

SUMMARY
The eight-subunit T cell receptor (TCR)-CD3 complex is the primary determinant for T cell fate decisions. Yet how it relays ligand-specific information across the cell membrane for conversion to chemical signals remains unresolved. We hypothesized that TCR engagement triggers a change in the spatial relationship between the associated CD3ζζ subunits at the junction where they emerge from the membrane into the cytoplasm. Using three in situ proximity assays based on ID-PRIME, FRET, and EPOR activity we determined that the cytosolic juxtamembrane regions of the CD3ζζ subunits are spread apart upon assembly into the TCR-CD3 complex. TCR engagement then triggered their apposition. This mechanical switch resides upstream of the CD3ζζ intracellular motifs that initiate chemical signaling as well as the polybasic stretches that regulate signal potentiation. These findings provide a framework from which to examine triggering events for activating immune receptors and other complex molecular machines.
INTRODUCTION
The eight-subunit αβ T cell receptor (TCR)-CD3 complex is essential for T cell fate decisions (Kuhns and Davis, 2012). This molecular machine consists of a ligand-binding module, the TCR (TCRα+TCRβ), coupled to the CD3γε, CD3δε, and CD3ζζ signaling modules via interactions in the transmembrane (TMD) and extracellular domains (ECD) (Call et al., 2002; Kuhns and Davis, 2007; Xu et al., 2006). The TCR binds composite surfaces of antigenic peptides embedded within major histocompatibility complex molecules (pMHC) on antigen-presenting cells (APCs) and relays specific information across the T cell membrane to the CD3 intracellular domains (ICDs) (Kuhns and Davis, 2012). There, mechanical information is converted to chemical information by Src kinase phosphorylation of the immune receptor tyrosine-based activation motifs (ITAMs) within the ICDs of the CD3 signaling modules (Kane et al., 2000; Reth, 1989). TCR multimerization, coincident changes in membrane composition, ITAM phosphorylation, and a rise in intracellular calcium are all proposed to potentiate signaling by relieving interactions between the CD3ε and CD3ζ ICDs with the inner leaf of the membrane (Aivazian and Stern, 2000; Gagnon et al., 2012; Shi et al., 2013; Zhang et al., 2011). Ultimately, these events instruct the fate decisions that drive T cell development, activation, differentiation, and the execution of effector functions (Guy and Vignali, 2009).
Despite a detailed understanding of TCR-pMHC interactions and intracellular signaling, the trigger that relays information from the TCR-pMHC interface to the CD3 ICDs remains poorly defined (Kuhns and Davis, 2012). The TCR-CD3 complex can function as a stand-alone molecular machine that generates transient signals in response to single TCR-pMHC interactions (Irvine et al., 2002; Ma et al., 2008; Manz et al., 2011). But, how this mechanical information is relayed across the membrane, for conversion to chemical information, requires a better understanding of how the complex subunits fit and work together upstream of the ICDs.
Data from less complex receptors indicate that changes in the proximity of the cytosolic juxtamembrane (JM) regions of receptor subunits can serve as a molecular trigger upon ligand engagement. For example, integrins hold their JM regions together until inside-out signaling triggers their divarication (i.e. spread apart) (Yang et al., 2009). Ligand engagement by epidermal growth factor receptor (EGFR) subunits is proposed to trigger a switch from an off to an on conformation by promoting interactions between JM segments that are normally sequestered away from each other (Endres et al., 2013). Furthermore, the JM regions of the homodimeric erythropoietin receptor (EPOR) are held apart until ligand engagement triggers their apposition (Livnah et al., 1999). Since evolution often converges on related principles to accomplish similar tasks, we hypothesized that the spatial relationship between the JM regions of the CD3ζζ homodimer is regulated to keep the TCR-CD3 complex in an inactive conformation, be that together or apart, until TCR engagement triggers a transition to an active conformation with an opposite spatial relationship.
We tested this hypothesis with three in situ reductionist systems designed to evaluate the spatial relationship of the JM regions of the CD3ζζ subunits on their own, when assembled within unengaged TCR-CD3 complexes, or after TCR engagement has triggered an active conformation. We focused on CD3ζζ because phosphorylation of the ITAMs within this module is key for most T cell fate decision (Guy and Vignali, 2009).
The solution structure of the TMDs of two disulfide-bonded CD3ζ helices shows a dimer with a small crossing angle, suggesting that the CD3ζζ TMDs emerge into the cytoplasm in close proximity when not assembled within a complex (Call et al., 2006). This provides a clear reference point for studying changes in the spatial relationship of the CD3ζζ subunits. Our hypothesis predicts that if the JM regions of the CD3ζζ subunits remain together within the TCR-CD3 complex then TCR engagement should trigger their divarication (Figure S1A). But, if assembly into the complex forces them apart then TCR engagement should trigger their juxtaposition (Figure S1B). If no changes occur between unengaged and engaged complexes then the hypothesis is incorrect (Figure S1C and S1D). Our data show that: the CD3ζζ cytosolic JM regions are in apposition in situ when not assembled within a TCR-CD3 complex; assembly into the complex divaricates these regions into what we propose to be an off conformation; and TCR engagement then triggers their juxtaposition into what we propose is an on conformation. We interpret these data as evidence of a pivot point in the linkage between the TCR and CD3ζζ that facilitates the transfer of mechanical information across the cell membrane.
RESULTS
In situ systems were engineered for monitoring the cytoplasmic juxtamembrane regions
Three complementary in situ reductionist systems were engineered for specific purposes and to cross-verify results. Their common properties and functional differences are briefly introduced here since they are integral to the hypothesis being tested (detailed in Supplemental Experimental Procedures, Figure S2 and S3). Live-cell systems were used since the TCR-CD3 complex must assemble within a cellular membrane (Sun et al., 2001; Sun et al., 2004). In addition, cells of non-T cell origin (M12 and Ba/F3 cells) were used to prevent competition between engineered and endogenous complex subunits prior to analysis in T cell hybridomas (Kuhns and Davis, 2007; Kuhns et al., 2010). Since our question focused on mechanical changes in the spatial proximity of the CD3ζζ cytosolic JM regions that occur upstream of intracellular events, one version of each system was built with truncated CD3γ, δ, and ε subunits (CD3T: CD3γT, δT, and εT) (Kuhns et al., 2010). These lacked the intracellular motifs that mediate signaling, signaling-feedback, and trafficking events but retained their TMDs and ECDs to allow assembly and stability within a TCR-CD3 complex (Aivazian and Stern, 2000; Call et al., 2002; Delgado and Alarcon, 2005; Fernandes et al., 2012; Gil et al., 2002; Kuhns and Davis, 2007; Lauritsen et al., 1998; Liu et al., 2000; Xu et al., 2006)(Figure S2D). A second version was built to address if concordant result would be obtained with full length CD3γ, δ, and ε subunits (CD3FL: CD3γFL, δFL, and εFL).
The key distinction between each system lay in the proximity probes fused to CD3ζ at the membrane-cytoplasm boundary. Testing our hypothesis required resolving intra- (cis) from inter-module (trans) interactions between CD3ζζ subunits. We addressed this directly through the use of a probe ligase (the W37V variant of E. coli lipoic acid ligase (LplAW37V)) in combination with its peptide substrate (ligase acceptor peptide 1 or LAP1) for Interaction-Dependent PRobe Incorporation Mediated by Enzymes (ID-PRIME) (Figure S2A, S3A-S3C)(Slavoff et al., 2011; Uttamapinant et al., 2013; Uttamapinant et al., 2010). The affinity between LplAW37V and LAP1 has been tuned such that probe transfer is efficient only when ligase and peptide are proximal, thus providing fine spatial resolution in live cells. The second system relied upon monomeric enhanced green fluorescent protein (mEGFP) and monomeric cherry (mCh) as Förster Resonance Energy Transfer (FRET) donor and acceptor probes, respectively (Figure S2B, S3A, S3B, S3D, S3E) (Vanderklish et al., 2000; Youvan et al., 1997; Zal and Gascoigne, 2004). Measuring FRET by live cell microscopy allowed us to assess interactions between CD3ζζ subunits at distinct cellular locations. Finally, we modified a previously described system in which the intracellular domain of the erythropoietin receptor (EPOR) was used as a proximity probe to drive proliferation of Ba/F3 cells (Figure S2C, S3A and S3B) (Kuhns et al., 2010). This provided a highly sensitive readout that relates interactions between CD3ζζ subunits to a biological outcome.
The CD3ζζ juxtamembrane regions divaricate within the TCR-CD3 complex at steady state
Our hypothesis was predicated on the CD3ζζ TMD solution structure, which predicts that the JM regions of unassembled CD3ζζ subunits lie in close proximity (Call et al., 2006). Confirming this in situ was necessary to establish a positive control as the reference point for comparison with CD3ζζ subunit interactions within TCR-CD3 complexes.
ID-PRIME allowed us to directly distinguish cis (intra-module) from trans (inter-module) interactions based on ligase activity, size, and abundance of the CD3ζζ modules (Figure 1A). The CD3ζ probe ligase chimera (CD3ζLplA) and the peptide substrate chimera (CD3ζLAP1) reported spatial proximity based on covalent picolyl azide (pAz) ligation to LAP1 by LplAW37V (Uttamapinant et al., 2010). pAz was subsequently derivatized by biotin in cell lysate via copper-catalyzed azide alkyne cycloaddition (CuAAC) for detection and quantification by streptavidin blotting (Figure S4A). Differences in size between the CD3ζζ modules allowed for each species to be identified and quantitated by non-reducing SDS-PAGE and blotting (Figure 1B).
Figure 1. Cis interactions between CD3ζLplAζLAP1 probes are reduced in TCR-CD3 complexes.

(A) Illustration of CD3ζζ modules. Arrows show cis (red) or trans (peach) interactions.
(B) Non-reducing SDS-PAGE of CD3ζζ modules from CD3T-only and TCR-CD3T M12 cell lysates. Two-color Western blot for HA (blue) and Myc (green) tagged CD3ζLplA and CD3ζLAP1 subunits, respectively (cyan = HA+Myc).
(C) Abundance of CD3ζζ species as a percent of total. Bars represent the mean +/−SEM for each species after band quantification (n=7 replicates).
(D) ID-PRIME after 0, 1, 2, and 5 minutes at 37°C. Ligated CD3ζLplAζLAP1 (cis) and CD3ζLAP1ζLAP1 (trans) modules is shown by two-color blot for immunoprecipitated (IP) CD3ζζ modules. Total LAP1 (Myc = green) and ligated LAP1 after biotinylation by CuAAC (SA = red) are shown overlaid or broken out individually in grey scale. See also Figure S4A.
(E) Cis interactions decrease in TCR-CD3 complexes. Bars represent the mean +/− SEM for cis modification (SA/Myc) of CD3T-only and TCR-CD3T M12 cell samples normalized to the cis CD3-only SA/Myc value at the indicated time points within an experiment (n=4 experiments at 0’, 1’, 2’; n=3 experiments at 5’; *p<0.05, Mann-Whitney).
(F) eak trans modification. Trans modification (SA/Myc) of CD3T-only and TCR-CD3T samples from individual experiments normalized to the cis CD3T-only SA/Myc value (as in E).
(G-I) Repeat with CD3FL-only and TCR-CD3FL M12 cells. (G) ID-PRIME for 5 minutes at 37°C with quantification of (H) cis and (I) trans modification (SA/Myc) of CD3FL-only and TCR-CD3FL M12 cell samples normalized to the cis CD3-only SA/Myc value (mean +/−SEM of three replicates from 2 independent experiments; *p<0.05, Mann-Whitney).
Within CD3T-only and TCR-CD3T M12 cell lines, the CD3ζLAP1ζLAP1 module was the most abundant species while CD3ζLplAζLAP1 and CD3ζLplAζLplA made up the remainder (Figure 1C). If cis interactions dominate then the low abundance CD3ζLplAζLAP1 module would contain the most heavily modified LAP1 at steady-state. But, if trans interactions dominate then the limiting pool of ligase would more heavily modify the abundant CD3ζLAP1ζLAP1module, which lacks intrinsic ligase activity. A minor band of free LplAW37V was observed just below the CD3ζLplA monomer due to translation initiation at an internal start codon (Figure 1B). This was removed for analysis of the CD3FL-only and TCR-CD3FL cell lines (Figure S3G).
ID-PRIME was performed as pulse experiments to measure the spatial proximity of the CD3ζζ JM regions alone or within TCR-CD3 complexes (Figure S4A). Modification of the CD3ζLplAζLAP1 module increased over time at 37°C more than the high abundance CD3ζLAP1ζLAP1 module in CD3T-only cells (Figure 1D), establishing that the CD3ζζ cytosolic JM regions were together in CD3T-only cells while trans interactions were rare. Cis interactions in CD3T-only cells thus provided a reference point for asking if changes occur upon assembly into the TCR-CD3T complex. Indeed, cis modification was lower in TCR-CD3T cells compared with CD3T-only cells, indicating that assembly into the complex divaricated the cytoplasmic JM regions (Figure 1D and 1E), while trans interactions were again rare (Figure 1D and 1F).
We next asked if the CD3ζζ subunits would divaricate upon assembly into complexes containing full length CD3 heterodimers. Cis modification in the CD3FL-only cells was reduced in the TCR-CD3FL cell lines (Figure 1G-1I), indicating that the cytoplasmic JM regions of the CD3ζζ subunits spread apart in a full TCR-CD3 complex.
Of note, modification of the CD3ζLplAζLAP1 modules in TCR-CD3 cells was not eliminated. The signal could arise from free CD3ζLplAζLAP1 modules that are not assembled within a complex. Also, equilibrium between juxtaposed and divaricated conformations of the CD3ζζ subunits could shift towards a divaricated state within a complex without being locked into this state. Finally, the CD3ζLplA and CD3ζLAP1 were engineered with linkers designed to allow the LAP1 substrate to reach the active site of an adjacent LplAW37V for modification. If the distance to which the subunits are spread apart reduces the local concentration of LAP1 and ligase but is insufficient to fully prevent LAP1 from reaching the enzyme active site then this would result in some signal. For example, experiments with CD28 and a TCR-based controls indicated that probe ligation is sensitive to the distance from an intra-chain bond to the LAP1 and resultant changes in the potential range of motion (Figure S4B-S4E). For the unassembled CD3ζζ subunits, which have N- and C-terminal TMD interactions (Call et al., 2006), the JM probes should be very close together. Given the N-terminal TMD disulfide bond between the CD3ζζ subunits, and the TMDs electrostatic TCRα-CD3ζζ interactions, the arch of movement for the chains to pivot around this linkage upon complex assembly is likely to be restricted. The possibilities listed above are not mutually exclusive. Still, the data point to divarication of the CD3ζζ cytoplasmic JM regions upon assembly into the TCR-CD3 complex (Figure S1B and S1D).
The CD3ζζ juxtamembrane regions are divaricated upon TCR-CD3 complex assembly
The spatial proximity of the CD3ζζ JM regions was independently assessed by FRET via live cell imaging to interrogate interactions at subcellular locations. CD3ζT subunits were fused via short, flexible linkers to mEGFP (CD3ζG) or mCh (CD3ζCh) to allow transient alignment of the probe dipoles since FRET depends upon both the distance (<100Å) and relative dipole orientation. Three CD3ζζ species (i.e. CD3ζGζG, CD3ζGζCh, and CD3ζChζCh) should be expressed in all the M12 lines (Figure S2B). Theoretically, the CD3ζGζCh species should be the most abundant (~50%) since proportional amounts of CD3ζG and CD3ζCh (diagonal by flow cytometry) were observed in CD3T/FL-only and TCR- CD3T/FL expressing M12 cells (Figure S3D and S3E).
Whole cell FRET was measured by three-channel sensitized emission using live wide-field fluorescence microscopy. Correction for spectral overlap (FRETc) produced values (Figure S5A) that were normalized to each cell’s donor intensity (nFRETc) as an index for cell-to-cell comparison within the cell lines and between the individual lines (Vanderklish et al., 2000; Youvan et al., 1997; Zal and Gascoigne, 2004). FRET in CD3T-only cells was two logs greater than a negative control line, but lower than a cell line in which all donor and acceptor molecules should be juxtaposed (Figure S5B-S5I). The signal in CD3T-only cells again served as the reference point for comparison of the nFRETc signal from TCR-CD3T cells, which was lower (Figure 2A-2C). Accounting for expression differences between the two populations by subset analysis of CD3T-only and TCR-CD3T cells based on CD3ζG and CD3ζCh expression (Figure 2D and 2E, shaded gates) also revealed a significant reduction in nFRETc in the TCR-CD3T cells (Figure 2F).
Figure 2. FRETc between CD3ζGζCh probes is reduced in TCR-CD3 complexes.

(A-J) FRETc is lower in TCR-CD3T cells. (A and B) Representative bright field images of (A) CD3T-only and (B) TCR-CD3T cells are shown at 60X with overlaid FRETc (pseudo color). Scale bar is 10μm. Arrows indicate cells shown in mEGFP (green), mCherry (red), and FRETc (pseudo color) overlaid on bright field images or alone after digital zoom (scale bar is 5 μm). Intracellular ROI of 3.6μm2 used for intracellular analysis in G-J is in yellow. Bright vesicles were excluded from intracellular analysis.
(C) FRETc values normalized on a per cell basis to mEGFP (nFRETc) for statistical comparison between CD3T-only and TCR-CD3T lines.
(D-F) Subset analysis of nFRETc. Expression of (D) mEGFP and (E) mCherry are shown. Shaded gates highlight subsets of cells analyzed for (F) nFRETc.
(G-J) Intracellular subset analysis. (G) nFRETc, (H) mEGFP, and (I) mCherry for the whole population are shown. Shaded gates highlight subsets of cells analyzed for (J) nFRETc. Representative of four experiments. Each dot represents a single cell (n). Green bars represent the median values (***p<0.0001; Mann-Whitney). See also Figure S5A-I.
The bulk of CD3ζG and CD3ζCh fluorescence in the CD3T-only and TCR-CD3T cells occurred intracellularly where subunits are synthesized, assembled, recycled and degraded. Analysis of intracellular nFRETc was performed with regions of interest (ROIs) positioned in areas of high intensity lacking bright vesicles (Figure 2A and 2B). Again, nFRETc values were higher for CD3T-only cells than TCR-CD3T cells in the total population (Figure 2G) and for subsets (Figure 2H and 2I, shaded gates, and 2J).
Live total internal reflection fluorescence microscopy (TIRFM) was performed on cells adhered to a glass coverslip to evaluate FRET at the cell surface where the TCR encounters its ligand. We measured donor recovery after acceptor bleaching (S6A-S6F, Movie S1 and S2) as an independent method of FRET quantitation in live cells (Xu et al., 2008). This yields a relative FRET efficiency (FRETE) value that is distinct from the index determined by excited emission, and was observed to be higher in positive control M12 cells than negative controls (Figure S6A-S6C). For TCR-CD3T cells, FRETE was lower than CD3T-only cells (Figure 3A and 3B). On average, acceptor bleaching and recovery was equivalent for both populations so any diffusion of unbleached species into the ROI would similarly impact the FRETE calculations for both populations (S6D and S6E). FRETE was also lower in TCR-CD3FL cells than cells expressing only CD3ζG and CD3ζCh (Figure 3C and S6F).
Figure 3. FRETE is lower in TCR-CD3T cells at the membrane.
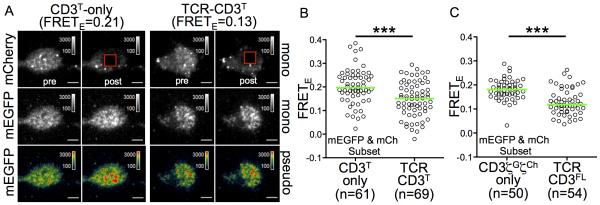
(A) TIRFM images pre and post mCherry bleaching. mCherry is shown in grey scale. mEGFP is shown in grey scale and pseudocolor. Scale bar is 5μm. Vesicles bright in mCherry are just below the membrane. See also Movie S1 and S2.
(B and C) FRETE was calculated for (B) CD3T-only and TCR-CD3T cell subsets or (C) CD3ζGζCh-only and TCR-CD3FL cell subsets by donor recovery after acceptor photo bleaching. Representative of (A and B) three or (C) two experiments. Each dot represents a single cell (n). Green bars represent the median values (***p<0.0001; Mann-Whitney). See also Figure S6A-S6F.
These data indicate that a measurable fraction of the CD3ζζ subunits emerge from the membrane into the cytoplasm in close proximity when not assembled within a complex. Assembly into TCR-CD3 complexes then forces their cytoplasmic JM regions apart, as in Figure S1B and S1D. FRET signals in TCR-CD3 cells could further be reduced due to a steric boundary created by the complex ECD that would impair trans FRET signal (Aivazian and Stern, 2000).
Functional divarication of the CD3ζζ subunits
In the EPOR system, if two EPOR ICDs are within ~39Å, their JM-associated JAK2 molecules undergo trans-activation that drives Ba/F3 cell proliferation (Livnah et al., 1999). Quantifying this proliferation is an accepted measure of the spatial relationship of the EPOR JM regions (Constantinescu et al., 2001; Kuhns et al., 2010; Yamasaki et al., 2006) that complements ID-PRIME and FRET by providing a readout for the biological consequences of changes in the spatial relationship of the CD3ζζ subunits.
The CD3ζEζE subunits drove Ba/F3 proliferation in CD3T-only and CD3FL-only cells, while complex assembly in TCR-CD3T and TCR-CD3FL cells abrogated proliferation (Figure 4A and 4B). These results indicate that the CD3ζζ JM regions are close together when not assembled within a complex and divaricated upon assembly into the complex (Figure S1B and S1D). Since the orientation of the EPOR cytosolic JM region can impact the efficiency by which juxtaposed EPOR ICDs signal (Constantinescu et al., 2001; Livnah et al., 1999; Lu et al., 2006), assembly of the CD3ζEζE chimeras into the TCR-CD3 complex could twist the TMDs to further alter the spatial relationship of these subunits. This cannot be ruled out. But, given that an N-terminal disulfide bond covalently links the CD3ζζ subunits and charge interactions link TCRα to CD3ζζ, any twisting of the TMD while constrained at one end would likely lead to divarication at the other end in the same way that twisting two sticks bound at one end by a pivot-point would result in spreading at the other.
Figure 4. CD3ζE-driven Ba/F3 cell proliferations is inhibited in TCR-CD3 cells.

CD3ζEζE drives Ba/F3 cell proliferation in CD3T/FL-only cells but not TCR-CD3T/FL cells. Bars represent mean proliferation of (A) four CD3T-only and TCR-CD3T or (B) three CD3FL-only and TCR-CD3FL independently generated sets of cell lines +/− SEM. Parental Ba/F3 cells for each line were included in each experiment (***p<0.0001; one-way ANOVA with Tukey’s post-test).
TCR engagement triggers the apposition of CD3ζζ juxtamembrane regions
The data from all systems indicate that the cytoplasmic JM regions of the CD3ζζ subunits spread apart within TCR-CD3 complexes (Figure S1B and S1D). We interpret this to be the off conformation, suggesting that TCR engagement brings the CD3ζζ cytoplasmic JM regions together into an on conformation (Figure S1B). Evidence for pMHC-driven proliferation of TCR-CD3T Ba/F3 cells was observed in the EPOR system; however, this system lacked the resolution to discriminate clustering from an intra-complex mechanical change (Figure S6G).
To evaluate changes by FRET, TCR-CD3T cells were imaged by TIRFM on glass coverslips coated with null (HB-Ek) or agonist (MCC-Ek) pMHC (Figure 5A and 5B) for the 2B4 TCR used in this study (Kuhns et al., 2010). Acquisition by three-channel sensitized emission revealed a significant increase in nFRETc for cells on agonist pMHC surfaces compared with null surfaces (Figure 5C), indicating that TCR engagement decreased the spatial distance between the JM probes.
Figure 5. TCR engagement of agonist pMHC increases FRET.

(A-F) Membrane associated FRETc on null and agonist pMHC surfaces. TCR-CD3T M12 cells on glass surfaces coated with (A) null pMHC or (B) agonist pMHC imaged by TIRFM. Representative fields are shown at 60x magnification in bright field overlaid with FRETc (scale bar is 10μm). Arrows indicate cells enlarged to show mEGFP, mCherry, and pseudocolored FRETc images overlaid on bright field images or alone (scale bar is 5μm).
(C) Single cell nFRETc values for the total population.
(D-F) TCR engagement increases contact area, complex density, and FRET. (D) The number of pixels analyzed per cell and (E) mean mCherry signal per pixel are shown for single cells.
(F) nFRETc for a subset of TCR-CD3T cells based on equivalent contact area (from D; shaded gate), mean mCherry intensity (from E; shaded gate), and mean mEGFP intensity (as in E; not shown). Representative of three experiments.
(G) FRETE is proportional to agonist pMHC concentration. TIRFM was performed on TCR-CD3T cells adhered to glass coverslips coated with pMHC. Agonist pMHC was diluted into null pMHC as indicated. FRETE was quantified as in Figure 2 for mEGFP and mCherry subsets. Circles represent means (+/−SEM) for at least 45 cells per pMHC concentration. Representative of two experiments.
(H and I) FRETE increases over background with 1% agonist pMHC in (H) TCR-CD3T M12 cells and (I) TCR-CD3FL M12 cells. Representative of two experiments. Dots are single cells and green bars are median values (**p<0.001; ***p<0.0001; Mann-Whitney).
FRETc was observed in diffuse puncta reminiscent of microclusters seen on T cells treated with the Src kinase inhibitor PP2 (Yokosuka et al., 2005), consistent with the absence of ITAMs and signaling in these complexes (Figure 5B). A larger contact area (Figure 5D) and increased accumulation of TCR-CD3T complexes was also observed on agonist surfaces (Figure 5E). The potential for multimer formation upon TCR engagement at high pMHC densities is presumed to occur within accumulated TCR clusters, although the diffraction limit of 250 nm prevents this from being established in such experiments (Huang et al., 2010). Still, some of the FRET signal may result from inter-complex (trans) juxtaposition of CD3ζζ subunits (Kuhns et al., 2010). We thus analyzed a subset of cells on the null and agonist pMHC surfaces that had equivalent contact area and mean TCR-CD3T complex density (shaded gate, Figure 5D and 5E). An increase in nFRETc still occurred upon engagement, demonstrating that at equivalent TCR-CD3T densities on the pMHC surfaces TCR engagement leads to a significant increase in CD3ζζ juxtaposition (Figure 5F).
TCR triggering of cis and trans interactions at high ligand densities are not mutually exclusive. To interrogate this further we diluted agonist pMHC into null pMHC on glass coverslips, while keeping the amount of pMHC constant, in order to quantitate engagement-induced FRET when single agonist pMHC should be surrounded by null pMHC. The surfaces were not mobile so TCR-driven accumulation of agonist pMHC at the contact interface could not occur or influence the results. FRETE decreased in a dose dependent manner for TCR-CD3T cells on agonist surfaces (Figure 5G). But, higher signal was still observed when only 1% of the pMHC were agonists compared with those on a completely null surface (Figure 5H). A similar result was observed for TCR-CD3FL cells on a 1% agonist surface (Figure 5I). These data suggest that isolated TCR-pMHC interactions induce an intra-complex conformational change in the CD3ζζ subunits to facilitate cis interactions at the JM region since any single agonist pMHC should, on average, be surrounded by ninety-nine null pMHC. This is consistent with TCR signaling in response to single agonist pMHC (Irvine et al., 2002; Ma et al., 2008; Manz et al., 2011).
TCR engagement triggers a mechanical change in the CD3ζζ module
Performing ID-PRIME with TCR-CD3T cells allowed us to assess if TCR triggering promotes a mechanical change in the spatial proximity of the CD3ζLplAζLAP1 JM probes that are independent of intracellular signaling. The experiments were conducted under conditions approximating those in which T cells naturally encounter antigenic pMHC on the surface of antigen presenting cells (APCs). In short, since the TCR-CD3T cells are M12 cells, we used a T cell hybridoma (58α−β− cells) as an APC by expressing I-Ek molecules with tethered MCC peptides and the parental 58α−β− cells, which do not express class II MHC, as the negative control (Figure S3F). TCR accumulation (TCRβ-mEGFP) was observed at the interface between TCR-CD3T cells and pMHC+ APCs, even though the CD3 molecules lack ITAMs and cannot signal, while accumulation did not occur between TCR-CD3T cells and control APCs (Figure 6A, Movie S3 and S4). These data provide evidence for TCR-pMHC interactions in this system.
Figure 6. A mechanical pivot in the linkage between TCRα and CD3ζζ.

(A) TCR-CD3T M12 cells accumulate TCRs at the interface with pMHC+ APCs. TCR-CD3T cells were incubated with control (top) or pMHC+ APCs (bottom). Live cell imaging (30 seconds per frame) is shown as TCRβ-mEGFP overlaid on DIC images or mEGFP alone. See also Movies S3 and S4 and Figure S7A and S7B
(B-D) Increased cis interactions after TCR engagement. (B) CD3ζLplAζLAP1 (cis) and CD3ζLAP1ζLAP1 (trans) modules are shown after ID-PRIME and CuAAC by two-color blot for Myc tag IPs of lysates from TCR-CD3T cells co-incubated with control or pMHC+ APCs. Change in (C) cis or (D) trans modification after TCR engagement relative to cis SA/Myc values from control APC samples are shown as the mean +/−SEM of 4 replicates from 4 independent experiments (*p<0.05; Mann-Whitney).
To assess TCR-triggered cis interactions between the CD3ζLplAζLAP1 probes, TCR-CD3T cells and APCs were co-pelleted prior to shifting to 37°C. Baseline modification of the CD3ζLplAζLAP1 module, as seen in Figure 1D, was observed for lysates of TCR-CD3T cells co-incubated with control APCs (Figure 6B). This served as the comparator for lysates from TCR-CD3T cells co-incubated with pMHC+ APCs. TCR engagement resulted in increased cis modification of the CD3ζLplAζLAP1 modules relative to the control (Figure 6B and 6C). Despite the high pMHC density and TCR accumulation at the cell-cell interface, trans modification of the CD3ζLAP1ζLAP1 module was minor (Figure 6B and 6D). These data provide direct evidence for a conformational change in the spatial relationship of the cytoplasmic JM regions of the CD3ζζ subunits upon TCR triggering.
The data in Figure 6C represent changes in CD3ζLplAζLAP1 modification normalized to the total LAP1 signal (SA/Myc) for the agonist pMHC lysates relative to the control, similarly to methods used to report changes in ITAM phosphorylation after TCR engagement (Kersh et al., 1998). Since TCR triggering is focused at a small area of cell-cell contact, unlike receptors for soluble ligands (e.g. EGFR), only a fraction of TCRs being engaged on any given cell lead to a change in signal. Thus, the fold change relative to the whole is small. To explore this further, we quantitated contacts between TCR-CD3T cells and APCs under conditions mimicking our ID-PRIME experiments. On average, any given TCR-CD3T cell made a single productive contact with an APC and the amount of TCR-CD3T complexes at an interface, relative to the total TCR (mEGFP) in a cell, was typically less than five percent (Figure S7A and S7B).
The affinity of TCR-pMHC interactions influences the CD3ζζ mechanical pivot
We next asked if we could observe this mechanical switch in the CD3ζζ subunits by ID-PRIME in cells expressing full length CD3γε and CD3δε subunits. Cis modification increased for TCR-CD3FL M12 cells co-incubated with APCs expressing agonist pMHC over the control, while trans interactions were absent (Figure 7A-7C). This difference was less than that observed with TCR-CD3T cells, but these cells did not as readily accumulate TCR at the contact interface (Figure S7C and S7D). This may be due to restored endocytosis of TCR-CD3FL complexes or signaling (Lauritsen et al., 1998; Liu et al., 2000).
Figure 7. TCR triggered cis interactions between CD3ζLplAζLAP1 probes.

(A-C) CD3ζζ cis modification in TCR-CD3FL M12 cells after TCR engagement. (A) ID-PRIME after 5 minutes co-culture of TCR-CD3FL cells with control or agonist-pMHC APCs at 37°C. Analysis of (B) cis and (C) trans SA/Myc signal relative to the control APC cis SA/Myc signal shown as the mean +/−SEM of 7 replicates from 4 independent experiments (*p<0.05, Mann-Whitney). See also Figure S7C and S7D.
(D-F) Divarication of the CD3ζζ subunits in 58α−β− T cell hybridomas. (D) ID-PRIME of CD3FL-only and TCR-CD3FL cells compared for (E) cis and (F) trans interactions between CD3ζζ relative to the CD3-only cis signal shown as mean +/−SEM of 3 replicates from 3 independent experiments (*p<0.05, Mann-Whitney).
(G-I) CD3ζζ cis modification in TCR-CD3FL 58α−β− T cells after TCR engagement with weak (T102S) or strong (MCC) agonist pMHC. (G) ID-PRIME after 5 minutes co-culture of TCR-CD3FL cells with control, (H) T102S, or (I) MCC APCs at 37°C. Cis interactions after T102S (n=3 replicates) or MCC (n=5 replicates) are shown relative to the control cis signal as mean +/− SEM of indicated replicates from 3 independent experiments (**p<0.001; Mann-Whitney). See also Figure S7E and S7F.
Finally, similar experiments were performed with CD3FL-only and TCR-CD3FL 58α−β− T cell hybridomas coupled to M12 cells expressing pMHC as APCs (Figure S7E and S7F). A reduction in cis and absence of trans interactions between CD3ζζ subunits was verified by comparing CD3FL-only and TCR-CD3FL cells (Figure 7D-7F). Upon TCR engagement, cis interactions trended higher with APCs presenting the weak agonist peptide, T102S, compared with the control (Figure 7G and 7H) while a greater increase was observed with agonist peptide (MCC) compared with controls (Figure 7G and 7I). Trans interactions were again absent (Figure 7G). Therefore, induction of this mechanical switch occurs in cells with full-length CD3 modules and is influenced by the affinity of TCR-pMHC interactions.
DISCUSSION
The mechanics by which the TCR-CD3 complex relays pMHC-specific information across the T cell membrane to initiate intracellular signaling has been enigmatic. Our data show that the CD3ζζ JM regions are forced apart upon assembly within a TCR-CD3 complex into what we propose represents an inactive conformation. TCR engagement then triggers a conformational change whereby the CD3ζζ cytosolic JM regions move together into what we propose is the active conformation.
The structural features most likely to facilitate these changes are the TMD charge interactions between CD3ζζ and TCRα (Call et al., 2002). These occur within the low dielectric environment of the lipid bilayer interior and are postulated to form a very strong bond that could serve as a pivot point around which the interacting subunits could move without risk of separation (Engelman, 2003). This idea cannot be tested by means such as mutagenesis since these charge interactions are essential for complex assembly (Call et al., 2002). But, the experiments performed here strongly support a model in which the negatively charged TMD residues of the CD3ζζ subunits interact with the positively charged residue in the TCRα TMD to create a pivot point. The N-terminal positioning of this hinge relative to the CD3ζζ cytosolic JM regions would maximize the arc of movement for these regions to diverge from each other upon assembly into mature TCR-CD3 complexes. This suggests an energetic input to divaricate the subunits that could be stored until released by TCR engagement. The hinge would then allow the TMDs to swing together upon TCR triggering to facilitate intra-complex CD3ζζ interactions, thus relaying information across the membrane. To what extent the CD3ζζ TMDs inhabit different states within different membrane environments, or induce membrane curvature in the inactive versus active conformations represent important questions for future studies.
Coupling TCR engagement to this pivot is mechanical. The CD3 motifs that connect the complex to the intracellular signaling machinery and those that potentiate signaling were removed in most of our experiments. Thus, it is possible that cytoskeletal movement actuates this mechanical switch (Kim et al., 2009; Li et al., 2010; Liu et al., 2014; Ma et al., 2008). Indeed, how engagement is coupled to this switch requires consideration of a number of key findings discussed elsewhere – we favor a model whereby the TCR acts as a lever that pivots over the top of the CD3γε and CD3δε signaling modules upon engagement and consequently applies an upward pull on the linkage between the TCR and CD3ζζ (Kuhns and Davis, 2012). But, a variety of scenarios can be envisioned whereby a push or pull on the TCR, or simple engagement, could induce the mechanical switch observed here (Kuhns et al., 2006; Sun et al., 2001; van der Merwe, 2001). These remain to be experimentally tested, refined, and resolved.
Once pMHC-specific information is relayed to the cytoplasm, it likely induces a change in the CD3ζζ ICDs that allows an initial connection between the TCR-CD3 complex and the intracellular signaling machinery. This could involve a change in interactions between the CD3ζζ ICDs and the inner leaf of the plasma membrane, as proposed for EGFR (Endres et al., 2013), which could facilitate Src kinase phosphorylation of an ITAM or interactions with other molecules (Zhang et al., 2011). Mechanical information would then be converted to chemical information via phosphorylation events to initiate signaling, whereby second messengers and/or changes in membrane composition could potentiate signaling via feedback mechanisms that further relieve interactions between CD3ζζ ICDs and the inner leaf of the membrane (Gagnon et al., 2012; Shi et al., 2013).
Trans interactions between CD3ζζ ICDs could also potentiate signaling by increasing the local ITAM concentration for phosphorylation by the Src kinases, or binding of phosphorylated ITAMs by the Syk kinase ZAP-70 (Aivazian and Stern, 2000; Brdicka et al., 2005; Kuhns and Davis, 2012; Kuhns et al., 2010). Accumulation of TCRs at the immunological synapse, and reports of TCR multimers, all point towards inter-complex TCR-CD3 interactions playing a role in chemical signal amplification after it is initiated (Kuhns and Davis, 2012). This would be particularly relevant for CD3ζζ, which we predict to sit below a TCR dimerization interface (Kuhns et al., 2010). However, the near absence of trans interactions in our ID-PRIME data appears to be inconsistent with such models.
The design of the experimental systems is relevant to this discussion. Each was engineered with proximity probes at the membrane-cytoplasm junction in order to measure the spatial relationship between the CD3ζζ JM regions and evaluate if TCR triggering involves regulating the intra-module distance at this region. As previously noted, the ECDs of the TCR-CD3 complex subunits may pose a steric boundary that prevents direct trans interactions between JM probes even though the natural ICDs of the CD3ζζ modules, with three ITAMs, are longer and might span the distance created by this boundary to potentiate signaling (Aivazian and Stern, 2000). The intense FRET signals observed at high ligand-densities on immobile surfaces could be explained by both intra- and inter-module FRET since this readout can occur without direct probe-probe contact. In contrast, ID-PRIME requires direct interactions between the probe ligase and substrate peptide. These may not reach one another, even if positioned in close proximity, due to the ECD steric boundary. An interesting implication of this is that the L19 residue in the CD3ζ TMD that is reported to mediate clustering in the T cell membrane may do so by influencing co-localization based on membrane lipid composition rather than direct interactions between distinct TCR-CD3 complexes (Kumar et al., 2011).
The conformational change reported here provides insights into how a TCR-CD3 complex can function as a stand-alone unit to generate transient chemical signals from mechanical information (Irvine et al., 2002; Ma et al., 2008; Manz et al., 2011). This would be relevant when T cells scan the surface of an APC for agonist TCR-pMHC interactions that would induce a stop signal and accumulation of higher concentrations of agonist pMHC in the immunological synapse. Sustained signaling is known to require higher pMHC densities (Irvine et al., 2002; Manz et al., 2011), supporting the idea that higher order TCR-pMHC structures can potentiate intracellular signaling once it is initiated (Aivazian and Stern, 2000; Kuhns and Davis, 2012; Kuhns et al., 2010). A modified experimental design will be required to specifically investigate trans interaction in future studies in order to further evaluate the distinct steps in the highly regulated and complex process of TCR-CD3 signaling that directs T cell fate decision (Kuhns and Davis, 2012).
Altogether, these data move our understanding of TCR triggering upstream of the CD3ζζ ICDs. The results also provide a framework from which to study the mechanics of this and other activating immune receptors in which the receptor and signaling modules are associated via transmembrane charge interactions. In addition, they provide a basis for cross-comparative studies with the functionally analogous receptor complexes, such as the B cell receptor (BCR)(Yang and Reth, 2010).
EXPERIMENTAL PROCEDURES
Constructs, standard experimental procedures, and more detailed descriptions of experimental protocols, imaging, and analysis are in Supplementary Experimental Procedures.
ID-PRIME
ID-PRIME procedures were adapted from published protocols (Uttamapinant et al., 2013). 3×106 cells were loaded in serum free media with 100μM (in early experiments) and 50μM (in later experiments) of membrane permeant pAz (pAz-acetoxymethyl ester (AM)) for 45 minutes at 4°C to minimize ligase activity. Cells were resuspended in pre-warmed HBSS+2% FCS and incubated at 37°C to initiate ID-PRIME. Ligase activity was terminated with a large volume of ice-cold wash buffer (HBS) containing 40μM Lipoic Acid. Cells were lysed in 1% n-Dodecyl-β-D-Maltopyranoside (DDM, Anatrace). For TCR engagement experiments, 1.5×107 APCs were pelleted with the TCR-CD3 cells at 4°C after loading with pAz-4AM. The pellet was overlaid with pre-warmed HBSS+2% FCS and incubated at 37°C without pellet disruption to initiate the reaction. The samples were treated the same as above thereafter. For steady state analysis of CD3FL-only cells in comparison with TCR-CD3FL cells, both were co-cultured with negative control APCs so that fold changes were more directly relatable to TCR-CD3FL cells with pMHC positive APCs. The CD3T-only steady state comparisons to TCR-CD3T cells were done in the absence of APCs. Myc tagged CD3ζζ species were immunoprecipitated with EZ view anti-myc beads (Sigma). CuAAC was performed on anti-myc beads after overnight immunoprecipitation in a final 30μl volume with 167μM CuSO4(Sigma), 830μM BTTAA, 1.67μM Na Ascorbate (Sigma), 83μM TEMPOL (Sigma), and 16.7μM BALK for 30 minutes at room temp. Samples were separated by non-reducing SDS-PAGE before transferring to Immobilon FL (Millipore) and blots were probed with anti-myc antibodies (mAb 9B11, Cell Signaling) followed by goat anti-mouse 2° antibodies DyLight 800 (Pierce) and streptavidin (SA) Alexa Fluor 680 (Invitrogen). Blots were scanned with an Odyssey two-laser (685nm and 785nm) infrared imaging system (LI-COR). Band intensities were quantitated after background subtraction with Image Studio Lite (LI-COR) software.
Microscopy
Live cell imaging was performed at 37°C, 5% CO2, and 50% relative humidity. Cells were dropped into chambered coverslips and incubated for 10 minutes prior to TIRF image acquisition to allow adhesion to glass coverslips or pMHC surfaces. Images were typically collected for 10-20 minutes after adhesion.
Image Analysis
FRETc was analyzed using SlideBook 5 software (3I) according to Youvan, et al. (Figure S5A)(Youvan et al., 1997). nFRETc values represent the mean FRETc values presented as a ratio of the mean mEGFP for a single cell (Vanderklish et al., 2000). FRETE analysis was performed using SlideBook 6 software (3I). The median intensity for mEGFP and mCherry were extracted from the region of interest (25.8μm2 ROI; 412 pixels) targeted for mCherry ablation. Background subtracted mEGFP values for the time points immediately before and after photobleaching were used to calculate FRETE= 1− (Q/DQ) where Q equals quenched and DQ equals dequenched fluorescence of the mEGFP as previously reported (Xu et al., 2008).
EPOR assay
Ba/F3 proliferation experiments were performed similarly to our previous description (Kuhns et al., 2010). 2.5×104 cells were cultured in 96 well plates in triplicate under drug selection for 3 days in the absence of IL-3. Cells were enumerated by flow cytometry with count beads.
Statistical Analysis
Statistical analyses were performed with Prism 5.0 (GraphPad Software, Inc). For the EPOR assays, one-way analysis of variance (ANOVA) was performed with a Tukey’s post-test for multiple comparisons. The ID-PRIME and imaging experiments involved normalized data or non-normally distributed cell populations so a Mann-Whitney t test and Kruskal-Wallis ANOVA with a Dunn’s multiple comparisons post-test were performed where appropriate.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jackson Egen, Felicia Goodrum, Matthew Krummel, Evan Newell, and Dominik Schenten for critical feedback on the manuscript,as well as Mark M. Davis, Yueh-Hsiu Chien, and K. Christopher Garcia for thoughtful comments and valuable reagents. We also thank Bentley Fane, Jeff Frelinger, Kurt Griffin, Anita Koshy, Lonnie Lybarger, Maggie So and members of the Frelinger, Kuhns, and Wu labs for feedback. M.S.K. is a Pew Scholar in Biomedical Sciences, supported by The Pew Charitable Trusts. This work was also supported by NIH 5R01CA186568 (AYT), The University of Arizona College of Medicine (MSK), the Bio5 Institute (MSK), and NIH/NIAID 1R01AI101053 (MSK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
MSK conceived of the project and wrote the manuscript. MSL, NRD, and MSK designed and built the ID-PRIME system with critical reagents and input from CU, PSS, and AYT. The ID-PRIME experiments were conducted by MSL and MSK with assistance from NRD and HLP and technical guidance from CU and AYT. HBB, MSL, NRD, CRG, and MSK designed and built the FRET systems. HBB conducted and analyzed all three-channel sensitized emission experiments. CRG and MSK conducted and analyzed all acceptor photo-bleaching experiments and cell-cell couple experiments. MSL and MSK designed and built the EPOR system. MSL and HLP conducted the EPOR experiments. All authors edited the manuscript.
REFERENCES
- Aivazian D, Stern LJ. Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat Struct Biol. 2000;7:1023–1026. doi: 10.1038/80930. [DOI] [PubMed] [Google Scholar]
- Brdicka T, Kadlecek TA, Roose JP, Pastuszak AW, Weiss A. Intramolecular regulatory switch in ZAP-70: analogy with receptor tyrosine kinases. Mol Cell Biol. 2005;25:4924–4933. doi: 10.1128/MCB.25.12.4924-4933.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call ME, Pyrdol J, Wiedmann M, Wucherpfennig KW. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell. 2002;111:967–979. doi: 10.1016/s0092-8674(02)01194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call ME, Schnell JR, Xu C, Lutz RA, Chou JJ, Wucherpfennig KW. The structure of the zetazeta transmembrane dimer reveals features essential for its assembly with the T cell receptor. Cell. 2006;127:355–368. doi: 10.1016/j.cell.2006.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu SN, Huang LJ, Nam H, Lodish HF. The erythropoietin receptor cytosolic juxtamembrane domain contains an essential, precisely oriented, hydrophobic motif. Mol Cell. 2001;7:377–385. doi: 10.1016/s1097-2765(01)00185-x. [DOI] [PubMed] [Google Scholar]
- Delgado P, Alarcon B. An orderly inactivation of intracellular retention signals controls surface expression of the T cell antigen receptor. J Exp Med. 2005;201:555–566. doi: 10.1084/jem.20041133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres NF, Das R, Smith AW, Arkhipov A, Kovacs E, Huang Y, Pelton JG, Shan Y, Shaw DE, Wemmer DE, et al. Conformational Coupling across the Plasma Membrane in Activation of the EGF Receptor. Cell. 2013;152:543–556. doi: 10.1016/j.cell.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman DM. Electrostatic fasteners hold the T cell receptor-CD3 complex together. Mol Cell. 2003;11:5–6. doi: 10.1016/s1097-2765(03)00016-9. [DOI] [PubMed] [Google Scholar]
- Fernandes RA, Shore DA, Vuong MT, Yu C, Zhu X, Pereira-Lopes S, Brouwer H, Fennelly JA, Jessup CM, Evans EJ, et al. T cell receptors are structures capable of initiating signaling in the absence of large conformational rearrangements. The Journal of biological chemistry. 2012;287:13324–13335. doi: 10.1074/jbc.M111.332783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon E, Schubert DA, Gordo S, Chu HH, Wucherpfennig KW. Local changes in lipid environment of TCR microclusters regulate membrane binding by the CD3epsilon cytoplasmic domain. J Exp Med. 2012;209:2423–2439. doi: 10.1084/jem.20120790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil D, Schamel WW, Montoya M, Sanchez-Madrid F, Alarcon B. Recruitment of Nck by CD3 epsilon reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell. 2002;109:901–912. doi: 10.1016/s0092-8674(02)00799-7. [DOI] [PubMed] [Google Scholar]
- Guy CS, Vignali DA. Organization of proximal signal initiation at the TCR:CD3 complex. Immunological Reviews. 2009;232:7–21. doi: 10.1111/j.1600-065X.2009.00843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Babcock H, Zhuang X. Breaking the diffraction barrier: super-resolution imaging of cells. Cell. 2010;143:1047–1058. doi: 10.1016/j.cell.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature. 2002;419:845–849. doi: 10.1038/nature01076. [DOI] [PubMed] [Google Scholar]
- Kane LP, Lin J, Weiss A. Signal transduction by the TCR for antigen. Curr Opin Immunol. 2000;12:242–249. doi: 10.1016/s0952-7915(00)00083-2. [DOI] [PubMed] [Google Scholar]
- Kersh GJ, Kersh EN, Fremont DH, Allen PM. High- and low-potency ligands with similar affinities for the TCR: the importance of kinetics in TCR signaling. Immunity. 1998;9:817–826. doi: 10.1016/s1074-7613(00)80647-0. [DOI] [PubMed] [Google Scholar]
- Kim ST, Takeuchi K, Sun ZY, Touma M, Castro CE, Fahmy A, Lang MJ, Wagner G, Reinherz EL. The alphabeta T cell receptor is an anisotropic mechanosensor. The Journal of biological chemistry. 2009;284:31028–31037. doi: 10.1074/jbc.M109.052712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhns MS, Davis MM. Disruption of extracellular interactions impairs T cell receptor-CD3 complex stability and signaling. Immunity. 2007;26:357–369. doi: 10.1016/j.immuni.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Kuhns MS, Davis MM. TCR signaling emerges from the sum of many parts. Frontiers in Immunology. 2012:3. doi: 10.3389/fimmu.2012.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhns MS, Davis MM, Garcia KC. Deconstructing the Form and Function of the TCR/CD3 Complex. Immunity. 2006;24:133–139. doi: 10.1016/j.immuni.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Kuhns MS, Girvin AT, Klein LO, Chen R, Jensen KD, Newell EW, Huppa JB, Lillemeier BF, Huse M, Chien YH, et al. Evidence for a functional sidedness to the alphabetaTCR. Proc Natl Acad Sci U S A. 2010;107:5094–5099. doi: 10.1073/pnas.1000925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Ferez M, Swamy M, Arechaga I, Rejas MT, Valpuesta JM, Schamel WW, Alarcon B, van Santen HM. Increased sensitivity of antigen-experienced T cells through the enrichment of oligomeric T cell receptor complexes. Immunity. 2011;35:375–387. doi: 10.1016/j.immuni.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Lauritsen JP, Christensen MD, Dietrich J, Kastrup J, Odum N, Geisler C. Two distinct pathways exist for down-regulation of the TCR. J Immunol. 1998;161:260–267. [PubMed] [Google Scholar]
- Li YC, Chen BM, Wu PC, Cheng TL, Kao LS, Tao MH, Lieber A, Roffler SR. Cutting Edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. J Immunol. 2010;184:5959–5963. doi: 10.4049/jimmunol.0900775. [DOI] [PubMed] [Google Scholar]
- Liu B, Chen W, Evavold BD, Zhu C. Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell. 2014;157:357–368. doi: 10.1016/j.cell.2014.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Rhodes M, Wiest DL, Vignali DA. On the dynamics of TCR:CD3 complex cell surface expression and downmodulation. Immunity. 2000;13:665–675. doi: 10.1016/s1074-7613(00)00066-2. [DOI] [PubMed] [Google Scholar]
- Livnah O, Stura EA, Middleton SA, Johnson DL, Jolliffe LK, Wilson IA. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science. 1999;283:987–990. doi: 10.1126/science.283.5404.987. [DOI] [PubMed] [Google Scholar]
- Lu X, Gross AW, Lodish HF. Active conformation of the erythropoietin receptor: random and cysteine-scanning mutagenesis of the extracellular juxtamembrane and transmembrane domains. J Biol Chem. 2006;281:7002–7011. doi: 10.1074/jbc.M512638200. [DOI] [PubMed] [Google Scholar]
- Ma Z, Sharp KA, Janmey PA, Finkel TH. Surface-anchored monomeric agonist pMHCs alone trigger TCR with high sensitivity. PLoS Biol. 2008;6:e43. doi: 10.1371/journal.pbio.0060043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manz BN, Jackson BL, Petit RS, Dustin ML, Groves J. T-cell triggering thresholds are modulated by the number of antigen within individual T-cell receptor clusters. Proc Natl Acad Sci U S A. 2011;108:9089–9094. doi: 10.1073/pnas.1018771108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reth M. Antigen receptor tail clue. Nature. 1989;338:383–384. [PubMed] [Google Scholar]
- Shi X, Bi Y, Yang W, Guo X, Jiang Y, Wan C, Li L, Bai Y, Guo J, Wang Y, et al. Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature. 2013;493:111–115. doi: 10.1038/nature11699. [DOI] [PubMed] [Google Scholar]
- Slavoff SA, Liu DS, Cohen JD, Ting AY. Imaging protein-protein interactions inside living cells via interaction-dependent fluorophore ligation. J Am Chem Soc. 2011;133:19769–19776. doi: 10.1021/ja206435e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZJ, Kim KS, Wagner G, Reinherz EL. Mechanisms contributing to T cell receptor signaling and assembly revealed by the solution structure of an ectodomain fragment of the CD3 epsilon gamma heterodimer. Cell. 2001;105:913–923. doi: 10.1016/s0092-8674(01)00395-6. [DOI] [PubMed] [Google Scholar]
- Sun ZY, Kim ST, Kim IC, Fahmy A, Reinherz EL, Wagner G. Solution structure of the CD3epsilondelta ectodomain and comparison with CD3epsilongamma as a basis for modeling T cell receptor topology and signaling. Proc Natl Acad Sci U S A. 2004;101:16867–16872. doi: 10.1073/pnas.0407576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttamapinant C, Sanchez MI, Liu DS, Yao JZ, Ting AY. Site-specific protein labeling using PRIME and chelation-assisted click chemistry. Nature protocols. 2013;8:1620–1634. doi: 10.1038/nprot.2013.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttamapinant C, White KA, Baruah H, Thompson S, Fernandez-Suarez M, Puthenveetil S, Ting AY. A fluorophore ligase for site-specific protein labeling inside living cells. Proc Natl Acad Sci U S A. 2010;107:10914–10919. doi: 10.1073/pnas.0914067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Merwe PA. The TCR triggering puzzle. Immunity. 2001;14:665–668. doi: 10.1016/s1074-7613(01)00155-8. [DOI] [PubMed] [Google Scholar]
- Vanderklish PW, Krushel LA, Holst BH, Gally JA, Crossin KL, Edelman GM. Marking synaptic activity in dendritic spines with a calpain substrate exhibiting fluorescence resonance energy transfer. Proc Natl Acad Sci U S A. 2000;97:2253–2258. doi: 10.1073/pnas.040565597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Call ME, Wucherpfennig KW. A Membrane-proximal Tetracysteine Motif Contributes to Assembly of CD3{delta}{epsilon} and CD3{gamma}{epsilon} Dimers with the T Cell Receptor. J Biol Chem. 2006;281:36977–36984. doi: 10.1074/jbc.M607164200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV, Chou JJ, Wucherpfennig KW. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell. 2008;135:702–713. doi: 10.1016/j.cell.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki S, Ishikawa E, Sakuma M, Ogata K, Sakata-Sogawa K, Hiroshima M, Wiest DL, Tokunaga M, Saito T. Mechanistic basis of pre-T cell receptor-mediated autonomous signaling critical for thymocyte development. Nat Immunol. 2006;7:67–75. doi: 10.1038/ni1290. [DOI] [PubMed] [Google Scholar]
- Yang J, Ma YQ, Page RC, Misra S, Plow EF, Qin J. Structure of an integrin alphaIIb beta3 transmembrane-cytoplasmic heterocomplex provides insight into integrin activation. Proc Natl Acad Sci U S A. 2009;106:17729–17734. doi: 10.1073/pnas.0909589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Reth M. The dissociation activation model of B cell antigen receptor triggering. FEBS letters. 2010;584:4872–4877. doi: 10.1016/j.febslet.2010.09.045. [DOI] [PubMed] [Google Scholar]
- Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, Dustin ML, Saito T. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat Immunol. 2005;6:1253–1262. doi: 10.1038/ni1272. [DOI] [PubMed] [Google Scholar]
- Youvan DC, Silva CM, Bylina EJ, Coleman WJ, Dilworth MR, Yang MM. Calibration of Fluorescnece Resonance Energy Transfer in Microscopy Using Genetically Engineered GFP Derivatives on Nickle Chelating Beads. Biotechnology et. alia. 1997;3:1–18. [Google Scholar]
- Zal T, Gascoigne NR. Using live FRET imaging to reveal early protein-protein interactions during T cell activation. Curr Opin Immunol. 2004;16:418–427. doi: 10.1016/j.coi.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cordoba SP, Dushek O, Anton van der Merwe P. Basic residues in the T-cell receptor zeta cytoplasmic domain mediate membrane association and modulate signaling. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:19323–19328. doi: 10.1073/pnas.1108052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.