

Abstract
A growing body of evidence suggests that the vascular actions of Ang-(1-7) appear to involve increased production of nitric oxide (NO), an important vasodilator, through the activation of MasR, thus indicating the involvement of the MasR in preventing endothelial dysfunction. However, it is unknown whether the MasR could be involved in the progression of the next step in atherosclerosis, neo-intimal formation. To determine whether the deletion of the MasR is involved in the development of intimal thickening in an in vitro model. Mice [three background controls (C57Bl/6) and 3 MasR (−/−)] were killed and the aortas excised and cleaned of connective tissue and cut into 3 mm rings. Rings were placed in an organ culture medium for 5 weeks, embedded in paraffin, cut at 5 μm and stained with haematoxylin and eosin and Masson’s trichrome. In addition, aortic reactivity was measured in organ baths. After 5 weeks of culture, the intima:media ratio increased in the aortas from MasR (−/−) mice compared to the control group by 4.5-fold (P < 0.01). However, no significant difference in nuclei area count (cell proliferation) between the MasR (−/−) mice and control group was observed (0.87 ± 0.29% vs. 0.94 ± 0.18%, respectively, P = ns). Functional studies showed only a minor vasoconstrictive and full vasodilative response. This study shows that the deletion of the MasR causes marked increase in the aortic intima:media ratio, which is not due to generalized cellular proliferation. These results provide a functional role for the MasR in atherogenesis.
Keywords: angiotensin (1-7) Mas receptor, atherosclerosis, endothelial function, intimal thickening
Coronary artery disease (atherosclerosis of the coronary arteries) is a multifactorial disease that is caused by the abnormal accumulation of low-density lipoprotein molecules in arteries (Andersson et al. 2010). It is well known that the endothelium layer exerts a number of vasoprotective effects, such as vasodilation and inhibition of inflammatory responses, which is mediated by the production of nitric oxide (NO) (Davignon & Ganz 2004). Accumulating evidence suggests that a defect in the production of NO could cause endothelial dysfunction, which is recognized as an early marker for atherosclerosis (Davignon & Ganz 2004). As NO prevents many of the cellular proliferative changes leading to atherosclerosis, the intima:media thickness ratio of arteries could be considered as a surrogate measure of atherosclerosis and follows endothelial dysfunction (Polak et al. 2011).
The renin–angiotensin system (RAS) is an endocrine system that has been shown to have a fundamental role in cardiovascular homoeostasis and atherogenesis (Dinh et al. 2001). Several therapeutic agents that inhibit the actions of RAS receptors and enzymes to treat CVD are available, such as ACE inhibitors (ACE-I) and angiotensin II type 1 receptor blockers (ARBs) (George et al. 2010). The Ang-(1-7) MasR is an important RAS factor and has been suggested to have a protective role in CVD (Polak et al. 2011). For example, Lemos et al. (2005) have reported that the vasodilator effect of the Ang-(1-7) abolished in the aorta of MasR (−/−) mice, indicating the involvement of the MasR in endothelial function (Lemos et al. 2005). As Ang(1-7) can have other non-target effects, it is unknown whether the MasR could be involved in the development of the next step in atherosclerosis, intimal thickening.
To this end, we employed the in vitro organ culture technique to determine whether the deletion of the MasR could increase the intima:media ratio and quantified aortic nuclei to determine whether cellular proliferation was affected.
Materials and methods
Thoracic aortas were excised from mice [three background controls (C57Bl/6) and three MasR (−/−)] and cleaned of connective tissue and fat, cut into 3 mm rings and placed in 24-well plates containing 2 ml of the completed media [Complete media were prepared by adding 50 ml of foetal bovine serum (FBS) to 450 ml of Dulbecco’s modified Eagle’s medium and 5 ml of antibiotic (10,000 units penicillin, 10 mg streptomycin and 25 μg amphotericin B per ml]. Plates were placed in a humidified incubator with 5% CO2 at 37°C. The media were changed every 3 days, and experiments were terminated at 5 weeks (Cable et al. 1999). The experiments were carried out according to the National Health and Medical Research Council ‘Australian Code of Practice for the Care and Use of Animals for Scientific Purposes’ (7th edition from 2004) and approved by the Austin Hospital Animal Ethics Committee.
After the organ culture was performed, aortic rings were either fixed in 4% paraformaldehyde/PBS solution (pH 7.4) overnight and processed for paraffin or collected for pharmacological assessment. Rings were embedded vertically in single paraffin blocks to maintain equal cutting thickness throughout all vessels. Paraffin ribbons were cut at 5 μm and placed on a 50°C water bath to allow the sections to expand to their original size before they were collected on microscope slides.
Haematoxylin and eosin staining
The slide was then immersed in 100% ethanol (two rinses, 10 min) to eliminate all the xylene and subsequently in 90% (one rinse, 1 min) to prepare it for rehydration. The slide was then immersed in 70%, 50% and 30% of ethanol (one rinse, 1 min). After that, the slide was rinsed in PBS for 5 min. Two-hundred microlitre of haematoxylin stain was then added to stain the nucleic acid (DNA) for 5 min at room temperature. The slide was washed and rinsed again in PBS for 5 min. After this, 400 μl of eosin stain was then added to stain cellular proteins and incubated for 30 s. The slide was washed and rinsed in PBS for 5 min. The slide was dehydrated in alcohol (two rinses, 2 min) and then in xylene (two rinses, 2 min). The slide was mounted in DPX medium and left to dry for 3 days.
Masson’s trichrome stain
Masson’ trichrome stain was also used to stain collagen. Slides were immersed in xylene for 5 min to dissolve all the wax. The xylene was then cleared by alcohol, and the slide rinsed with distilled water. Next the slide was immersed in Weigert’s iron haematoxylin for 5–10 min to stain nuclei and then rinsed with distilled water. Then the slide was stained with Biebrich scarlet for 5 min to allow the acidophilic tissues to bind. The slide was rinsed in distilled water and treated with phosphotungstic and phosphomolybdic acid for 5 min to let the Biebrich scarlet dye to diffuse out of the collagen, but remain in other tissues. Next the slide was rinsed with distilled water and stained with light green dye to detect the collagen. Finally the slide was dehydrated in alcohol, cleared in xylene and then mounted in DPX medium. The slides were left to dry for 3 days.
Image analysis
For intimal thickening, each blood vessel (n = 3) was photographed in quadruplicate, using a Leica camera (DFC450) attached to an imaging program (100× magnification) and one image that encompassed the whole artery was taken. For nuclei and collagen quantification, four images per artery were obtained (400× magnification). Images were then further analysed using the microcomputer imaging device analysis software (MCID; InterFocus, Linton, UK). Both of the intima:media ratio and nuclei count were taken. The proportional area for each image was measured: for intima:media ratio, the intimal area was divided by the medial area; for nuclei count, the area of blue nuclei was divided by total scanned area.
Pharmacological assessment
After 5 weeks of incubation, one ring from each MasR (−/−) mice was placed in the organ bath system (OB8; Zultek Engineering, Victoria, Australia). The bath was then filled with Krebs solution, kept at a constant temperature of 37°C and continuously bubbled with 95% oxygen/5% carbon dioxide. After that, the ring was mounted between two metal hooks in an organ bath attached to a force displacement transducer coupled to a data acquisition system. After 2 h equilibration period, the rings were then stretched to 0.5 g. After 1 h, rings were incubated with 1 μM U46619 (TX) to induce constriction and then 1 μM acetylcholine (ACh) to induce relaxation.
Statistical analysis
Data were analysed using Students’ t-test. Data are presented as mean ± SEM, and significance was taken at P < 0.05.
Results
Intima:media ratio and intimal thickening
The intima:media ratio was analysed to investigate whether the deletion of the MasR is involved in intimal thickening. It was shown that the deletion of the MasR significantly increased intimal thickening in MasR (−/−) mice compared to the control group by 4.5-fold (P < 0.01) (Figure1 insert). Magnified excerpts of sections A and B clearly show the extent of intimal thickening (red arrows).
Figure 1.
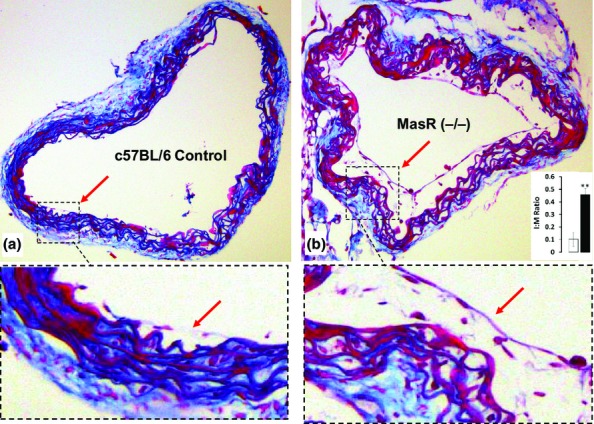
Coronal section of thoracic aorta after 5 weeks of culture. Control mice showed minimal intimal proliferation (a, magnified excerpt, red arrow) compared with MasR (−/−) mice (b, magnified excerpt, red arrow). Quantitation of intima:media ratio showed nearly five-fold increase in proliferation (b, insert, **P < 0.01).
Additionally, nuclei quantification using haematoxylin- and eosin-stained sections showed no significant difference in nuclei area:total area between the MasR (−/−) mice and control (0.87 ± 0.29% vs. 0.94 ± 0.18%, respectively, P = ns).
Vasoactivity studies
To investigate whether the 5 weeks of incubation time affected the function of the arteries, the organ bath was performed and this was determined only in the MasR (−/−) mice. A small (10 mg) constrictive response was observed in response to 1 μM U46619 (TX), yet the rings relaxed completely to acetylcholine indicating responsive arteries (Figure2, typical image).
Figure 2.
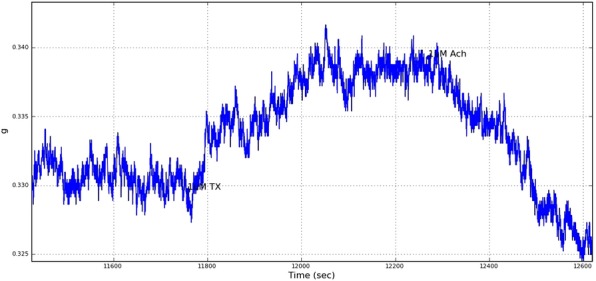
Typical real-time image of aortic segment in organ bath. U46619 causes a small increase in contraction (10 mg, arrow), and acetylcholine relaxed vessel beyond baseline.
Discussion
Major findings from the present study demonstrate that the deletion of the MasR significantly increased the intimal thickening in MasR (−/−) mice in an organ culture model. However, no role was observed for the MasR in generalized cell proliferation. The first evidence for a functional role for the MasR as the mediator of the Ang-(1-7) effects in the vascular system was suggested by Santos et al. (2003). It has been shown that relaxation induced by Ang-(1-7) in mice aorta was absent in the Mas-deficient mice, indicating involvement of the MasR in this effect. Peiró et al. (2007) have further suggested that the relaxation effects of the Ang-(1-7) were correspondingly impaired in both wild-type arteries pretreated with A779, a Mas receptor blocker, and arteries isolated from the MasR (−/−) mice, suggesting an essential MasR role in preserving normal vascular relaxation. Accordingly, Ping et al. (2008) have further indicated in MasR (−/−) mice that the deletion of the MasR results in increased blood pressure and endothelial dysfunction, indicating a significant role for the MasR in resolving endothelial dysfunction.
Ang-(1-7) MasR in intimal thickening, cellular proliferation
Ang-(1-7) has been shown to have a fundamental role in resolving endothelial function, which is mediated by MasR activity. As endothelial dysfunction is the prerequisite for intimal thickening, in our results, the deletion of the MasR increased the intimal thickening in the MasR (−/−) mice compared to the control group, which indicates the involvement of the MasR in this stage, suggesting its fundamental role in the development of atherosclerosis. However, it is uncertain which cells are involved in this phenomenon; the cells might originate from endothelial cells, smooth muscle cells or resident stem cells.
In line with our results, Jaiswal et al. (1992) have shown in rat carotid artery that Ang-(1-7) infusion reduces neo-intimal formation and smooth muscle cell proliferation after vascular injury, indicating a possible role for the MasR in this effect. As well, a recent study by Tesanovic et al. (2010) has suggested that in apolipoprotein E-deficient mice that the intima:media ratio was significantly decreased with Ang-(1-7) treatment, which is mediated by MasR activity. This observation was demonstrated through resolving endothelial function as the result of an increase in NO production, which resulted from increased eNOS expression and decreased superoxide levels (Tesanovic et al. 2010). Ang-(1-7) MasR has also shown to have anti-proliferative effects in the cardiovascular system. For example, a previous study has shown in animal models that thrombus formation and smooth muscle cell proliferation are diminished by Ang-(1-7) (Kucharewicz et al. 2002), indicating the anti-proliferative role for the Ang-(1-7). As well, Tallant et al. (1999) have reported in rat models that the proliferative response of the injured artery was inhibited by Ang-(1-7), which could be mediated by MasR activity. This observation was suggested through the participation of Ang-(1-7) in the reversal of vascular proliferation through inhibition of Ang II formation or activity. In line with these results, Zhang et al. (2010) have reported that a MasR blocker, A779, blocks the inhibitory effect of Ang-(1-7) on vascular smooth muscle cell (VSMC) growth, suggesting the participation of the MasR in the anti-proliferative action of Ang-(1-7). In contrast, our results showed that the deletion of the MasR indicated no significant difference in the nuclei numbers in the media between the MasR (−/−) mice and the control groups. This suggests that the deletion of the MasR might not be involved in generalized cell proliferation in this type of experimental model.
In atherogenesis, it is well known that a group of cytokines stimulate VSMCs to migrate into the intima to secrete collagen, which leads to the production of the fibrous matrix which subsequently matures into atherosclerotic plaque with a fibrous cap (Barter et al. 2004).
The aortic reactivity after 5 weeks of incubation
The aortic reactivity in the MasR (−/−) mice model in response to 1 μM U46619 and ACh was measured. U46619 is a potent vasoconstrictor in several vascular beds. For example, Gunnett et al. (1999) have shown in mice models that U46619 contracted the carotid artery by 110 ± 20 and 380 ± 30 mg in wild-type mice and 30 ± 10 and 190 ± 30 mg in interleukin-10-deficient mice. As well, Lamping and Faraci (2001) have indicated in mice models that contraction of the carotid artery in response to U46619 was 140 ± 10 and 50 ± 10 mg in male versus female mice. Additionally, U46619 has also shown in rats models to have vasoactive role. For example, Cogolludo et al. (2003) have shown in isolated pulmonary arteries of rats that U46619 simultaneously increased the contractile force for these arteries, which produced ≈50% of the maximal response induced a sustained contractile response of 185 ± 23 mg.
In contrast our results, a small constrictive response (10 mg) was observed in response to U46619, which indicates that the 5 weeks of incubation affected the contractile force for these arteries. However, the arteries completely relaxed to ACh, indicating responsive arteries. Thus, future studies aimed at determining whether the deletion of the MasR reduces endothelial function compared to control after 5 weeks of incubation should be performed, which would support the role for MasR in endothelial dysfunction.
Conclusion
In summary, findings from this study suggest that the MasR is involved in intimal thickening. Our results highlight the importance of the MasR in intimal thickening, indicating that the MasR could be targeted to reduce the burden of cardiovascular disease.
Acknowledgments
This study was elaborated within the grant of the European Regional Development Fund—Project FNUSA-ICRC (No. CZ.1.05/1.1.00/02.0123).
Conflict of interest
Authors declare no conflict of interest.
References
- Andersson J, Libby P, Hansson GK. Adaptive immunity and atherosclerosis. Clin. Immunol. 2010;134:33–46. doi: 10.1016/j.clim.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Nicholls S, Rye KA, Anantharamaiah G, Navab M, Fogelman AM. Antiinflammatory properties of HDL. Circ. Res. 2004;95:764–772. doi: 10.1161/01.RES.0000146094.59640.13. [DOI] [PubMed] [Google Scholar]
- Cable DG, Caccitolo JA, Caplice N, et al. The role of gene therapy for intimal hyperplasia of bypass grafts. Circulation. 1999;100:II392–II396. doi: 10.1161/01.cir.100.suppl_2.ii-392. [DOI] [PubMed] [Google Scholar]
- Cogolludo A, Moreno L, Bosca L, Tamargo J, Perez-Vizcaino F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction role of protein kinase Cζ. Circ. Res. 2003;93:656–663. doi: 10.1161/01.RES.0000095245.97945.FE. [DOI] [PubMed] [Google Scholar]
- Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–III32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- Dinh DT, Frauman AG, Johnston CI, Fabiani ME. Angiotensin receptors: distribution, signalling and function. Clin. Sci. 2001;100:481–492. [PubMed] [Google Scholar]
- George AJ, Thomas WG, Hannan RD. The renin–angiotensin system and cancer: old dog, new tricks. Nat. Rev. Cancer. 2010;10:745–759. doi: 10.1038/nrc2945. [DOI] [PubMed] [Google Scholar]
- Gunnett CA, Berg DJ, Faraci FM. Vascular effects of lipopolysaccharide are enhanced in interleukin-10–deficient mice. Stroke. 1999;30:2191–2196. doi: 10.1161/01.str.30.10.2191. [DOI] [PubMed] [Google Scholar]
- Jaiswal N, Diz DI, Chappell MC, Khosla MC, Ferrario CM. Stimulation of endothelial cell prostaglandin production by angiotensin peptides. Characterization of receptors. Hypertension. 1992;19:II49. doi: 10.1161/01.hyp.19.2_suppl.ii49. [DOI] [PubMed] [Google Scholar]
- Kucharewicz I, Pawlak R, Matys T, Pawlak D, Buczko W. Antithrombotic effect of captopril and losartan is mediated by angiotensin-(1-7) Hypertension. 2002;40:774–779. doi: 10.1161/01.hyp.0000035396.27909.40. [DOI] [PubMed] [Google Scholar]
- Lamping KG, Faraci FM. Role of sex differences and effects of endothelial NO synthase deficiency in responses of carotid arteries to serotonin. Arterioscler. Thromb. Vasc. Biol. 2001;21:523–528. doi: 10.1161/01.atv.21.4.523. [DOI] [PubMed] [Google Scholar]
- Lemos VS, Silva DM, Walther T, Alenina N, Bader M, Santos RA. The endothelium-dependent vasodilator effect of the nonpeptide Ang (1-7) mimic AVE 0991 is abolished in the aorta of mas-knockout mice. J. Cardiovasc. Pharmacol. 2005;46:274–279. doi: 10.1097/01.fjc.0000175237.41573.63. [DOI] [PubMed] [Google Scholar]
- Peiró C, Vallejo S, Gembardt F, et al. Endothelial dysfunction through genetic deletion or inhibition of the G protein-coupled receptor Mas: a new target to improve endothelial function. J. Hypertens. 2007;25:2421–2425. doi: 10.1097/HJH.0b013e3282f0143c. [DOI] [PubMed] [Google Scholar]
- Polak JF, Pencina MJ, Pencina KM, O’Donnell CJ, Wolf PA, D’Agostino RB., Sr Carotid-wall intima–media thickness and cardiovascular events. N. Engl. J. Med. 2011;365:213–221. doi: 10.1056/NEJMoa1012592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos RA, e Silva ACS, Maric C, et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl Acad. Sci. USA. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallant EA, Diz DI, Ferrario CM. Antiproliferative actions of angiotensin-(1-7) in vascular smooth muscle. Hypertension. 1999;34:950–957. doi: 10.1161/01.hyp.34.4.950. [DOI] [PubMed] [Google Scholar]
- Tesanovic S, Vinh A, Gaspari TA, Casley D, Widdop RE. Vasoprotective and atheroprotective effects of angiotensin (1-7) in apolipoprotein E–deficient mice. Arterioscler. Thromb. Vasc. Biol. 2010;30:1606–1613. doi: 10.1161/ATVBAHA.110.204453. [DOI] [PubMed] [Google Scholar]
- Xu P, Costa-Goncalves AC, Todiras M, Rabelo LA, Sampaio WO, Moura MM, Santos SS, Luft FC, Bader M, Gross V, Alenina N, Santos RA. Endothelial dysfunction and elevated blood pressure in MAS gene-deleted mice. Hypertension. 2008;51:574–580. doi: 10.1161/HYPERTENSIONAHA.107.102764. [DOI] [PubMed] [Google Scholar]
- Zhang F, Hu Y, Xu Q, Ye S. Different effects of angiotensin II and angiotensin-(1-7) on vascular smooth muscle cell proliferation and migration. PLoS One. 2010;5:e12323. doi: 10.1371/journal.pone.0012323. [DOI] [PMC free article] [PubMed] [Google Scholar]