

Abstract
Background
Left ventricular noncompaction (LVNC) is an autosomal dominant, genetically heterogeneous cardiomyopathy with variable severity, which may co-occur with cardiac hypertrophy.
Methods and Results
Here, we generated whole exome sequence (WES) data from multiple members from five families with LVNC. In four out of five families, the candidate causative mutation segregates with disease in known LVNC genes MYH7 and TPM1. Subsequent sequencing of MYH7 in a larger LVNC cohort identified seven novel likely disease causing variants. In the fifth family, we identified a frameshift mutation in NNT, a nuclear encoded mitochondrial protein, not implicated previously in human cardiomyopathies. Resequencing of NNT in additional LVNC families identified a second likely pathogenic missense allele. Suppression of nnt in zebrafish caused early ventricular malformation and contractility defects, likely driven by altered cardiomyocyte proliferation. In vivo complementation studies showed that mutant human NNT failed to rescue nnt morpholino-induced heart dysfunction, indicating a probable haploinsufficiency mechanism.
Conclusions
Together, our data expand the genetic spectrum of LVNC and demonstrate how the intersection of WES with in vivo functional studies can accelerate the identification of genes that drive human genetic disorders.
Keywords: noncompaction cardiomyopathy, genetics, human, genomics, left ventricular noncompaction
Left ventricular noncompaction/hypertrabeculation (LVNC/LVHT) is a clinically distinct, primary cardiomyopathy1. LVNC is characterized by a spongy, noncompacted myocardium layer clearly distinct from the underlying, compacted myocardium2, as well as increased trabeculation and deep intertrabecular recesses in the left ventricle3. LVNC has variable clinical severity, ranging from benign to early-onset, severe heart failure and death4. LVNC may be first detected prenatally or as late as 95 years of age, and appears to affect a greater proportion of males than females5. Although readily diagnosable by echocardiogram6, the true prevalence is estimated to be between 0.014% and 0.24%2,7.
Although LVNC may occur in isolated cases, familial LVNC is recognized as a genetic disorder that is inherited in an autosomal dominant (AD) manner8. LVNC is associated frequently with mitochondrial disorders and cardiac hypertrophy. It is most commonly attributed to mutations in seven genes (TAZ, DTNA, LDB3, LMNA, SCN5A, MYH7 and MYBPC3), with additional contributing variants reported in rare instances (ACTC1, TNNT2, MIB1, PRDM16, and TPM1); all LVNC loci encode proteins involved in cellular energy, muscle development, ion channel formation, or are components of the muscle filaments9–13. Despite these advances, many of the causative LVNC genes have yet to be identified1. Mutations in MYH7 are the single most common cause of LVNC, accounting for 8–13% of cases, with the remainder of the genes reported to be mutated in rare cases10,12. The large number of causative loci, as well as overlap with other cardiomyopathy genes, suggests that hypertrabeculation is a common compensatory mechanism of impaired or injured myocardium1. To improve our understanding of the genetic and molecular basis of this disorder we used whole exome sequencing14,15 (WES) to conduct an unbiased investigation of the genetic basis of LVNC.
Methods
IRB approval for this study was obtained from Baylor College of Medicine. All participants provided informed consent prior to participating in this study.
DNA capture sequencing
Hybridization was conducted with a custom capture reagent15. Precapture libraries for SOLiD (2 ug) were hybridized in solution according to the NimbleGen Seqcap EZ protocol with minor revisions. Specifically, hybridization enhancing oligos TrTA-A and SOLiD-B were used in the hybridization reactions to block the common TrTA adaptor sequences for increased capture efficiency. Following sequence capture, post-capture LM-PCR was performed using 12 cycles. Capture libraries were quantified using PicoGreen (Cat. No. P7589) and their size distribution was analyzed using the Agilent Bioanalyzer 2100 DNA Chip 7500 (Cat. No. 5067-1506). Capture efficiency of each capture library was evaluated by performing a qPCR-based SYBR Green assay (Applied Biosystems; Cat. No. 4368708) with built-in controls (RUNX2, PRKG1, SMG1, and NLK). Capture library enrichment was estimated at 7 to 9-cycles over background by qPCR. Captured libraries were processed further for sequencing, with approximately 6–12 Gbs of sequence generated per capture library on SOLiDv4 instruments. Illumina library preparation was conducted as described previously15.
Whole Exome Sequencing Analysis
Sequence data were aligned to the human genome (hg19) and variants were identified and filtered for quality as described14,15. Briefly, read qualities were recalibrated with GATK and a minimum quality score of 30 was required; the variant must have been present in at least 15% of the reads that cover the position. In addition, prior to variant calling, reads with low (<11) mapping qualities (MQ) (a value based on the ratio of the best alignment score to the second best alignment score) were removed. Common variants were filtered based on minor allele frequency and previous association with disease using previously established parameters16. Variant effect on protein was established in both RefSeq and ENSEMBL (v78) gene sets. Variants were prioritized based on phenotypic overlap, deleteriousness of the variant (e.g. truncating/splice affecting and in silico predictions), and known mutational spectrum of the gene in unaffected populations.
Capillary Sequencing
Validation and segregation of discovered variants, and sequencing of the NNT coding exons in the expanded cohort was conducted on an ABI3730 capillary sequencer. Amplification and sequencing primers were designed by an automated pipeline; variants were automatically called using SNPDectector (version 3). Variants were subsequently validated visually.
Ion Torrent Sequencing
PCR primers were designed for each coding exon of MYH7 using primer3. Sixteen molecular barcodes were generated and appended to each of the forward PCR primers. Each exon for each subject was amplified separately, and then pooled into groups of 16 prior to sequencing on the Ion Torrent PGM. Reads were mapped to the genome and variants called as described above for the Illumina data.
Zebrafish embryo injections and live phenotypic assessment
We identified two zebrafish orthologs of human NNT protein, referred to as nnt-a and nnt-b on D. rerio chromosomes 21 and 18 respectively using reciprocal BLAST. We then designed splice-blocking morpholinos (MO)s against the splice donor site of exon 4 of each transcript (Gene Tools). We injected 9ng of each MO into wild-type embryos at the one- to two-cell stage and determined MO efficiency by RT-PCR of cDNA generated from whole embryos (n=25 embryos/injection batch; Quantitect Reverse Transcription kit, Qiagen) harvested at 3 days post-fertilization (dpf) in Trizol (Invitrogen). Specific targeting of each of nnt-a and nnt-b is evidenced by the semi-quantitative decrease in correctly spliced transcript. Phenotype specificity experiments were conducted by targeting hacl1, nav1, rab28, rbm28 and notch1a. We conducted live embryo phenotypic scoring at 2, 3, or 5 dpf using either wild-type (EK/AB outcross); or cmlc2:GFP 17, a myocardium-specific GFP transgene. MO dose response curves for individual and combined suppression of nnt-a and nnt-b (3ng, 6ng, and 9ng MO) were generated with EK/AB embryos at 3 dpf; n=50–100 embryos/injection, repeated three times with masked scoring. For rescue experiments, we generated a full-length human NNT ORF construct by PCR amplification from lymphocyte cDNA, cloning into a pCR8/GW vector (Invitrogen), and LR recombinase-mediated cloning into the pCS2+ backbone. We conducted mutagenesis using the QuikChange site directed mutagenesis kit (Agilent); all vectors were sequence confirmed. Capped mRNA was in vitro transcribed using the mMessage mMachine kit (Ambion); 200 pg RNA and 5 ng MOs were used for in vivo complementation experiments. To assess cardiac heartbeat and morphology, we anesthetized 2 dpf cmlc2:GFP larvae with Tricaine, and conducted live video imaging of ventral views using a Nikon AZ100 microscope and NIS Elements AR software at 12x magnification (10 second videos at 8.77 frames/second; n=10 embryos/injection, replicate batches; see Supplementary Movies).
Zebrafish histology
We generated cardiomyocyte cell counts at 2 and 3 dpf using cmlc2:FUCCI18, a double transgenic line enabling the detection of non-proliferating (cmlc2:mCherry-zCdt1, red) and proliferating (cmlc2:Venus-hGeminin, green) cardiomyocytes. Larval batches were fixed overnight in paraformaldehyde (PFA), and 50 μM-thick sections were imaged using a Zeiss LSM 700 confocal microscope and ZEN software at 20x magnification. Maximum intensity projections were analyzed using Imaris Version 7.6 software; mCherry+ and Venus+ nuclei were quantified using surface analysis (n=12 larvae/batch; repeated once). Red+green+ and red+green− nuclei were scored as non-proliferating cells, and red− green+ nuclei were scored as proliferating.
Statistical Analyses
Mutation burden
To test whether the frequency of rare variants in MYH7 differed between ARIC and our LVNC cohort we used a two-tailed Fisher’s exact test. The Atherosclerosis Risk In Communities (ARIC)19 cohort consists of 1,650 individuals who have undergone WES using the NimbleGen capture reagent followed by Illumina sequencing15. On average, >99% of the coding bases in MYH7 have at least 20x redundant coverage in this dataset.
In vivo complementation
To test for differences in cardiac edema in 3 dpf morphant zebrafish larvae, we compared qualitative scoring data (normal or affected) in a pairwise manner across embryo batches using χ2 tests (n=46–15 embryos/batch; three biological replicates).
Contractile dysfunction and cardiomyocyte counts
We used a student’s t-test to determine the statistical significance of differences in heart beat (2 dpf controls and morphants; n=10 larvae/injection batch; two biological replicates). Differences in proliferating and non-proliferating cardiomyocytes were also determined using a two-tailed, homoscedastic Student’s t-test (2 and 3 dpf controls and morphants; n=12 larvae/injection batch; two biological replicates).
Results
We identified five families (A–E) with apparent hereditary LVNC (Supplementary Figure 1A–E). Initial presentation of the index case for each family was secondary to either an abnormal electrocardiogram or a screening echocardiogram that led to the diagnosis of LVNC. The remaining family members in each pedigree underwent transthoracic echocardiography for indicated screening. Varying degrees of coexisting myocardial involvement were identified, including evidence of dilation of the left ventricle (LV) with depressed LV systolic function (dilated phenotype); or increased left ventricular thickness and increased LV systolic function (hypertrophic phenotype). One member of family C had severe dilated cardiomyopathy (DCM) with associated LVNC that required mechanical assist and cardiac transplantation.
We generated WES data from multiple affected and unaffected members, when available, from all families (Table 1). In total, 228 Gbp of sequence data were produced and aligned to the human genome, producing an average depth of coverage of 79x (Supplementary Figure 1A–E). Bioinformatic analyses were used to identify candidate causative alleles in each family. Variants were prioritized for validation based on a) minor allele frequency (MAF); b) whether they were predicted to alter protein function (either through a predicted change to mRNA splicing or truncation/substitution of the amino acid sequence); c) previous association with LVNC or genes with functional similarity to known LVNC genes; d) whether they were shared by all affected individuals, but not present in unaffected individuals; and e) whether mutants in other organisms had been reported with relevant heart pathologies. In all cases, the small size of the pedigree and AD inheritance of the disease made it difficult to identify a single variant by segregation alone. However, when a predicted pathogenic allele was identified in a known LVNC gene, it was considered the most likely causative variant (Supplementary Table 1). We then segregated all LVNC-associated variants reported here by capillary sequencing in all available members of the pedigree.
Table 1.
LVNC Family sequencing results - for each of the five LVNC families in this study, the number of affected and unaffected individuals sequenced, the implicated candidate disease causing gene, mutation and in silico predictions are given. Pedigrees are available in Supplementary Figure 1.
| Family ID | Affected Individuals Sequenced/Total in pedigree | Unaffected Individuals Sequenced/Total in pedigree | Gene | Mutation (HG19 Coordinates) | PolyPhen-2 Prediction (score) | Mutation Taster Prediction |
|---|---|---|---|---|---|---|
| A | 4/5 | 1/4 | TPM1 | p.D275H (chr15:g.23857430C>C) | Probably damaging (0.992) | Disease causing |
| B | 5/7 | 0/2 | MYH7 | p.S648L (chr14:g.23896462G>A) | Possibly damaging (0.906) | Disease causing |
| C | 2/2 | 2/6 | NNT | c.638_639insT (chr5:g.43619172insT) | N/A | Disease causing |
| D | 2/2 | 0/2 | MYH7 | p.Q163P (chr14:g.23901862T>G) | Probably damaging (0.979) | Disease causing |
| E | 4/4 | 3/4 | MYH7 | p.E700G (chr14:g.23895236T>C) | Probably damaging (0.997) | Disease causing |
In four out of five families we identified novel nonsynonymous changes in known LVNC genes. In three families (B, D, E) we identified rare, protein changing mutations in MYH7, encoding myosin heavy chain beta, p.S648L, p.Q163P and p.E700G respectively, none of which have been previously reported but all of which were predicted to be pathogenic with multiple in silico prediction algorithms (PolyPhen-2 HDIV/HVAR scores >0.9; Mutation Taster, Mutation Assessor; Table 1). MYH7 consists of 40 exons, encoding a protein of 1,935 residues that is present primarily in slow-type skeletal muscle and in the ventricles20. Mutations in MYH7 are associated with other cardiomyopathies, including familial hypertrophic cardiomyopathy type 1 and dilated cardiomyopathy type 1S21. In family A, we discovered a p.D275H variant in another known LVNC gene22,23, tropomyosin 1 (TPM1). TPM1 encodes an actin-binding tropomyosin, is involved in the contractile system in both smooth and striated muscle24 and accounts for a small proportion of LVNC cases.
In some cohorts, mutations in MYH7 are thought to account for a minority of LVNC cases but were found in three of the five families we studied. We hypothesized that the difficulty inherent in sequencing and interpreting data derived from a gene as large as MYH7 might lead to under-reporting of its association with LVNC. The capture sequencing reagent used here provides high, relatively even coverage of every coding exon in MYH7 and subsequently we found 60% of our families had rare, protein changing variants in MYH7. We investigated the prevalence of MYH7 mutations in a cohort of 49 individuals diagnosed with LVNC using a PCR-amplicon high-throughput sequencing strategy. In total, we produced >350Mbp of sequence data from the Ion Torrent sequencing platform, generating an average depth of coverage >200x across all coding exons. Eight individuals harbored a single, rare (MAF <0.001), protein changing mutation in MYH7 and one (L030) harbored two rare mutations (Supplementary Table 2). The proportion (18.4%) of subjects with MYH7 mutations is modestly higher compared to similar studies10,12 in which 7.9–13% of individuals harbored pathogenic mutations in MYH7. Although we cannot be certain that all discovered variants are deleterious without functional testing, we compared the frequency of rare, protein changing MYH7-mutations in LVNC to a cohort of unselected individuals from the Atherosclerosis Risk In Communities (ARIC)19 cohort. We found that only 2.5% of ARIC individuals had rare, protein changing mutations in MYH7, a 7.4 fold enrichment that, despite the relatively modest case sample size in our study, was nonetheless significant (p<0.0001). Although the MYH7 sequencing data produced from the ARIC samples is of high quality (>99% of coding bases at >20x coverage; see Methods), we cannot rule out the possibility that different sequencing methods may contribute to this result. Even so, these findings may imply that MYH7 plays a greater role in the development of LVNC either as the sole cause of the disease, or possibly acting in concert with other genes. Of note, four (50%) of the mutations we discovered in our LVNC cohort were reported previously25–28 and in one case (p.L061P in L022) a mutation affecting the same amino acid (p.L061R) was reported21. This may indicate that the set of pathogenic mutations in MYH7, at least in European populations, is relatively well surveyed.
In family C, neither known LVNC genes, nor other genes associated with human cardiomyopathy were found to contain rare, protein changing mutations. Of the 25 genes that remained after analysis, the majority either had no known function or no shared function with other known LVNC genes (e.g. related to the heart, musculature, or cellular energy). Cross-referencing this gene set with human (OMIM) or mouse (MGI) phenotype databases reduced the priority of most of these genes on the basis that loss of function mutations induced phenotypes irrelevant to the cardiac defects in family C, with two exceptions: MAP1S and NNT (Supplementary Table 3). However, MAP1S contains numerous homozygous frameshift alleles in control individuals, suggesting that its loss of function is tolerated in humans. In contrast, NNT, encoding nicotinamide nucleotide transhydrogenase, remained a strong candidate for several reasons. First, the mutation detected was a frameshift allele within the fourth coding exon of the gene that introduces a premature codon that likely renders the transcript subject to nonsense-mediated decay (Figure 1A). Second, we found a single heterozygous truncating or stop mutation at any position of the coding sequence in 13,000 control chromosomes; this frequency is consistent with the prevalence of LVNC and suggests that severe truncating mutations at this locus are generally intolerable. Third, mouse Nnt mutants have revealed a direct relevance of NNT to cardiac development and function: Nnt null29 mice have a mild cardiovascular background phenotype including lower LV shortening, higher aortic ejection time, and increased LV weight30. Moreover, when these mice are rendered null for a second mitochondrial gene, MnSOD, they develop lethal prenatal cardiac-hypertrophy29. This is in contrast to MnSOD-null mice on other (i.e. NNT normal) backgrounds which are long lived with no signs of cardiac-hypertrophy29. Finally, all these observations are consistent with a critical role for NNT in cellular health: NNT is an evolutionarily-conserved, nuclear-encoded mitochondrial protein that is involved in cellular respiration31; it is responsible for coupling hydride transfer between NAD(H) and NADP(+) to proton translocation across the inner mitochondrial membrane (Figure 1B)32. In mitochondria, NADPH is used for the regeneration of glutathione and theoredoxin, two important antioxidant molecules.
Figure 1.

Genetic and Functional evaluation of NNT in LVNC. A. Schematic of the human NNT locus on human chromosome 5 (top) showing non-coding exons (white boxes), and coding exons (blue boxes). Double slash indicates large introns (> 5kb). A schematic of NNT protein is shown below with functional domains Alanine dehydrogenase/PNT, N-terminal domain (AlaDh_PNT_N, shown in green), Alanine dehydrogenase/PNT, C-terminal domain (AlaDh_PNT_N, shown in purple) and twelve transmembrane domains (orange). The two mutations identified in LVNC families are shown with asterisks. B. Schematic of NNT function in mitochondria to regenerate NADPH in the cell; NADPH is then used to regenerate Thioredoxin Reductase 2 (TrxR-2) and glutathione reductase (GR). C. Lateral view of representative 3 dpf control (top) and nnt-a/b morphant (bottom) larvae showing cardiac edema (arrow). D. In vivo complementation assay of NNT p.D277Y; quantification of cardiac edema in 3 dpf morphant zebrafish larvae (5 ng nnt-a/b MOs) with and without addition of 200 pg human wild type NNT RNA (WT), and mutant RNA (p.D277Y). Statistically significant differences were calculated with χ2 tests and p<0.0001 are indicated (*); n=46–51 embryos/injection batch, repeated three times with masked scoring.
Despite the known cellular and in vivo roles of NNT relevant to heart development and function, we were cautious in interpreting a pathogenic mutation in a single family. Therefore, we expanded our genetic study and Sanger sequenced all coding exons of NNT in a cohort of 56 unrelated individuals with LVNC (Supplementary Table 2). We identified a missense mutation that affected an evolutionarily conserved residue (p.D277Y) in a single individual (L011), who was diagnosed at infancy with LVNC and cyanotic congenital heart disease. This mutation was predicted in silico to be pathogenic (PolyPhen-2: possibly damaging, score 0.686; Mutation Taster: disease causing; Mutation Assessor: high impact) and was absent from all available controls.
Cumulatively, the two families with NNT mutations and the previously-reported phenotypes in mouse strengthened the candidacy of NNT in LVNC. Nonetheless, even though the mutation in the second family was unique to the patient and was predicted to be deleterious, we sought to test directly its effect on protein function. We therefore employed an in vivo complementation strategy in a transient zebrafish model system, which we have used previously to study the effect of missense mutations for a range of human phenotypes33,34 Using reciprocal BLAST, we detected two NNT orthologs in the Danio rerio genome, nnt-a and nnt-b (83% identical, 91% similar (a); and 75% identical, 85% similar (b), versus human respectively; Supplementary Figure 2A), of which our in-house RNAseq data from the anterior structures of 5 dpf zebrafish larvae indicated that nnt-a has markedly increased expression when compared to nnt-b (456 vs. 2 counts per million mapped reads; (GSE #63191)35. Individual injection of morpholinos (MO) targeting either nnt-a or nnt-b and scoring blind to injection cocktail (n=50–100 embryos per injection) resulted in reduced endogenous transcript (Supplementary Table 4; Supplementary Figure 2B) and impaired heart function as evident by cardiac edema at 3-days post fertilization (Figure 1C). Importantly, whereas individual MO injections gave rise to 30%-40% affected embryos at the highest dose tested (9 ng) for nnt-a and nnt-b respectively, combined suppression of nnt-a/b yielded 90% affected embryos (n=46–51 embryos/injection; (Supplementary Figure 2C); this may be the result of varying dose thresholds for each of the two encoded zebrafish NNT proteins. Moreover, the cardiac edema phenotype of nnt morphants was reminiscent of those observed for other morphants modeling known human LVNC genes such as taz, ldb3, lmna, and prdm16 9,36–38 and was consistent with the observed phenotype of our positive experimental control (notch1a)39. Additionally, as a test of specificity of the assay, suppression of four randomly selected genes harboring rare variants in LVNC Family C did not produce a cardiac edema phenotype (Supplementary Figure 2A, B, D; Supplementary Table 4).
The edema phenotype was specific to the nnt-a/nnt-b MOs, since we were able to rescue the MO-induced defects by co-injecting 200 pg of capped wild-type (WT) human NNT mRNA (45% vs. 12% affected embryos for MO vs. WT rescue; p<0.0001; Figure 1D). Next, we compared the rescue efficiency of NNT mRNA harboring the 277Tyr-encoding change found in the LVNC patient to that of WT. Blind scoring of injected embryos showed that the mutant rescue was significantly worse than WT (p=0.0027), and was ameliorated significantly from that of MO (p<0.0001) (Figure 1D), indicating that the variant results in a partial loss of protein function in this assay and is a likely hypomorph. We saw no significant cardiac defects in embryos injected with either WT or p.D277Y message in the absence of MO.
To determine the cellular and morphological basis underlying the nnt-a/nnt-b MO-induced cardiac edema, we utilized transgenic reporter zebrafish lines to refine the relevance of the phenotype to LVNC in humans. Although humans and zebrafish display key differences in cardiac structure (most notably, zebrafish do not have separate left-right circulation), the highly conserved signaling pathways in cardiac development make the zebrafish a useful surrogate model to evaluate the candidacy of NNT in LVNC (reviewed in ref 33). First, we conducted live imaging of 2 dpf cmlc2:GFP control and nnt-a/nnt-b MO-injected larvae to visualize cardiac chamber morphology, contraction, and heart beat (n=10 larvae/injection batch, repeated twice). First, we noted that nnt-a/nnt-b morphants display significant brachycardia in comparison to their control counterparts (mean 91.5 vs. 66.8 beats/minute for control vs. morphants; p<0.0001). Second, we observed marked contractile dysfunction in morphant hearts, as evidenced by a lack of ventricular expansion during atrial contraction, with a concomitant generalized enlargement of the atrium, a likely compensatory result of the ventricular defect (Figure 2; Supplementary Movies).
Figure 2.
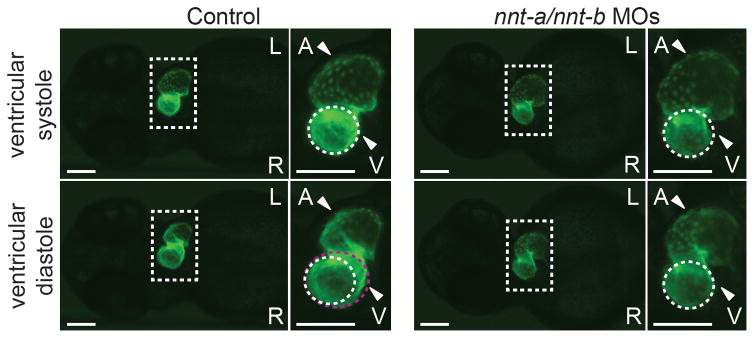
nnt-a/nnt-b morphants display contractile dysfunction. Representative live ventral views of 2 dpf cmlc2:GFP larvae at ventricular systole (top panels) and ventricular diastole (bottom panels). Ventricles in control larvae expand normally as the atrium contracts (compare white dashed circle to violet dashed circle in the bottom inset); ventricles in nnt-a/nnt-b morphants fail to expand during atrial contraction (5 ng each MO injected/embryo). Dashed box corresponds to the magnified image to the right of each large panel; L, left; R, right; A, atrium; V, ventricle; scale bars, 100 μm. nnt-a/nnt-b morphants also display brachycardia (mean 91.5 vs. 66.8 beats/minute, controls vs. morphants; p<0.0001; student’s t-test; n=10 larvae/injection, repeated twice; see Supplemental movies).
Previous studies have shown that depletion of bona fide LVNC genes in zebrafish models can result in cardiomyocyte proliferation defects9. Therefore, to investigate the cellular basis for the contractile defects of nnt-a/nnt-b MO-injected larvae, we quantified the numbers of proliferating versus non-proliferating cardiomyocytes. We utilized cmlc2:FUCCI, a dual transgenic reporter line that indicates non-proliferating cardiomyocytes (mCherry-zCdt1, a G1 marker), and proliferating cardiomyocytes (Venus-hGeminin, an S/G2/M phase marker)18. Confocal microscopy and semi-automated cell counting of larval heart sections revealed significantly more non-proliferating cardiomyocytes in at 2 dpf in nnt-a/nnt-b morphants in comparison to controls (Figure 3A, B; p<0.0001; n=12 larvae/injection batch, replicated). However, the difference in cardiomyocyte counts did not persist until 3 dpf, a time point at which morphants and controls did not display marked differences in non-proliferating cardiomyocytes; this was consistent with a significant reduction in proliferating cardiomyocytes in 3 dpf morphants vs. controls (Figure 3A, C; p<0.0001; n=12 larvae/injection batch, repeated with similar results). These data suggest that the ventricular contractile defects may be the result of aberrant cardiomyocyte proliferation in the developing heart.
Figure 3.

Suppression of nnt-a/nnt-b in zebrafish results in altered cardiomyocyte proliferation. A. Representative maximum intensity projections of cardiac sections from control and nnt-a/nnt-b morphants (5 ng each MO injected/embryo) at 2 dpf and 3 dpf were used to monitor non-proliferating (cmlc2:mCherry-zCdt1, red, G1 phase of the cell cycle) and proliferating (cmlc2:Venus-hGeminin, green, S/G2/M phase of the cell cycle) cardiomyocyte counts. Cardiomyocytes with only the green signal were counted as proliferating (exemplified by 2 dpf nnt-a/nnt-b or 3 dpf control panels). Scale bars, 50 μm; arrowheads point to the ventricle (V); or atrium (A). B. Quantification of non-proliferating cardiomyocytes as indicated by red+green− or red+green+ cells. C. Quantification of proliferating cardiomyocytes as indicated by red−green+ cells. p<0.0001; student’s t-test; n=12 larvae/injection batch, repeated once with similar results. Error bars represent standard error of the mean (sem).
Discussion
Disorders with variable presentation can be challenging to diagnose. Although LVNC can be diagnosed by echo-cardiogram, such tests are expensive and are not warranted without strong prior indication that the patient has impaired cardiac function. Further, many cases may be considered border-line for a noncompaction diagnosis by imaging. Single-gene tests can provide an exact diagnosis but are likewise difficult and expensive to conduct on genetically heterogeneous diseases where there are myriad potential genes to sequence. WES is delivered less expensively than multiple gene tests and provides a relatively unbiased method to scan the coding portions of the genome for causative mutations. Using this technique we were able to identify mutations in TPM1 in one family, a rare cause of LVNC. Further, we found that 3/5 of our families had mutations in MYH7 which likely caused their LVNC. The proportion of families carrying MYH7 mutations was higher than expected based on previous reports. Investigation into an expanded cohort of individuals found that 18.4% of them harbored mutations in MYH7 suggesting a potentially greater contribution of this locus than appreciated previously.
The combination of our human genetics data, the in vivo data from zebrafish embryos and the prior observations in the mouse implicates mutations in NNT as contributory to LVNC. Interestingly, homozygous mutations in NNT have been associated with glucocorticoid deficiency, in which the adrenal cortex is unable to produce cortisol in response to stimulation by ACTH40. Although no cardiac defects were reported in these subjects, it is not surprising that defects in mitochondrial genes may lead to multiple phenotypes and that presentation may be influenced by other factors. Multiple lines of evidence from in vivo functional testing in zebrafish indicate that nnt suppression results in malformation of the ventricle, likely resulting in contractility defects and brachycardia; our data suggest altered cell proliferation contributes to the cellular basis of this malformation. These data are consistent with other mouse and zebrafish models of LVNC. First, Ki67-immunostaining of E13.5 Fkbp12−/− (also known as Fkbp1a) mouse hearts demonstrated increased cell cycle activity in the myocardium in comparison to matched controls41. Moreover, a recent study in which zebrafish were used to model LVNC showed that suppression of prdm16 resulted in decreased cardiomyocyte counts and significantly reduced proliferation from 2–4 dpf, an observation that was coincident with increased apoptosis9. Finally, and most relevant to NNT, mouse models of Tafazzin, a protein critical to mitochondrial function that when mutated gives rise to the syndromic LVNC phenotype of Barth syndrome42, display altered cellular proliferation in embryonic hearts43. We do not know the precise developmental mechanisms that link cardiomyocyte proliferative defects with ventricular noncompaction defects. However, NNT is important for supporting the antioxidant capacity of mitochondria and NNT deficient cells are likely under severe oxidative stress, a cellular context with documented influence on cardiomyocyte cell cycle44. The implication of this pathway in the pathogenesis of this disorder merits additional study, not least because drugs that reduce oxidative stress may have a potential therapeutic role in select cases of LVNC. Furthermore, our findings of brachycardia and abnormal contractility in the nnt in vivo model highlight potential clinical implications. Numerous reports have associated LVNC in humans with depressed LV systolic function; this has been associated independently with increased mortality in both children and adults. Moreover, LVNC is reported increasingly in conjunction with congenital heart disease 45–47. The implication of NNT in brachycardia may offer future understanding of the mechanisms behind LVNC and concomitant CHD.
Identifying novel genetic loci in AD diseases is challenging because many genes contain single, rare, protein changing mutations while far fewer have multiple mutations as would be the case in autosomal recessive diseases. For AD cases, such as those in the present study, segregation in a single family likely will not provide enough statistical-power to identify a single, novel causative gene. These approaches are limited in showing causality and biological relevance to disease. In that regard, we and others have shown that functional studies are a powerful and often necessary tool to both implicate novel causative genes in instances where genetic arguments are underpowered and offer the added benefit of informing the direction of effect of missense alleles32,33. Further development of in vivo or in vitro methods for which a robust phenotypic readout is available will become increasingly important for the dissection of disorders underscored by extreme genetic heterogeneity, and for which few members of the pedigree are available for genetic analysis.
Supplementary Material
Acknowledgments
We would like to thank the families for their participation. We thank Joseph Yost for the cmlc2:GFP transgenic zebrafish line; Kenneth Poss for helpful discussions and critical reading of the manuscript; Amalia Kondyles, Kelly McKnight, and Kavita Praveen for assistance in determining morpholino efficiency; and Ben Carlson at the Duke University Light Microscopy Core Facility for assistance with image analysis. NK is a distinguished George W. Brumley Professor.
Funding Sources: Supported in part by grants from the National Human Genome Research Institute (5 U54 HG003273, to Dr. Gibbs).
Footnotes
Conflict of Interest Disclosures: MNB is the founder of Codified Genomics LLC.
References
- 1.Finsterer J. Cardiogenetics, neurogenetics, and pathogenetics of left ventricular hypertrabeculation/noncompaction. Pediatr Cardiol. 2009;30:659–681. doi: 10.1007/s00246-008-9359-0. [DOI] [PubMed] [Google Scholar]
- 2.Sarma RJ, Chana A, Elkayam U. Left ventricular noncompaction. Prog Cardiovasc Dis. 2010;52:264–273. doi: 10.1016/j.pcad.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Chin TK, Perloff JK, Williams RG, Jue K, Mohrmann R. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation. 1990;82:507–513. doi: 10.1161/01.cir.82.2.507. [DOI] [PubMed] [Google Scholar]
- 4.Stöllberger C, Blazek G, Wegner C, Winkler-Dworak M, Finsterer J. Neuromuscular and cardiac comorbidity determines survival in 140 patients with left ventricular hypertrabeculation/noncompaction. Int J Cardiol. 2011;150:71–74. doi: 10.1016/j.ijcard.2010.02.049. [DOI] [PubMed] [Google Scholar]
- 5.Pignatelli RH, McMahon CJ, Dreyer WJ, Denfield SW, Price J, Belmont JW, et al. Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation. 2003;108:2672–2678. doi: 10.1161/01.CIR.0000100664.10777.B8. [DOI] [PubMed] [Google Scholar]
- 6.Paterick TE, Umland MM, Jan MF, Ammar KA, Kramer C, Khandheria BK, et al. Left ventricular noncompaction: a 25-year odyssey. J Am Soc Echocardiogr. 2012;25:363–375. doi: 10.1016/j.echo.2011.12.023. [DOI] [PubMed] [Google Scholar]
- 7.Stöllberger C, Finsterer J. Trabeculation and left ventricular hypertrabeculation/noncompaction. J Am Soc Echocardiogr. 2004;17:1120–1121. doi: 10.1016/j.echo.2004.06.009. author reply 1121. [DOI] [PubMed] [Google Scholar]
- 8.Towbin JA, Lipshultz SE. Genetics of neonatal cardiomyopathy. Curr Opin Cardiol. 1999;14:250–262. doi: 10.1097/00001573-199905000-00010. [DOI] [PubMed] [Google Scholar]
- 9.Arndt A-K, Schafer S, Drenckhahn J-D, Sabeh MK, Plovie ER, Caliebe A, et al. Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am J Hum Genet. 2013;93:67–77. doi: 10.1016/j.ajhg.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, Schuler P, et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. 2008;117:2893–2901. doi: 10.1161/CIRCULATIONAHA.107.746164. [DOI] [PubMed] [Google Scholar]
- 11.Luxán G, Casanova JC, Martínez-Poveda B, Prados B, D’Amato G, MacGrogan D, et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med. 2013;19:193–201. doi: 10.1038/nm.3046. [DOI] [PubMed] [Google Scholar]
- 12.Probst S, Oechslin E, Schuler P, Greutmann M, Boyé P, Knirsch W, et al. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet. 2011;4:367–374. doi: 10.1161/CIRCGENETICS.110.959270. [DOI] [PubMed] [Google Scholar]
- 13.Xing Y, Ichida F, Matsuoka T, Isobe T, Ikemoto Y, Higaki T, et al. Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab. 2006;88:71–77. doi: 10.1016/j.ymgme.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Bainbridge MN, Wang M, Burgess DL, Kovar C, Rodesch MJ, D’Ascenzo M, et al. Whole exome capture in solution with 3 Gbp of data. Genome Biol. 2010;11:R62. doi: 10.1186/gb-2010-11-6-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bainbridge MN, Wang M, Wu Y, Newsham I, Muzny DM, Jefferies JL, et al. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol. 2011;12:R68. doi: 10.1186/gb-2011-12-7-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang C-J, Tu C-T, Hsiao C-D, Hsieh F-J, Tsai H-J. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev Dyn Off Publ Am Assoc Anat. 2003;228:30–40. doi: 10.1002/dvdy.10356. [DOI] [PubMed] [Google Scholar]
- 18.Choi W-Y, Gemberling M, Wang J, Holdway JE, Shen M-C, Karlstrom RO, et al. In vivo monitoring of cardiomyocyte proliferation to identify chemical modifiers of heart regeneration. Dev Camb Engl. 2013;140:660–666. doi: 10.1242/dev.088526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrison AC, Bare LA, Chambless LE, Ellis SG, Malloy M, Kane JP, et al. Prediction of coronary heart disease risk using a genetic risk score: the Atherosclerosis Risk in Communities Study. Am J Epidemiol. 2007;166:28–35. doi: 10.1093/aje/kwm060. [DOI] [PubMed] [Google Scholar]
- 20.Liew CC, Sole MJ, Yamauchi-Takihara K, Kellam B, Anderson DH, Lin LP, et al. Complete sequence and organization of the human cardiac beta-myosin heavy chain gene. Nucleic Acids Res. 1990;18:3647–3651. doi: 10.1093/nar/18.12.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 22.Chang B, Nishizawa T, Furutani M, Fujiki A, Tani M, Kawaguchi M, et al. Identification of a novel TPM1 mutation in a family with left ventricular noncompaction and sudden death. Mol Genet Metab. 2011;102:200–206. doi: 10.1016/j.ymgme.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 23.Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358:1899–1908. doi: 10.1056/NEJMoa075463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng G, Porter JD. Transcriptional profile of rat extraocular muscle by serial analysis of gene expression. Invest Ophthalmol Vis Sci. 2002;43:1048–1058. [PubMed] [Google Scholar]
- 25.Arad M, Penas-Lado M, Monserrat L, Maron BJ, Sherrid M, Ho CY, et al. Gene mutations in apical hypertrophic cardiomyopathy. Circulation. 2005;112:2805–2811. doi: 10.1161/CIRCULATIONAHA.105.547448. [DOI] [PubMed] [Google Scholar]
- 26.Frisso G, Limongelli G, Pacileo G, Del Giudice A, Forgione L, Calabrò P, et al. A child cohort study from southern Italy enlarges the genetic spectrum of hypertrophic cardiomyopathy. Clin Genet. 2009;76:91–101. doi: 10.1111/j.1399-0004.2009.01190.x. [DOI] [PubMed] [Google Scholar]
- 27.Kaneda T, Naruse C, Kawashima A, Fujino N, Oshima T, Namura M, et al. A novel beta-myosin heavy chain gene mutation, p.Met531Arg, identified in isolated left ventricular non-compaction in humans, results in left ventricular hypertrophy that progresses to dilation in a mouse model. Clin Sci Lond Engl 1979. 2008;114:431–440. doi: 10.1042/CS20070179. [DOI] [PubMed] [Google Scholar]
- 28.Waldmüller S, Erdmann J, Binner P, Gelbrich G, Pankuweit S, Geier C, et al. Novel correlations between the genotype and the phenotype of hypertrophic and dilated cardiomyopathy: results from the German Competence Network Heart Failure. Eur J Heart Fail. 2011;13:1185–1192. doi: 10.1093/eurjhf/hfr074. [DOI] [PubMed] [Google Scholar]
- 29.Huang T-T, Naeemuddin M, Elchuri S, Yamaguchi M, Kozy HM, Carlson EJ, et al. Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum Mol Genet. 2006;15:1187–1194. doi: 10.1093/hmg/ddl034. [DOI] [PubMed] [Google Scholar]
- 30.Hoit BD, Kiatchoosakun S, Restivo J, Kirkpatrick D, Olszens K, Shao H, et al. Naturally occurring variation in cardiovascular traits among inbred mouse strains. Genomics. 2002;79:679–685. doi: 10.1006/geno.2002.6754. [DOI] [PubMed] [Google Scholar]
- 31.Zieger B, Ware J. Cloning and deduced amino acid sequence of human nicotinamide nucleotide transhydrogenase. DNA Seq J DNA Seq Mapp. 1997;7:369–373. doi: 10.3109/10425179709034058. [DOI] [PubMed] [Google Scholar]
- 32.Hoek JB, Rydström J. Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem J. 1988;254:1–10. doi: 10.1042/bj2540001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis EE, Frangakis S, Katsanis N. Interpreting human genetic variation with in vivo zebrafish assays. Biochim Biophys Acta. 2014;1842:1960–1970. doi: 10.1016/j.bbadis.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niederriter AR, Davis EE, Golzio C, Oh EC, Tsai I-C, Katsanis N. In vivo modeling of the morbid human genome using Danio rerio. J Vis Exp JoVE. 2013:e50338. doi: 10.3791/50338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borck G, Hög F, Dentici ML, Tan PL, Sowada N, Medeira A, et al. BRF1 mutations alter RNA polymerase III-dependent transcription and cause neurodevelopmental anomalies. Genome Res. 2015;25:155–166. doi: 10.1101/gr.176925.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khuchua Z, Yue Z, Batts L, Strauss AW. A zebrafish model of human Barth syndrome reveals the essential role of tafazzin in cardiac development and function. Circ Res. 2006;99:201–208. doi: 10.1161/01.RES.0000233378.95325.ce. [DOI] [PubMed] [Google Scholar]
- 37.Van der Meer DLM, Marques IJ, Leito JTD, Besser J, Bakkers J, Schoonheere E, et al. Zebrafish cypher is important for somite formation and heart development. Dev Biol. 2006;299:356–372. doi: 10.1016/j.ydbio.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 38.Vogel B, Meder B, Just S, Laufer C, Berger I, Weber S, et al. In-vivo characterization of human dilated cardiomyopathy genes in zebrafish. Biochem Biophys Res Commun. 2009;390:516–522. doi: 10.1016/j.bbrc.2009.09.129. [DOI] [PubMed] [Google Scholar]
- 39.Yeo S-Y, Kim M, Kim H-S, Huh T-L, Chitnis AB. Fluorescent protein expression driven by her4 regulatory elements reveals the spatiotemporal pattern of Notch signaling in the nervous system of zebrafish embryos. Dev Biol. 2007;301:555–567. doi: 10.1016/j.ydbio.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 40.Meimaridou E, Kowalczyk J, Guasti L, Hughes CR, Wagner F, Frommolt P, et al. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat Genet. 2012;44:740–742. doi: 10.1038/ng.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen H, Zhang W, Li D, Cordes TM, Mark Payne R, Shou W. Analysis of ventricular hypertrabeculation and noncompaction using genetically engineered mouse models. Pediatr Cardiol. 2009;30:626–634. doi: 10.1007/s00246-009-9406-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet. 1996;12:385–389. doi: 10.1038/ng0496-385. [DOI] [PubMed] [Google Scholar]
- 43.Phoon CK, Acehan D, Schlame M, Stokes DL, Edelman-Novemsky I, Yu D, et al. Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J Am Heart Assoc. 2012:1. doi: 10.1161/JAHA.111.000455. pii: jah3-e000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brescia ST, Rossano JW, Pignatelli R, Jefferies JL, Price JF, Decker JA, et al. Mortality and sudden death in pediatric left ventricular noncompaction in a tertiary referral center. Circulation. 2013;127:2202–2208. doi: 10.1161/CIRCULATIONAHA.113.002511. [DOI] [PubMed] [Google Scholar]
- 46.Aras D, Tufekcioglu O, Ergun K, Ozeke O, Yildiz A, Topaloglu S, et al. Clinical features of isolated ventricular noncompaction in adults long-term clinical course, echocardiographic properties, and predictors of left ventricular failure. J Card Fail. 2006;12:726–733. doi: 10.1016/j.cardfail.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 47.Vermeer AMC, van Engelen K, Postma AV, Baars MJH, Christiaans I, De Haij S, et al. Ebstein anomaly associated with left ventricular noncompaction: an autosomal dominant condition that can be caused by mutations in MYH7. Am J Med Genet C Semin Med Genet. 2013;163C:178–184. doi: 10.1002/ajmg.c.31365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.