 weeks of age (Figure 3A) and showed early-onset severe motor (Figure 3B) and grip (Figure S3B) deficits. Alternative splicing patterns were compared for wild-type (Mbnl1+/+; Mbnl2+/+; Nestin-Cre+/−), Mbnl1 KO (Mbnl1−/−; Mbnl2c/c; Nestin-Cre−/−), Mbnl2 KO (Mbnl1+/+; Mbnl2c/c; Nestin-Cre+/−) and Nestin-Cre DKO brains (Figures 3C–D and S3C). As anticipated, compound loss of MBNL1 and MBNL2 in the nervous system significantly enhanced mis-splicing of alternative exons, such as Add1 E15, Kcnma1 E25a and Clasp2 E16 (Figure 3C), previously identified in Mbnl2 KO and DM1 (Charizanis et al., 2012). Importantly, the Nestin-Cre DKO adult and WT fetal/neonatal splicing patterns were similar in these targets and in Camk2d (Suenaga et al., 2012) indicating that compound loss of MBNL1 and MBNL2 is required for loss of adult exon splicing in the mammalian brain (Figure 3D). Moreover, comparison of the Nestin-Cre DKO with DM1 and DM2 brains showed the same alternative splicing trend. For example, splicing of mouse Cacna1d exon 12a in the brain was almost completely blocked in Nestin-Cre DKOs while splicing of the orthologous human exon was reduced in DM1 and DM2 (Figure 4A). A striking concordance was also observed for enhanced inclusion of Grin1/GRIN1 exon 5 between Nestin-Cre DKOs and DM1 (Figure 4B). We conclude that Mbnl depletion in the mouse brain recapitulates the aberrant CNS splicing patterns characteristic of DM1 and DM2.
Figure 3. Reversal to Fetal CNS Splicing due to Combined Loss of MBNL1 and MBNL2.

(A) Kaplan-Meier analysis of Mbnl1ΔE3/ΔE3, Mbnl2c/c; Nestin-Cre+/− (Nestin-Cre DKO) mice, wild type (WT, Mbnl1+/+, Mbnl2+/+) and Cre controls (Mbnl1+/+, Mbnl2+/+; Nestin-Cre+/−; Mbnl1+/+, Mbnl2c/c; Nestin-Cre+/−; Mbnl1ΔE3/ΔE3, Mbnl2c/c; Nestin-Cre−/−) (n = 21 per group).
(B) Accelerating rotarod performance of Nestin-Cre DKO mice and controls (5 weeks of age) over the four day training course (n ≥ 8 per group, data are reported ± SEM, ***p < 0.001).
C) RT-PCR analysis of splicing patterns in WT, Mbnl1ΔE3/ΔE3, Mbnl2 ΔE2/ΔE2, and Nestin-Cre DKO brain showing several targets (Add1, Kcnma1, Clasp2) with increased mis-splicing after compound loss of Mbnl function.
(D) Reversal to fetal splicing patterns in Nestin-Cre DKO brain. The splicing patterns of four Camk2d isoforms in WT to Mbnl1ΔE3/ΔE3, Mbnl2 ΔE2/ΔE2, and Nestin-Cre DKO brain are compared to the splicing patterns of WT postnatal day (P)6 and P42 mice.
Figure 4. DM-relevant mis-splicing in Nestin-Cre DKO brain.

(A) RT-PCR splicing analysis of Cacna1d in WT, Mbnl1ΔE3/ΔE3, Mbnl2 ΔE2/ΔE2 and Nestin-Cre DKO brain. Splicing of CACNA1D in human control, DM2, and DM1 brain shown for comparison (n = 3 per group, data are reported ± SEM, ***p < 0.001, **p < 0.01).
(B) Same as (A) but splicing analysis of mouse Grin1 compared to human GRIN1 (n = 3 per group, data are reported ± SEM, *p < 0.05).
(C) RNA splicing maps using human MBNL2 CLIP tags near exons mis-spliced in the DM1 frontal cortex (included exons, red; skipped exons, blue; coverage ≥ 20, |dI| ≥0.1, FDR ≤ 0.05). Also included is MBNL binding motif data, or YGCY motifs in the human genome near the mis-spliced exons, using a previously described computational procedure (included exons, light red; skipped exons, light blue) (Zhang et al., 2013).
To extend these results and examine the global relationship between human and mouse MBNL protein binding and alternative splicing regulation, RNA-seq data was generated from human DM1 and control frontal cortex (Table S3). We identified 596 alternative exons with lower inclusion, and 335 exons with higher inclusion, in DM1 corresponding to cassette exons activated and repressed by MBNL proteins, respectively. Next, we generated normalized complexity maps to correlate these changes on a global level with MBNL binding, using both HITS-CLIP data (Table S1) and predicted functional YGCY motifs (Zhang et al., 2013) (Figure 4C). Compared to mouse Mbnl2−/− KO brain (Charizanis et al., 2012), the human exon inclusion pattern indicates that MBNL binding proximal to both the 5′ ss and 3′ ss promotes alternative cassette splicing. The exon exclusion pattern is similar between mouse and human although a larger peak was observed near the downstream flanking exon 3′ ss. Surprisingly, this RNA-seq analysis revealed that human microtubule-associated protein tau (MAPT) exons 2, 3 and 10 were some of the most mis-spliced cassette exons in the DM1 frontal cortex (Table S3). Since a prominent manifestation of DM1 CNS disease is the progressive appearance of neurofibrillary tangles (NFTs) composed of intraneuronal aggregates of hyper-, and abnormally, phosphorylated tau protein as well as expression of fetal MAPT isoforms (Sergeant et al., 2001), we next investigated the effect of loss of MBNL activity on MAPT pre-mRNA splicing regulation.
Aberrant Tau Processing in DM1 and Mbnl Compound Knockouts
In the human adult brain, MAPT encodes six tau isoforms through the alternative splicing of exons 2, 3 and 10 (exons 3, 4 and 8 in mouse), but in DM all three of these exons are skipped so that fetal tau is preferentially expressed (Dhaenens et al., 2008; Jiang et al., 2004). Although MAPT minigene studies have shown that synergistic interactions between MBNL1 and MBNL2 activate the splicing of MAPT exons 2 and 3, it is not clear if these MBNL interactions are important in vivo (Carpentier et al., 2014). To address this point, Mapt exon 2 and 3 splicing in the brain was examined using Mbnl2−/− single KO versus Nestin-Cre DKO and compared to control and DM1 human MAPT splicing. The results indicated that Mbnl1 and Mbnl2 interactions were also essential in vivo since the DM1 pattern of predominant skipping of Mapt exons 2 and 3 was only observed in the DKO mice, similar to the enhanced skipping of these exons in DM1 compared to control brain (Figures 5A and S3D).
Figure 5. Tau Isoform Mis-regulation in DM1 and Mbnl DKO Brain.

(A) RT-PCR splicing analysis showing shift toward skipping of MAPT exons 2 and 3 in DM1 brain compared to controls and in Nestin-Cre DKO brain relative to WT.
(B) MBNL2 binding is reduced near MAPT exon 10 in DM1 brain compared to controls. UCSC browser view of MBNL2 dCLIP binding profiles near MAPT exon 10 (alternative exon, red box; flanking exons, thick black boxes; introns, gray lines) in control (orange) and DM1 (green) brain (n = 3). Bottom panel shows the mouse Mbnl2 HITS-CLIP binding profile near exon 10.
(C) RT-PCR analysis of MAPT/Mapt exon 10 splicing for human control versus DM1 and mouse wild-type (WT), Mbnl2 KO (Mbnl2−/−) and Nestin-Cre DKO (DKO).
(D) Two-dimensional gel electrophoresis (1st dimension, 3 to 11 non-linear pH gradient strips; 2nd dimension SDS-PAGE) and immunoblot of tau isoforms (2N4R, 1N4R, 0N4R and 0N3R) in WT, Mbnl1ΔE3/ΔE3 (Mbnl1 KO), Mbnl2 ΔE2/ΔE2 (Mbnl2 KO) and Nestin-Cre DKO brain. Bottom panel corresponds to tau staining after treatment with lambda phosphatase. Tau protein was stained with the α-TauCter antibody. The N-terminal inserts correspond to inclusion/exclusion of alternative exon 2 and exon 2 + 3. The 3R and 4R isoforms correspond to isoforms without/with the exon 10 encoding sequence.
In contrast to MAPT exons 2 and 3, MBNL-mediated regulation of MAPT exon 10 has not been reported previously (Carpentier et al., 2014). Interestingly, dCLIP and crosslinked induced mutation site (CIMS) analysis of MBNL2 binding in hippocampus revealed that this MBNL protein bound primarily to YGCY clusters within intron 10 (Figures 5B and S4). Although the mouse ortholog also recognized YGCY motifs in intron 10, a major Mbnl2 binding peak was also detectable in Mapt intron 9 upstream of the exon 10 3′ ss (Figure 5B). This difference in binding distribution is intriguing because in human adult brain, equivalent inclusion and exclusion of MAPT exon 10 occurs in control brain although in adult mouse brain exon 10 inclusion is favored (Figure 5C). While knockout of Mbnl2 produced only a modest shift towards exclusion of this exon, combinatorial loss of Mbnl1 and Mbnl2 in the DKO brain triggered a strong change towards exclusion (Figures 5C and S3D), suggesting that both MBNL1 and MBNL2 synergize to control MAPT exon 10 splicing.
Since MAPT mis-splicing should result in abnormal expression of tau protein isoforms, MBNL-dependent changes in tau expression and phosphorylation status were assessed. The relative amounts of tau protein isoforms and post-translational modification were compared for wild-type, Mbnl1 knockout, Mbnl2 knockout, and Nestin-Cre DKO mouse brains using 2D gel electrophoresis (2D-GE) coupled with immunoblotting (Figure 5D). The alternative splicing of exons 2, 3, and 10 results in six tau protein isoforms in the adult brain that differ based on inclusion of two 29 amino acid N-terminal inserts (0N, 1N, or 2N) encoded by exons 2 and 3 and an additional microtubule binding domain encoded by exon 10 that generates the tau 4R isoform. As expected, loss of Mbnl1 alone did not significantly alter Mapt splicing or protein isoform expression. Mbnl1 KO tau expression patterns resembled wild-type including isoforms with 0, 1, and 2 N-terminal inserts and all four microtubule binding domains (0N4R, 1N4R, and 2N4R). In Mbnl2 KO brain, a shift towards diminished expression of 1N4R and 2N4R isoforms occurred with more acidic isovariants of the 0N4R isoform. More strikingly, in the Nestin-Cre DKO brain the 1N4R and 2N4R isoforms were undetectable and this change was accompanied by the emergence of a 0N3R isoform lacking the fourth microtubule-binding domain (Figure 5D, fourth panel). Specific anti-tau 4R and 3R antibodies confirmed loss of 2N4R and 1N4R expression and expression of both the 0N4R and 0N3R isoforms (Figure S5A). Noteworthy, the amount of 0N4R isoform was strongly reduced in DKOs compared to WT.
To determine whether phosphorylation accounts for the post-translational modifications responsible for the acidic isoforms, Nestin-Cre DKO brain homogenate was treated with λ-phosphatase, which resulted in the loss of the more acidic isoforms (Figure 5D). The additional band at 70 kDa was recognized by α-tauCter and could correspond to hyperphosphorylated tau, but this band was not detected by other anti-tau antibodies (Figure S5B) identifying it as a non-tau protein. Overall, these results support the hypothesis that loss of MBNL binding to MAPT intron 10 in DM results in reversion of MAPT pre-mRNA splicing to the fetal pattern.
Alternative PolyA Site Selection Following MBNL Sequestration in the Brain
While the alternative splicing function of MBNL proteins has been well characterized, MBNL activity is also required for skeletal muscle APA regulation (Batra et al., 2014). The high proportion of MBNL2 binding sites in target RNA 3′ UTRs revealed by HITS-CLIP (Figure 2B) led us to speculate that C(C)UGexp-induced MBNL2 sequestration also results in APA dysregulation in the brain. To test for APA changes in DM1, we employed a high-throughput PolyA-seq strategy (Derti et al., 2012). PolyA-seq libraries were prepared from controls, DM1 and DM2 frontal cortex and sequencing reads were mapped to the human reference genome followed by computational removal of templated A-rich tracts (Table S4) (Batra et al., 2014). To assess Mbnl-dependent APA changes in the brain, PolyA-seq libraries were also prepared for control and Nestin-Cre DKO frontal cortex. Scatter plots were generated to quantify polyA site (pA) use, and significant changes in utilization of alternative pA sites were recorded (FDR ≤0.05 and dI ≥ I0.15I). This analysis identified APA changes for thousands of genes in both DM1 (n= 6647 events, 2826 genes) and DM2 (n= 5563 events, 2425 genes) (Figure 6A) as well as Nestin-Cre DKO frontal cortex (n= 3195 events, 1556 genes) (Figure 6B) with 502 genes misregulated in both species (Figures S6A and S6B, Table S4). Shifts to more proximal and distal pA sites were observed in the Nestin-Cre DKO and DM1 (48% and 58%, respectively) with a slight bias towards more proximal sites in the DM brain (Figure 6A–B). Thus, MBNL activity promotes both utilization and skipping of alternative pA sites in the brain. Interestingly, some genes showed abnormal regulation of both APA and AS in DM1 hippocampus (Figure S6C). Similar to the reported splicing targets of MBNL2, many of the APA targets are involved in pathways associated with neuronal functions and the neurotrophin signaling pathway as determined by gene ontology analysis (Figure S6D). In addition, pathway analysis identified enriched terms associated with ubiquitin-mediated proteolysis and the mTOR pathway, remarkably similar to enriched terms identified by PolyA-seq analysis of DM skeletal muscle (Batra et al., 2014).
Figure 6. Disrupted Polyadenylation in Human DM and Mouse Mbnl DKO Brain.

(A) Scatter plot representation of PolyA-seq data showing APA shifts to more distal (blue) or proximal (red) pA sites relative to the coding region in DM1 (top) and DM2 (bottom) versus control brain (FDR < 0.05, |dI| > 0.15). The data represents distal (n = 2,794), proximal (3,853), total (6,647), and no shift (25,357) in DM1 and distal (2,273), proximal (3,290), total (5,563), and no shift (27,683) in DM2.
(B) Scatter plot illustrating shifts to more distal (blue) or proximal (red) pA sites in Nestin-Cre DKO versus WT brain (FDR < 0.05, |dI| > 0.15). The data represent distal (1,668), proximal (1,528), total (3,195) and no shift (47,944).
(C) PolyA-seq wiggle plots showing shifts to distal polyA sites in the FZR1 3′ UTR (3′ UTR, thin black box; coding region, thick black boxes; intron, gray line) in DM1 versus control (top) and Nestin-Cre DKO versus WT (middle). RT-PCR validation (bottom) of FZR1/Fzr1 switches (distal, D; total, T; n = 3 per group, data are reported ± SEM, *p < 0.05, ***p < 0.001).
(D) Wiggle plots (left) of PolyA-seq data for Sptb and Rgs9 comparing APA patterns in WT versus Nestin-Cre DKO brain. RT-PCR validation (middle) of APA changes with quantification (right) of distal (D) versus total (T) pA utilization (n = 3 per group, data are reported ± SEM, **p < 0.01, ***p < 0.001).
Analysis of PolyA-seq profiles in individual genes revealed examples of APA shifts in the DM brain that were reproduced in Nestin-Cre DKOs. For example, two alternative pA sites exist in the 3′ UTR of the FZR1 gene, and a shift towards distal site utilization occurs in the DM1 frontal cortex (Figure 6C). Similarly, this APA shift occurs in mouse DKO frontal cortex. In many cases, APA shifts resulted in the selection of an intronic pA site (Figure 6D), which in some cases was conserved between human and mouse (Figure S6E). Taken together, the results from HITS-CLIP, dCLIP, RNA-seq and PolyA-seq support the model that sequestration of MBNL proteins by C(C)UGexp RNA and subsequent loss of binding from endogenous pre-mRNA targets leads to dysregulated RNA processing in the DM brain.
DISCUSSION
RNA-mediated pathogenesis has emerged as an important disease mechanism for a number of neurological and neuromuscular disorders caused by microsatellite expansions (Mohan et al., 2014; Nelson et al., 2013). This unusual pathogenic process has been implicated in diseases in which the expansion mutation originates in a non-coding gene, such as ATXN8/ATXN8OS, or in the non-coding regions (introns, 5′ UTR, 3′ UTR) of a protein-encoding gene. Current disease models propose that these microsatellite expansion RNAs are toxic because they either directly sequester, and/or indirectly activate, cellular factors or generate RAN peptides via an unconventional translational mechanism (Cleary and Ranum, 2014). Here, we tested if HITS-CLIP, splicing and polyadenylation analyses could be combined to assess the binding and sequestration of proteins by microsatellite expansion RNAs, and corresponding depletion from endogenous RNA targets, isolated from affected human brains. Our results demonstrate that MBNL proteins are directly sequestered by mutant DMPK and CNBP RNAs in DM1 and DM2 brains, respectively, and the resulting depletion of MBNL activity reverts specific pre-mRNA processing events to a fetal regulatory pattern.
MBNL2 Entrapment by Toxic RNAs in the Myotonic Dystrophy Brain
MBNL loss-of-function in myotonic dystrophy is a prominent example of factor sequestration by disease-associated microsatellite expansion RNAs and the resulting downstream effects on cell function. In this study, we demonstrate that the major MBNL protein in the brain, MBNL2, interacts directly with DM1 CUGexp, and DM2 CCUGexp, RNAs in the brain. As anticipated, CTG and CCTG sequence reads also mapped to other repeat-containing genes, including the antisense strands of AR and MAML3 with 22 and 19 CTG repeats, respectively) (Table S2D). Mapping to C(C)TG/CA(G)G repeats at these other genomic loci is due to misalignment of pure repeat reads, which results in an underestimation of mutant DMPK and CNBP binding events. Improved sequencing and mapping technologies may overcome this challenge in future HITS-CLIP studies and will likely reveal more robust MBNL sequestration by DM1 and DM2 expansion mutation RNAs.
We also observed loss of MBNL2 binding proximal to mis-spliced exons in both DM1 and DM2, supporting the model that MBNL2 sequestration compromises its function as a splicing regulator in DM. Similarly, coupling PolyA-seq analysis and dCLIP revealed loss of MBNL2 binding in 3′ UTRs and APA changes in the same targets supporting the hypothesis that MBNL2 controls normal APA regulatory function and this function is altered in the DM CNS. These pre-mRNA processing changes are strikingly similar to those observed in Nestin-Cre DKO mice indicating that disease-associated changes in AS and APA are due to direct depletion of MBNL proteins from their normal target RNAs.
Recently, another MBNL activity has been proposed. RNA and RAN toxicity may be coupled in some microsatellite expansion diseases since MBNL1 promotes nuclear accumulation of mutant CUGexp and CAGexp RNAs that, in turn, represses synthesis of the corresponding RAN proteins in the cytoplasm (Kino et al., 2014). Alternatively, MBNL proteins may also bind repeats in the cytoplasm and directly block recruitment of the translational machinery. Of course, these potential mechanisms are not mutually exclusive. While it remains important to discriminate the toxic effects of protein sequestration and RAN translation (Mizielinska et al., 2014), both mechanisms might be susceptible to therapeutic strategies designed to upregulate MBNL protein levels (Kanadia et al., 2006).
Regulation of MAPT Splicing by MBNL Proteins
Abnormal expression of tau fetal isoforms is a characteristic feature of the adult DM1 brain and compound loss of MBNL1 and MBNL2 recapitulates this reversion to fetal Mapt RNA and protein expression in the Nestin-Cre DKO model. Moreover, tau isoform expression is profoundly modified in the Nestin-Cre DKO brain but not associated with a major change in tau phosphorylation status. However, the changes in tau post-translational modifications (PTMs) occur with aging in the human disease while the DKO mice are young adults. Therefore, we cannot exclude the possibility that tau PTMs could occur in older animals if they survived beyond 23 weeks of age. Noteworthy, tau isovariants were more acidic in the Nestin-Cre DKO mice when compared with wild type suggesting that tau PTMs may also be deregulated in this disease model. Interestingly, MBNL1 alone does not significantly alter Mapt splicing and MBNL2 primarily regulates exon 2 and exon 2/3 splicing (Carpentier et al., 2014) while loss of both MBNL proteins caused skipping of exons 2, 3 and 10. Although the tau 3R isoform is expressed in the mouse fetal, but not adult, brain (Liu and Gotz, 2013), both the 3R and 4R tau isoforms are found in the human brain where both MBNL1 and MBNL2 are expressed. Therefore, the regulation of MBNL1 or MBNL2 expression, or their splicing activity, may control tau isoform expression. For instance, in DM1 the loss of exons 2 and 10 inclusion indicates dual loss of MBNL1 and MBNL2 function since tau protein isoforms expressed in DM1 brains consist mainly of tau 0N3R and 0N4R, which is reproduced in Nestin-Cre DKO mice. This deregulation may also depend on the relative level of MBNL expression, and thus polymorphisms associated with MBNL expression could also contribute to disease severity (Huin et al., 2013).
A remaining question is how MBNL proteins control MAPT exon 10 splicing. Interestingly, HITS-CLIP identified sites for MBNL2 binding that are located within intron 10 but downstream of the exon 10-intron 10 junction region previously implicated in the mis-regulation of MAPT exon 10 in frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) (Niblock and Gallo, 2012). Since prior studies have proposed that additional splicing factors, including RBM4, CELF3 and CELF4, also bind to intron 10 to promote exon 10 splicing, it will be important to map these binding sites in the human brain and compare their binding patterns to MBNL2.
In Situ Validation Assay for Protein Sequestration in RNA-Mediated Disease
An RNA-mediated pathogenic mechanism has been proposed for additional microsatellite diseases with different non-coding expansion motifs, including FXTAS, SCA10, SCA12, Huntington disease-like 2 (HDL2) and C9orf72 ALS/FTS (Echeverria and Cooper, 2012; Goodwin and Swanson, 2014). For C9 ALS/FTD, multiple GGGGCCexp binding proteins have been identified including ADARB2, HNRNPA2B1, HNRNPA3, HNRNPH1, NCL, PURA and SRSF1 (Lee et al., 2013b; Mori et al., 2013a; Reddy et al., 2013). Unfortunately, it is not clear if any of these factors are effectively sequestered by the corresponding expansion RNAs in human tissues. Here, we demonstrate that HITS-CLIP provides an in situ validation technique for proteins that crosslink directly to tandem repeat expansions in frozen autopsy tissue. While some protein-RNAexp interactions may be less susceptible to UV light-induced crosslinking and indirect binding events would not be captured by HITS-CLIP, this in situ method provides an important tool that complements studies based on RNA binding characteristics in vitro and co-localization with RNA foci in cells and tissues. Since the development of genetic models is both time-consuming and costly, we recommend HITS-CLIP be performed prior to embarking on full-scale animal projects designed to investigate the role of candidate sequestered factors in microsatellite expansion disease.
EXPERIMENTAL PROCEDURES
Human Tissues and Genotyping
Autopsy tissues were obtained from brain (frontal cortex, hippocampus) of DM1, DM2 and control patients (Table S5). Protocols were approved by the Institutional Ethics Committee, University of Minnesota and the University of Florida Human Subjects Review Boards and all patients provided written informed consent. Genotyping for repeat expansions was performed using either genomic blot analysis or PCR.
Mouse Mbnl Compound Knockout Generation and Characterization
Constitutive and conditional Mbnl2 (Mbnl2 ΔE2/ΔE2, Mbnl2c/c) and constitutive Mbnl1 (Mbnl1 ΔE3/ΔE3) KO mice have been described (Charizanis et al, 2012; Kanadia et al, 2003). Transgenic Nestin-Cre mice (B6.Cg-Tg(Nes-cre)1Kln/J Strain 003771, JAX) were used to generate Nestin-Cre DKOs (Mbnl1ΔE3/ΔE3, Mbnl2c/c; Nestin-Cre+/−) and controls (Mbnl1+/+; Mbnl2+/+; Nestin-Cre−/−, Mbnl1+/+; Mbnl2+/+; Nestin-Cre+/−, Mbnl1ΔE3/ΔE3; Mbnl2+/+; Nestin-Cre−/−, Mbnl1ΔE3/ΔE3; Mbnl2+/+; Nestin-Cre+/−, Mbnl1+/+, Mbnl2c/c; Nestin-Cre−/−; Mbnl1+/+, Mbnl2c/c; Nestin-Cre+/− and Mbnl1ΔE3/ΔE3, Mbnl2c/c; Nestin-Cre−/−). Motor function (accelerating rotarod) and grip strength were assessed as described (Lee et al., 2013a). All animal procedures were approved by the University of Florida IACUC.
HITS-CLIP and dCLIP
HITS-CLIP was performed as described (Charizanis et al., 2012; Jensen and Darnell, 2008) with the following modifications. Autopsy-derived frozen brain tissues (hippocampus, frontal cortex) from control, disease control (ALS), DM1 and DM2 (~25 mg frozen tissue, n=3 each) were pulverized in liquid nitrogen, UV crosslinked and fragmented using RNase A (553 μg/μL, high; 5.5 x 103 μg/μL, low). For immunoprecipitation, lysates were treated at 90°C for 10 min in 1% SDS, 5 mM EDTA, and 2.5 mM EGTA (final concentration) followed by dilution to 0.1% SDS in PXL wash buffer (Chi et al., 2009). Lysates were incubated on ice for 10 min, followed by addition of anti-MBNL2 monoclonal (mAb) 3B4 (Santa Cruz sc-136167) and immunoprecipitation at 4°C for 2 hr. CLIP libraries were prepared using linkers for Illumina sequencing, including a modified barcoded 5′-linker (AGGGAGGAC GAUGCGNNNNG). Libraries were sequenced (36 cycles) using an Illumina Genome Analyzer IIx. The dCLIP analysis was performed as described previously (Wang et al., 2014) with modifications for comparative analysis of MBNL2 binding in control versus DM1 and DM2 brain (see Supplemental Experimental Procedures).
RNA-seq and PolyA-seq
For RNA-seq, 101 nt paired-end reads were generated from polyA-selected DM1 and control frontal cortex RNA (n = 3 each). Reads were aligned to the hg19 reference and a database of known exon junctions using OLego (Wu et al., 2013) and processed using Quantas (Zhang et al., 2014) to identify differentially spliced alternative exons. Exons with coverage ≥ 20, |ψ| ≥ 0.1 and FDR ≤ 0.05 were selected for analysis. Normalized complexity maps were generated as described previously (Charizanis et al., 2012) using RNA-seq, MBNL2 HITS-CLIP (Table S1) and genome-wide (hg19) binding sites identified using mCarts (Zhang et al., 2013). PolyA-seq libraries were prepared from frontal cortex tissue from wild-type, Nestin-Cre DKO, human control, DM1, and DM2 frontal cortex (Table S5) as described (Derti et al., 2012) with several modifications (see Supplemental Experimental Procedures for details).
Splicing and Polyadenylation Validation Assays
To validate AS changes, RNA was isolated from dissected mouse and human autopsy brain tissues (n=3). RNA integrity values were obtained using the Agilent 2100 Bioanalyzer (Table S5). RT-PCR splicing assays were performed as previously described (Lee et al., 2013a) using gene specific primers in flanking exons (Table S6). Validation of APA changes was performed using qRT-PCR (Batra et al., 2014) or a modified RT-PCR protocol, in which two alternative pA sites were simultaneously amplified using gene-specific forward primers and hybrid gene-specific/oligo(dT) reverse primers. Statistically significant AS and APA changes were identified using unpaired Student’s t-test.
Two-dimensional Gel Electrophoresis and Immunoblotting
Brain tissue was processed based on a previously published protocol (Fernandez-Gomez et al., 2014). For isoelectrofocusing (IEF), strips (pH 3-11NL) were rehydrated with the protein homogenate overnight at room temperature and IEF performed with an IPGphor III Isoelectrofocusing unit (GE Heathcare) at 20°C (see Supplemental Experimental Procedures for details). For phosphatase treatment, brain tissue was added to 10 volumes of 10 mM Tris and 320 mM sucrose supplemented with a cocktail of protease inhibitors (Roche Complete Mini EDTA-free) and 30 nM of okadaic acid (Calbiochem) and homogenized followed by sonication. Dephosphorylation was performed with 50 μg of protein in a final volume of 30 μL of Tris-sucrose buffer with 4 μl of 50 mM HEPES pH 7.5, 10 mM NaCl, 2mM DTT, 0.01% Brj35 × buffer (New England Biolabs), 4 μl of a 10 mM MnCl2 solution and 2 μl of lambda phosphatase (New England Biolabs). The mixture was mixed, incubated for 3 hr at 30°C and the reaction was stopped by heating the homogenate and addition of lysis buffer.
Supplementary Material
Acknowledgments
We thank J. Lewis for discussions on tauopathies, UF Research Computing for computational resources and J. Cleary and E. Wang for comments on the manuscript. This study was supported by grants from the NIH (AR06799 and NS058901) to M.S.S, Inserm to L.B. and N.S., CNRS to L.B., France Alzheimer and AFM to N.S., DN2M and ANR NeuroSplice to L.B., N.S., F.J.F.G. and S.E., LabEx and DISTALZ to L.B., N.S. and S.E. and the University of Lille, CHR of Lille and Region Nord Pas-de Calais to N.S.
Footnotes
AUTHOR CONTRIBUTIONS
M.G., A.M., N.S. and M.S.S. designed the study and prepared the manuscript, M.G. and R.B. performed HITS-CLIP and PolyA-seq, M.G., R.B. and K.C. contributed the computational analysis, M.G., A.M., K.Y.L. and R.B. performed AS and APA assays, S.W-V and C.Z. performed the human RNA-seq analysis, K.Y.L produced and characterized the control and Nestin-Cre mice, F.J.F.G, L.B. and N.S. analyzed tau isoform expression, S.E. developed the dephosphorylation assay, T.R., L.P.W.R, T.K. and A.M. determined DMPK CTG and CNBP CCTG expansion lengths, and T.K., J.D., K.T., and J.W.D. contributed human control and disease samples.
ACCESSION NUMBERS
Sequencing data from HITS-CLIP and polyA-seq were deposited in GEO under accession number GSE68890.
Supplemental Information is available online (___) and includes Supplemental Experimental Procedures, six figures, and six tables and can be found at______.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, Goodwin M, Zhang C, Sobczak K, Thornton CA, et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell. 2014;56:311–322. doi: 10.1016/j.molcel.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier C, Ghanem D, Fernandez-Gomez FJ, Jumeau F, Philippe JV, Freyermuth F, Labudeck A, Eddarkaoui S, Dhaenens CM, Holt I, et al. Tau exon 2 responsive elements deregulated in myotonic dystrophy type I are proximal to exon 2 and synergistically regulated by MBNL1 and MBNL2. Biochim Biophys Acta. 2014;1842:654–664. doi: 10.1016/j.bbadis.2014.01.004. [DOI] [PubMed] [Google Scholar]
- Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron. 2012;75:437–450. doi: 10.1016/j.neuron.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JD, Ranum LP. Repeat associated non-ATG (RAN) translation: new starts in microsatellite expansion disorders. Curr Opin Genet Dev. 2014;26C:6–15. doi: 10.1016/j.gde.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derti A, Garrett-Engele P, Macisaac KD, Stevens RC, Sriram S, Chen R, Rohl CA, Johnson JM, Babak T. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012;22:1173–1183. doi: 10.1101/gr.132563.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaenens CM, Schraen-Maschke S, Tran H, Vingtdeux V, Ghanem D, Leroy O, Delplanque J, Vanbrussel E, Delacourte A, Vermersch P, et al. Overexpression of MBNL1 fetal isoforms and modified splicing of Tau in the DM1 brain: two individual consequences of CUG trinucleotide repeats. Expt Neurol. 2008;210:467–478. doi: 10.1016/j.expneurol.2007.11.020. [DOI] [PubMed] [Google Scholar]
- Echeverria GV, Cooper TA. RNA-binding proteins in microsatellite expansion disorders: mediators of RNA toxicity. Brain Res. 2012;1462:100–111. doi: 10.1016/j.brainres.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, Brook JD. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- Gemayel R, Vinces MD, Legendre M, Verstrepen KJ. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Ann Rev Genet. 2010;44:445–477. doi: 10.1146/annurev-genet-072610-155046. [DOI] [PubMed] [Google Scholar]
- Goodwin M, Swanson MS. RNA-binding protein misregulation in microsatellite expansion disorders. Adv Expt Med Biol. 2014;825:353–388. doi: 10.1007/978-1-4939-1221-6_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA. Muscleblind proteins regulate alternative splicing. EMBO J. 2004;23:3103–3112. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huin V, Vasseur F, Schraen-Maschke S, Dhaenens CM, Devos P, Dupont K, Sergeant N, Buee L, Lacour A, Hofmann-Radvanyi H, et al. MBNL1 gene variants as modifiers of disease severity in myotonic dystrophy type 1. J Neurol. 2013;260:998–1003. doi: 10.1007/s00415-012-6740-y. [DOI] [PubMed] [Google Scholar]
- Jensen KB, Darnell RB. CLIP: crosslinking and immunoprecipitation of in vivo RNA targets of RNA-binding proteins. Meth Mol Biol. 2008;488:85–98. doi: 10.1007/978-1-60327-475-3_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, Swanson MS. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci USA. 2006;103:11748–11753. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Langlois MA, Lee KB, Riggs AD, Puymirat J, Rossi JJ. HnRNP H inhibits nuclear export of mRNA containing expanded CUG repeats and a distal branch point sequence. Nucl Acids Res. 2005;33:3866–3874. doi: 10.1093/nar/gki698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino Y, Washizu C, Kurosawa M, Oma Y, Hattori N, Ishiura S, Nukina N. Nuclear localization of MBNL1: splicing-mediated autoregulation and repression of repeat-derived aberrant proteins. Hum Mol Genet. 2014;24:740–756. doi: 10.1093/hmg/ddu492. [DOI] [PubMed] [Google Scholar]
- Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345:1139–1145. doi: 10.1126/science.1254917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent FX, Sureau A, Klein AF, Trouslard F, Gasnier E, Furling D, Marie J. New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats. Nucl Acids Res. 2012;40:3159–3171. doi: 10.1093/nar/gkr1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Li M, Manchanda M, Batra R, Charizanis K, Mohan A, Warren SA, Chamberlain CM, Finn D, Hong H, et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol Med. 2013a;5:1887–1900. doi: 10.1002/emmm.201303275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013b;5:1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Gotz J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PloS One. 2013;8:e84849. doi: 10.1371/journal.pone.0084849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345:1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan A, Goodwin M, Swanson MS. RNA-protein interactions in unstable microsatellite diseases. Brain Res. 2014;1584:3–14. doi: 10.1016/j.brainres.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Lammich S, Mackenzie IR, Forne I, Zilow S, Kretzschmar H, Edbauer D, Janssens J, Kleinberger G, Cruts M, et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013a;125:413–423. doi: 10.1007/s00401-013-1088-7. [DOI] [PubMed] [Google Scholar]
- Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013b;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- Nelson DL, Orr HT, Warren ST. The unstable repeats--three evolving faces of neurological disease. Neuron. 2013;77:825–843. doi: 10.1016/j.neuron.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niblock M, Gallo JM. Tau alternative splicing in familial and sporadic tauopathies. Biochem Soc Trans. 2012;40:677–680. doi: 10.1042/BST20120091. [DOI] [PubMed] [Google Scholar]
- Pettersson OJ, Aagaard L, Andrejeva D, Thomsen R, Jensen TG, Damgaard CK. DDX6 regulates sequestered nuclear CUG-expanded DMPK-mRNA in dystrophia myotonica type 1. Nucle Acids Res. 2014;42:7186–7200. doi: 10.1093/nar/gku352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos MG, Batra R, Li M, Yuan Y, Zhang C, Darnell RB, Swanson MS. Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice. Hum Mol Genet. 2013;22:3547–3558. doi: 10.1093/hmg/ddt209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nature structural & molecular biology. 2011;18:840–845. doi: 10.1038/nsmb.2067. [DOI] [PubMed] [Google Scholar]
- Ravel-Chapuis A, Belanger G, Yadava RS, Mahadevan MS, DesGroseillers L, Cote J, Jasmin BJ. The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J Cell Biol. 2012;196:699–712. doi: 10.1083/jcb.201108113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy K, Zamiri B, Stanley SY, Macgregor RB, Jr, Pearson CE. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J Biol Chem. 2013;288:9860–9866. doi: 10.1074/jbc.C113.452532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant N, Sablonniere B, Schraen-Maschke S, Ghestem A, Maurage CA, Wattez A, Vermersch P, Delacourte A. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum Mol Genet. 2001;10:2143–2155. doi: 10.1093/hmg/10.19.2143. [DOI] [PubMed] [Google Scholar]
- Suenaga K, Lee KY, Nakamori M, Tatsumi Y, Takahashi MP, Fujimura H, Jinnai K, Yoshikawa H, Du H, Ares M, Jr, et al. Muscleblind-like 1 knockout mice reveal novel splicing defects in the myotonic dystrophy brain. PloS One. 2012;7:e33218. doi: 10.1371/journal.pone.0033218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–724. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Xie Y, Xiao G. dCLIP: a computational approach for comparative CLIP-seq analyses. Genome Biol. 2014;15:R11. doi: 10.1186/gb-2014-15-1-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB, et al. Antisense Proline-Arginine RAN Dipeptides Linked to C9ORF72-ALS/FTD Form Toxic Nuclear Aggregates that Initiate In Vitro and In Vivo Neuronal Death. Neuron. 2014;84:1213–1225. doi: 10.1016/j.neuron.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Anczukow O, Krainer AR, Zhang MQ, Zhang C. OLego: fast and sensitive mapping of spliced mRNA-Seq reads using small seeds. Nucl Acids Res. 2013;41:5149–5163. doi: 10.1093/nar/gkt216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Lee KY, Swanson MS, Darnell RB. Prediction of clustered RNA-binding protein motif sites in the mammalian genome. Nucl Acids Res. 2013;41:6793–6807. doi: 10.1093/nar/gkt421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci USA. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA. 2013;110:E4968–4977. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.