

Abstract
Cardiometabolic disease, a global health threat, has been linked to chronic inflammation, in which activated macrophages play a key role. Macrophages are highly heterogeneous hematopoietic cells found in nearly every tissue in the body. Various stimuli recruit monocytes into cardiovascular system and metabolic organs, where they differentiate to macrophages, and activate these pro-inflammatory phagocytes, leading to the initiation and development of inflammation in these organs. Key regulators of macrophage activation therefore may serve as therapeutic targets for the cardiometabolic disease. The Notch signaling pathway, involving five ligands and four receptors, regulates the differentiation of various cell types during development, and also contributes to the disease processes in adults. We found that the Notch ligand delta-like 4 (Dll4) activates macrophages in vitro as determined by the induction of genes and pathways associated with cardiovascular and metabolic disorders. Our recent study demonstrated in vivo that blockade of Dll4 by a neutralizing antibody attenuates key features typical of cardiovascular and metabolic diseases, such as accumulation of activated macrophages in arteries and fat, chronic atherosclerosis, arterial and valvular calcification, insulin resistance, and fatty liver. These results suggest that Dll4-mediated Notch signaling participates in the shared disease mechanisms for cardiovascular and metabolic disorders. This review summarizes the role of macrophages and Dll4/Notch signaling in the development of inflammation in both cardiovascular system and metabolic organs.
Introduction
Chronic inflammation, in which macrophages play a key role1, associates with a disease complex “cardiometabolic syndrome,” characterized by atherosclerosis, obesity, insulin resistance, and fatty liver2-5. Macrophages accumulate in the vascular system and metabolic organs, and are activated through various cellular and molecular pathways. Despite much effort taken to elucidate these mechanisms, knowledge remains limited, and no satisfactory therapeutic strategies are available. Notch signaling involves the highly preserved pathway among species and influences cell fate decisions, cell proliferation, differentiation, and apoptosis6. Notch receptors (Notch1, Notch2, Notch3 and Notch4) and ligands (Jagged1, Jagged2, Delta-like 1 [Dll1], Dll3, Dll4) have diverse functions in physiological and biological conditions7-9. We demonstrated that in cultured human primary macrophages, the Notch ligand Dll4 triggers various pro-inflammatory effects associated with atherosclerosis and metabolic disorders10. We further demonstrated that blockade of Dll4-mediated Notch signaling inhibited the development of atherosclerosis and insulin resistance in vivo11. Dll4 was discovered as an endothelial cell–specific Notch ligand and have been known to contribute to angiogenesis. The roles of Dll4-mediated Notch signaling in vascular development in many disease contexts have been reviewed extensively12-15. This article, therefore, aims to summarize the role of macrophages in the development of inflammation in both cardiovascular system and metabolic organs, and to present our recent progress that demonstrates the role of Dll4-mediated Notch signaling in macrophage activation.
The role of macrophages in the development of cardiovascular and metabolic disorders
Accumulating evidence supports the premise that chronic inflammation is central to the pathobiology of atherosclerotic vascular disease and metabolic disorders1, 16. In the context of atherosclerosis, activated macrophages participate critically in every stage of lesion progression, from early fatty streak formation to the acute onset of plaque rupture and thrombosis. Extensive studies demonstrated that monocyte chemoattractant protein-1 (MCP-1), a potent chemokine secreted from activated endothelial cells, promotes the recruitment of circulating monocytes into the arterial vessel wall as one of the initial major events leading to atherosclerotic vascular diseases17. Matrix-degrading enzymes and pro-thrombotic molecules elaborated from activated macrophages promote plaque disruption and subsequent thrombosis16, 18, 19. Moreover, macrophage proliferation may contribute to development of the inflamed plaque20. Additionally, macrophages secrete various pro-inflammatory cytokines such as interleukin 1β (IL-1β) that induce atherothrombosis-associated molecules through the activation of endothelial cells, smooth muscle cells, and macrophages themselves. Macrophage-derived MCP-1 may recruit more monocytes into inflamed plaques. Thus, macrophages participate in an amplification cascade that sustains inflammatory responses in the atherosclerotic plaque and promote its structural instability and thrombogenicity.
The macrophage is also the primary cell type responsible for inflammatory responses that occur in adipose tissue21. Many cellular and molecular responses, accompanied by the increase of adiposity — such as the elevation of saturated fatty acid levels, endoplasmic reticulum (ER) stress, and the decrease of local oxygen pressure — increase chemokine expression in adipose tissue. MCP-1 and its receptor CC chemokine receptor 2 (CCR2) are the most critical chemokines in the development of insulin resistance22, 23. Many previous studies using loss-of-function and gain-of-function models revealed that MCP-1 triggers macrophage accumulation, which reached to 40%–50% of the total stromal vascular fraction (SVF). Once accumulated in adipose tissues, macrophages respond to many inflammatory milieus and secrete pro-inflammatory molecules, such as MCP-1, tumor necrosis factor-α (TNF-α), and IL-6. These inflammatory molecules further promote inflammation in adipose tissues. Recent studies also demonstrated a paracrine loop involving saturated fatty acids and TNF-α derived from adipocytes and macrophages, respectively, that establishes a vicious cycle that augments the inflammatory changes in adipose tissue24, leading to the development of insulin resistance.
Thus, chronic inflammation, especially macrophage accumulation and activation, participates in the development of both atherosclerotic diseases and metabolic disorders. Several studies have suggested that inflammatory changes in adipose tissue can influence systemic inflammatory states such as atherosclerosis25, but the precise mechanisms by which adipose tissue inflammation causes systemic inflammation remain unknown, and the shared mechanisms that initiate or accelerate the inflammatory milieu in both vasculature and metabolic organs have not been fully investigated. Exploring possible links between atherosclerosis and metabolic disorders may help to establish new therapeutic strategies for this global health threat.
Macrophage Heterogeneity
Macrophages are a highly heterogeneous cell population26-28. Originating from bone marrow cells, they reside in nearly every tissue and organ in the human body. Tissue-resident macrophages, such as Kupffer cells in the liver and microglia in the neuronal system, adapt to their local environment and have distinct characteristics, including functional and morphological phenotypes. These residential macrophages sense signs of infection or tissue damage. On the other hand, the characteristics and functions of macrophages that infiltrate into cardiovascular system and metabolic organs in response to the inflammatory milieu and cause cardiovascular and metabolic disorders are different from those of residential macrophages.
In the presence of cardiovascular and/or metabolic dysfunction, cells in metabolic organs (i.e., adipocytes and endothelial cells) express chemokines, including MCP-1, which recruit circulating monocytes to organs and stimulate differentiation to macrophages. These recruited macrophages are activated by different stimuli and exhibit various activation states. Recent advances in immunology have suggested a more complicated classification of macrophage activation, but two well-established and commonly-used polarization patterns include classically activated (M1) macrophages and alternately activated (M2) macrophages29, 30. Bacterial lipopolysaccharide (LPS) or Th1 cytokines (e.g., interferon γ [IFN-γ]) induce M1 polarization. M1 macrophages produce pro-inflammatory mediators such as TNF-α, IL-1β, IL-6, matrix metalloproteinase 9 (MMP-9), and inducible nitric oxide synthase (iNOS). In contrast, Th2 cytokines (e.g., IL-4) induce M2 polarization, leading to the production of anti-inflammatory mediators, notably IL-10.
Emerging evidence has suggested close links between the M1/M2 paradigm and vascular and metabolic diseases. In 2007, several groups used hyperlipidemic mice to demonstrate that hyperlipidemia polarizes monocytes/macrophages to an M1-activated phenotype and associates with the development of atherosclerosis26, 28. Other studies demonstrated that pro-inflammatory macrophage polarization may induce atherogenesis and plaque destabilization through collagen loss and calcification in plaques16, 31. Dietary fatty acids also polarize macrophages towards M1 or M2 activation states, depending on the presence of carbon-carbon double bonds (saturated/unsaturated) or their length, thus providing a molecular basis for the crosstalk between metabolic and inflammatory pathways that associates with the development of insulin resistance32. These studies demonstrated the pivotal role of macrophages and their polarization in the development and prevention of cardiometabolic disorders. Excessive polarization of macrophages toward a pro-inflammatory M1 state in the organ may contribute to the pathogenesis of cardiovascular and metabolic diseases33-35, while a microenvironment with a dominance of M2 macrophage polarization may inhibit these diseases32.
Recent advances in immunology dissect molecular and cellular mechanisms for the development of cardiometabolic inflammation, although information related to precise mechanisms for regulation of the M1/M2 macrophage balance remains limited. Furthermore, recent studies have suggested that the balance of M1/M2 activation is not steady29. Foster et al. reported that macrophages lose the ability to produce pro-inflammatory cytokines following re-stimulation after repeated LPS treatment, even though they can produce other genes, including IL-1036. This phenomenon may associate with the mechanism for endotoxin tolerance37. Furthermore, whether M1 or M2 activated macrophages can switch their phenotypes remains obscure, particularly in vivo. Because the polarization shift to M1 macrophages closely relates to the pathophysiology of inflammation in cardiovascular system and metabolic organs, finding key molecular switches may provide potential therapeutic strategies. Despite accumulating evidence and its large clinical impact, in vivo mechanisms for macrophage polarization remain incompletely understood38. Thus, further studies are needed to reveal the molecular basis of macrophage polarization.
Overview of Notch signaling
The Notch pathway, one of the most fundamental cell signal transduction mechanisms, regulates embryonic development and differentiation of various cell types and organs6. Activation of Notch signaling requires cell-to-cell contact (Figure 1). In mammals, the pathway involves five ligands (Jagged1, Jagged2, Dll1, Dll3, and Dll4) and four receptors (Notch1, Notch2, Notch3, and Notch4). Notch signaling occurs when a ligand (e.g., Dll4) of a sending cell binds to the extracellular domain of a receptor (e.g., Notch3) expressed on a receiving cell. This binding triggers a cascade of enzymatic cleavages of the receptor by ADAM family members and the γ-secretase complex, and the release of its intracellular domain of receptor (ICD), which translocates to the nucleus. The ICD of the Notch receptor interacts with the DNA-binding protein RBP-J transcriptional repressors, and converts them to transcriptional activators. The outcome of signals transmitted by the Notch receptor is highly pleiotropic in a context-specific manner, and profoundly affects differentiation, proliferation, and apoptotic events throughout development. Such critical cell signaling pathways for survival often play a role in normal homeostasis and disease processes in adults. In addition to embryonic development, Notch mutations cause various diseases in adults. For example, Notch1 mutations associate with T-cell leukemia, aortic valve disease, and cardiomyopathy39, 40. Many studies have revealed links between mutations of Notch receptors/ligands and various diseases41, 42(Table). Many studies also have shown that Notch signaling contributes to tumorigenesis, angiogenesis, and tissue regeneration after injury43, 44 — a recent series showed that Dll4 is normally induced by VEGF, and is a negative-feedback regulator that restrains vascular sprouting and branching45. Consistent with this role, the deletion or inhibition of Dll4 results in excessive, non-productive angiogenesis. This unrestrained angiogenesis unexpectedly and paradoxically decreases tumor growth, even in tumors resistant to anti-VEGF therapies46, 47. Furthermore, among previous studies on diverse roles of Notch signaling in physiology and pathology, recent reports have suggested that Notch signaling has metabolic functions, and that Notch inhibition is beneficial in the treatment of insulin resistance48, 49. Taken together, Notch signaling components appear to have expanded roles in disease mechanisms, offering possible therapeutic targets for various disorders.
Figure 1. Notch signaling pathway.
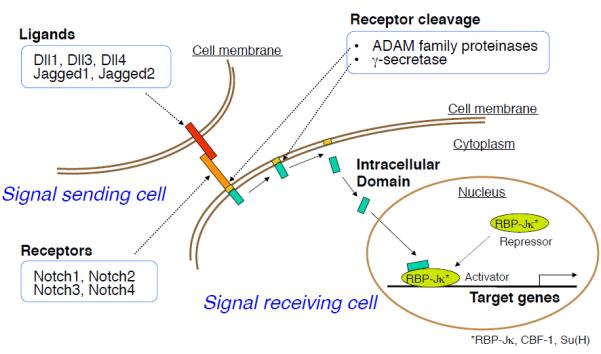
Interaction of Notch receptors with their ligands triggers a cascade of enzymatic cleavages of the receptor by ADAM family proteinases and the γ-secretase complex, which allow the Notch intracellular domain (NICD) to migrate into the nucleus. In the nucleus, NICD associates with a transcription factor, RBP-Jκ (also known as CSL for CBF1/Su(H)/Lag-1) and activates transcription from the RBP-Jκ DNA binding site.
Table.
Involvement of Notch signaling in adult diseases
| Cell/Organ type | Related disease |
|---|---|
| Macrophages | Atherosclerosis, metabolic diseases |
| T lymphocytes | Acute T-lymphoblastic leukemia, Autoimmune disease |
| B lymphocytes | Chronic B-lymphoblastic leukemia |
| Smooth muscle cells | Remodeling after vascular injruy, Alagille syndrome |
| Endothelial cells | Angiogenesis |
| Neural system | Alzheimer's disease, CADASIL, Multiple sclerosis |
| Cardiovascular system | Valvular disease, Cardiomyopathy |
| Muscle | Muscular dystrophy |
| Skin | Cancer (basal cell carcinoma, squamous cell carcinoma) |
Notch signaling in cardiovascular diseases
Notch signaling may participate in the pathogenesis of cardiovascular diseases. Several studies demonstrated that Notch signaling contributes to smooth muscle cell differentiation and proliferation, leading to vascular calcification and neointimal hyperplasia after vascular injury50, 51. Doi et al. showed that the Notch pathway regulates the differentiation of bone marrow-derived cells into smooth muscle-like cell during neointimal formation52. In addition, Notch signaling promotes pro-inflammatory responses and induces senescence in endothelial cells, leading to the development of atherosclerosis53. In this review article, we mainly discuss the contribution of macrophage activation by Notch signaling to cardiovascular inflammation, although other inflammatory cell types such as T-lymphocytes may also have roles in the pathogenesis of inflammation of cardiovascular system. The number of previous studies demonstrated that the Notch signaling is necessary for maintenance and differentiation of hematopoietic stem cells54. Particularly, T-lymphocyte commitment and maturation has been extensively studied. Several studies reported that Dll1 or Dll4 expressed on antigen presenting cells (APCs) promote Th1 cell differentiation8. Th1 cells and Th1 cytokines produced by these cells accelerate cardiovascular inflammation. On the other hand, Jagged ligands on APCs promote Th2 cell differentiation, which may suppress inflammatory responses in cardiovascular system8. Various cell types and biological processes contribute to the pathogenesis of cardiovascular disease, therefore, investigating the role of these Notch signaling may provide new insight into the mechanisms of these diseases.
Dll4-mediated Notch signaling and macrophage activation
The Notch pathway mediates juxtacrine signaling that requires cell-to-cell contact 6. Previous immunohistochemical and ultrastructural studies have clearly demonstrated direct membrane contact between adjacent macrophages55, which supports a role for homotypic juxtacrine communication between macrophages in inflamed tissues. We therefore tested the hypothesis that the activation of macrophages, a critical process for the initiation and development of atherosclerosis, involves Notch signaling. In our previous study, quantitative immunohistochemical analyses in human carotid atherosclerotic lesions localized Dll4 and other Notch components in macrophages10. We found that macrophages in human atherosclerotic plaques express various Notch pathway components. Although Dll4 had been considered as an endothelial cell–specific Notch ligand, we demonstrated in vitro that human primary macrophages express Dll4 upon activation. Dll4 expression increased in macrophages exposed to pro-inflammatory stimuli such as LPS, IL-1β, or minimally modified low-density lipoprotein (LDL) in a toll-like receptor 4– and nuclear factor (NF)-κB–dependent fashion10. What is the functionality of Dll4 in macrophages? Co-incubation of macrophages with cells expressing Dll4 triggered Notch proteolysis and activation, leading to the induction of pro-inflammatory molecules and pathways such as the typical M1 gene iNOS, mitogen-activated protein kinase, Akt, and NF-κB. Interestingly, Dll4 ligation to macrophages promoted the expression of Dll4 itself10. These findings led to our working hypothesis that Dll4-triggered Notch signaling mediates inflammatory responses by accelerating a positive feedback loop of macrophage activation, leading to the development of atherosclerotic vascular diseases, metabolic disorders, and other inflammatory diseases. More recent studies also demonstrated the contribution of the Notch signaling to the pathogenesis of inflammation56, 57. Our previous study prompted us to explore in vivo evidence for the role of Dll4-mediated Notch signaling in chronic inflammatory diseases.
Dll4-mediated Notch signaling and cardiovascular and metabolic disorders
The Notch pathway regulates embryonic development and differentiation of various cell types and organs9. Such critical cell signaling pathways often play a role in normal adult homeostasis and disease processes in major organs. As mentioned above, we found that in cultured macrophages, Notch ligand Dll4 triggers various pro-inflammatory effects associated with atherosclerosis and metabolic disorders10. We therefore hypothesized that Dll4-mediated Notch signaling participates in shared mechanisms for inflammation in cardiovascular system and metabolic organs, thus serving as a novel therapeutic target. Investigation of the in vivo role of Dll4-mediated Notch signaling in the pathogenesis of cardiovascular and metabolic disorders employed administration of the neutralizing anti-Dll4 antibody58-64 to LDL receptor-deficient (Ldlr−/−) mice fed a high-fat, high-cholesterol diet, an established model for atherosclerosis and metabolic disturbances in humans65. Unlike pan-Notch inhibitors (e.g., γ-secretase inhibitors), which cause severe and potentially fatal gut toxicity and thymus atrophy66, our Dll4 antibody did not exert any obvious adverse effects and was well tolerated. This approach enabled us to circumvent the embryonic lethality of Dll4 deficiency67, and to provide clinically translatable evidence for the pro-inflammatory role of Dll411.
Dll4 blockade reduced the size of atherosclerotic lesions in the aorta. Furthermore, Dll4 antibody treatment markedly decreased MCP-1 expression and macrophage accumulation in atherosclerotic lesions11. Macrophages play a pivotal role in the destabilization of atherosclerotic plaques, leading to the disruption of atherosclerotic plaques and acute thrombotic complications18. We and others have established the role of macrophage-derived proteinases, including matrix metalloproteinase 13 (MMP-13), in such plaque “vulnerability” by inducing loss of fibrillar collagen, a critical determinant of arterial integrity16, 68, 69. The early stage of calcification in arteries (“microcalcification”) may decrease plaque stability and cause plaque disruption70. Several studies, including our own, have shown that macrophages promote arterial calcification31, 71, 72. Other lines of evidence indirectly suggest that Notch signaling also regulates calcification50. Dll4 antibody treatment decreased collagen loss and calcification of plaques11, suggesting the role of Notch signaling in two major processes for plaque instability and disruption. Consistent with reduction of collagen loss and calcification, Dll4 antibody treatment reduced the expression of MMP-9 and MMP-13, enzymes responsible for collagen degradation in plaques, and reduced osteogenic regulators such as Cbfa-1 and osteocalcin and bone morphogenetic proteins (BMPs) in the aorta. Dll4 antibody also decreased the expression of BMP-2 and MMP-9 in peritoneal macrophages. Furthermore, in macrophages, siRNA against Dll4 reduced MMP-9 expression, whereas overexpression of Dll4 or exogenous immobilized recombinant Dll4 (rDll4)45 tended to increase this matrix-degrading enzyme. These results suggest that Dll4-mediated Notch signaling serves as an important instigator for the instability and clinical complications of atherosclerotic plaques11.
The same treatment also decreased MCP-1 expression and macrophage accumulation in adipose tissue, which associated with reduced excessive fat deposition and decreased insulin resistance11. Dll4 blockade did not affect food intake. Accompanied with the improvement of insulin sensitivity, the expression of adiponectin, GLUT4, C/EBPα, and IRS-1 — each of which correlates with insulin sensitivity — increased in the adipose tissue of antibody-treated mice. To our surprise, Dll4 blockade also improved major features of fatty liver without modulating lipid profile, as examined via decreased fat deposition, MCP-1 expression, and macrophage accumulation11.
Dll4-mediated Notch signaling regulates MCP-1 expression
Blockade of Dll4 reduced MCP-1 expression in important cardiometabolic tissues/organs, such as atherosclerotic lesions, adipose tissues, and the liver, resulted in the reduction of macrophage infiltration11. Several in vitro experiments confirmed these in vivo results. Loss-of-function experiments using siRNA against Dll4 reduced MCP-1 expression in the murine macrophage cell line RAW264.7 and differentiated 3T3-L1 adipocytes. On the other hand, gain-of-function experiments using overexpression Dll4 plasmid and immobilized rDll4 promoted MCP-1 expression in these cell types. These results clearly demonstrate that Dll4-mediated Notch signaling regulates MCP-1 expression in macrophages and adipocytes. Furthermore, Dll4-mediated Notch signaling in these cell types seems to link with the NF-κB pathway, an important regulator of MCP-1 expression11. Several previous studies have suggested a close link between Notch signaling and NF-κB pathway, but the precise mechanisms are still obscure73. Although further investigations are needed to reveal these mechanisms, the Dll4-Notch-MCP-1 axis may be a new target for effective therapies for inflammation in both cardiovascular system and metabolic organs.
Dll4-mediated Notch signaling shifts macrophage polarization to M1 polarity
Evidence suggests the heterogeneity of macrophages, and has identified at least two subpopulations: pro-inflammatory M1 macrophages, and anti-inflammatory/non-inflammatory M2 macrophages26. Multiple studies have associated M1 macrophages with atherogenesis and plaque instability through collagen loss and calcification in plaques16, 31, 74 and the development of adipose inflammation and insulin resistance32. The concept of M1/M2 macrophage balance was developed in vitro, as gauged by the expression of inflammatory mediators. Recent evidence suggests a wide range of monocyte/macrophage heterogeneity in response to either innate or adaptive immune signals30. Despite accumulating in vitro evidence and its large clinical impact, in vivo mechanisms for macrophage activation remain incompletely understood38. To explore the role of Dll4-mediated Notch signaling in macrophage polarization, we investigated whether Dll4 antibody treatment affects monocyte/macrophage polarization using a stromal vascular fraction obtained from epididymal fat. Flow cytometry analyses demonstrated that Dll4 blockade tended to decrease the Ly6C-high population, which is generally considered to be pro-inflammatory. F4/80-positive macrophages collected from the stromal vascular fraction of antibody-treated animals tended to express lower levels of pro-inflammatory mediators, including the typical M1 marker iNOS, and slightly higher levels of anti-inflammatory M2 mediators, such as IL-10. Furthermore, peritoneal macrophages obtained from Dll4-antibody treated animals took up fewer lipids and expressed lower levels of scavenger receptor-A (SR-A) RNA. Accumulation of lipids and SR-A expression are important features of plaque macrophages. Silencing with Dll4 siRNA decreased the expression of typical pro-inflammatory M1 mediators (e.g., iNOS and TNF-α) and increased the expression of the M2 marker mannose receptor 1 in RAW264.7 cells. In contrast, enforced Dll4 expression increased iNOS expression and suppressed IL-10 in this cell type. Furthermore, stimulation with immobilized rDll4 increased pro-inflammatory IL-1β and iNOS and decreased IL-10. Collectively, these in vivo and in vitro results suggest that Dll4-mediated Notch signaling skews macrophage polarization toward a pro-inflammatory phenotype11.
Assuming that macrophages have plasticity, local microenvironmental cues may tip the M1/M2 balance. Alternatively, distinct subsets of circulating monocytes may be committed to particular M1/M2 fates. Although Notch signaling is required for the appearance of hematopoietic stem cells during early development, canonical Notch signaling is not required for the generation of myeloid cells from hematopoietic stem cells75. Similarly, in this study, Dll4 blockade did not alter circulating monocyte numbers and/or the Ly6C-high monocyte subpopulation in the blood and bone marrow. Thus, Dll4 may trigger the pro-inflammatory activation of macrophages in lesions.
Dll4-mediated Notch signaling as a therapeutic option
Dissecting the multiple intertwined mechanisms for cardiovascular and metabolic disorders, including atherosclerotic vascular diseases, insulin resistance, and fatty liver, should offer insight into potential new therapeutic options 16, 76. Regulation of the circulatory, metabolic, and immune systems is highly integrated. Exploring the shared and unique mechanisms for inflammation in this disease complex will provide important insight into the development of new therapeutic strategies. We identified Dll4-Notch signaling as a key mediator for inflammation in cardiovascular system and metabolic organs. Dll4 may control central elements of inflammatory and metabolic responses in arteries, fat, and liver, and thus constitutes a unique therapeutic target in cardiovascular and metabolic disorders (Figure 2).
Figure 2. The role of the Dll4-Notch axis in cardiovascular and metabolic disorders.
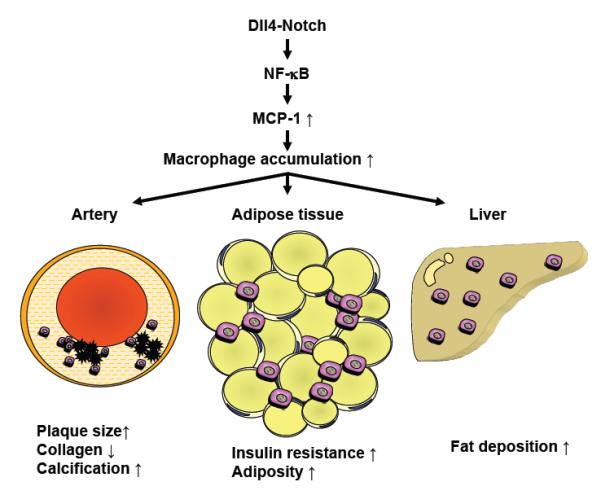
The Dll4-mediated Notch signaling activates the NF-κB pathway. Activation of NF-κB increases the expression of MCP-1, which leads to the accumulation and pro-inflammatory activation of macrophages in organs such as arteries, adipose tissue, and the liver. Accumulation of activated macrophages contributes to the pathogenesis of cardiovascular and metabolic disorders, including atherosclerosis, insulin resistance, and fatty liver disease. (Partly modified with permission from Fukuda D, et al8.)
Acknowledgments
The studies described in this article were supported by grants from the National Institutes of Health (R01HL107550, to M.A.) and from the American Heart Association (Grant-In-Aid, 0655878T, to M.A), by a Postdoctoral Fellowship Award for Research Abroad from the Japanese Heart Foundation (to D.F.), by the Japan Society for the Promotion of Science (to D.F.), and by the Uehara Memorial Foundation (to D.F.). We thank Sara Karwacki for her editorial assistance.
References
- 1.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 2.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 3.Kirk EP, Klein S. Pathogenesis and pathophysiology of the cardiometabolic syndrome. J Clin Hypertens (Greenwich) 2009;11:761–765. doi: 10.1111/j.1559-4572.2009.00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Govindarajan G, Whaley-Connell A, Mugo M, Stump C, Sowers JR. The cardiometabolic syndrome as a cardiovascular risk factor. Am J Med Sci. 2005;330:311–318. doi: 10.1097/00000441-200512000-00009. [DOI] [PubMed] [Google Scholar]
- 5.Castro JP, El-Atat FA, McFarlane SI, Aneja A, Sowers JR. Cardiometabolic syndrome: pathophysiology and treatment. Curr Hypertens Rep. 2003;5:393–401. doi: 10.1007/s11906-003-0085-y. [DOI] [PubMed] [Google Scholar]
- 6.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 7.Aster JC, Pear WS, Blacklow SC. Notch signaling in leukemia. Annu Rev Pathol. 2008;3:587–613. doi: 10.1146/annurev.pathmechdis.3.121806.154300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radtke F, Fasnacht N, Macdonald HR. Notch signaling in the immune system. Immunity. 2010;32:14–27. doi: 10.1016/j.immuni.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11:338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 10.Fung E, Tang SM, Canner JP, Morishige K, Arboleda-Velasquez JF, Cardoso AA, et al. Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation. 2007;115:2948–2956. doi: 10.1161/CIRCULATIONAHA.106.675462. [DOI] [PubMed] [Google Scholar]
- 11.Fukuda D, Aikawa E, Swirski FK, Novobrantseva TI, Kotelianski V, Gorgun CZ, et al. Notch ligand Delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc Natl Acad Sci U S A. 2012;109:E1868–1877. doi: 10.1073/pnas.1116889109. doi: 10.1073/pnas.1116889109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dufraine J, Funahashi Y, Kitajewski J. Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene. 2008;27:5132–5137. doi: 10.1038/onc.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hellstrom M, Phng LK, Gerhardt H. VEGF and Notch signaling: the yin and yang of angiogenic sprouting. Cell Adh Migr. 2007;1:133–136. doi: 10.4161/cam.1.3.4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li JL, Harris AL. Crosstalk of VEGF and Notch pathways in tumour angiogenesis: therapeutic implications. Front Biosci. 2009;14:3094–3110. doi: 10.2741/3438. [DOI] [PubMed] [Google Scholar]
- 15.Yan M, Plowman GD. Delta-like 4/Notch signaling and its therapeutic implications. Clin Cancer Res. 2007;13:7243–7246. doi: 10.1158/1078-0432.CCR-07-1393. [DOI] [PubMed] [Google Scholar]
- 16.Aikawa M, Libby P. The vulnerable atherosclerotic plaque: pathogenesis and therapeutic approach. Cardiovasc Pathol. 2004;13:125–138. doi: 10.1016/S1054-8807(04)00004-3. [DOI] [PubMed] [Google Scholar]
- 17.Martinovic I, Abegunewardene N, Seul M, Vosseler M, Horstick G, Buerke M, et al. Elevated monocyte chemoattractant protein-1 serum levels in patients at risk for coronary artery disease. Circ J. 2005;69:1484–1489. doi: 10.1253/circj.69.1484. [DOI] [PubMed] [Google Scholar]
- 18.Deguchi JO, Aikawa E, Libby P, Vachon JR, Inada M, Krane SM, et al. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation. 2005;112:2708–2715. doi: 10.1161/CIRCULATIONAHA.105.562041. [DOI] [PubMed] [Google Scholar]
- 19.Fukumoto Y, Deguchi JO, Libby P, Rabkin-Aikawa E, Sakata Y, Chin MT, et al. Genetically determined resistance to collagenase action augments interstitial collagen accumulation in atherosclerotic plaques. Circulation. 2004;110:1953–1959. doi: 10.1161/01.CIR.0000143174.41810.10. [DOI] [PubMed] [Google Scholar]
- 20.Aikawa M, Rabkin E, Sugiyama S, Voglic SJ, Fukumoto Y, Furukawa Y, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation. 2001;103:276–283. doi: 10.1161/01.cir.103.2.276. [DOI] [PubMed] [Google Scholar]
- 21.Bhargava P, Lee CH. Role and function of macrophages in the metabolic syndrome. Biochem J. 2012;442:253–262. doi: 10.1042/BJ20111708. [DOI] [PubMed] [Google Scholar]
- 22.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol. 2005;25:2062–2068. doi: 10.1161/01.ATV.0000183883.72263.13. [DOI] [PubMed] [Google Scholar]
- 25.Hirata Y, Tabata M, Kurobe H, Motoki T, Akaike M, Nishio C, et al. Coronary atherosclerosis is associated with macrophage polarization in epicardial adipose tissue. J Am Coll Cardiol. 2011;58:248–255. doi: 10.1016/j.jacc.2011.01.048. [DOI] [PubMed] [Google Scholar]
- 26.Gordon S. Macrophage heterogeneity and tissue lipids. J Clin Invest. 2007;117:89–93. doi: 10.1172/JCI30992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 28.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 30.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aikawa E, Aikawa M, Libby P, Figueiredo JL, Rusanescu G, Iwamoto Y, et al. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation. 2009;119:1785–1794. doi: 10.1161/CIRCULATIONAHA.108.827972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chawla A. Control of macrophage activation and function by PPARs. Circ Res. 2010;106:1559–1569. doi: 10.1161/CIRCRESAHA.110.216523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang CP, Han S, Senokuchi T, Tall AR. The macrophage at the crossroads of insulin resistance and atherosclerosis. Circ Res. 2007;100:1546–1555. doi: 10.1161/CIRCRESAHA.107.152165. [DOI] [PubMed] [Google Scholar]
- 34.Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. 2009;29:1419–1423. doi: 10.1161/ATVBAHA.108.180497. [DOI] [PubMed] [Google Scholar]
- 35.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 37.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 38.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harper JA, Yuan JS, Tan JB, Visan I, Guidos CJ. Notch signaling in development and disease. Clin Genet. 2003;64:461–472. doi: 10.1046/j.1399-0004.2003.00194.x. [DOI] [PubMed] [Google Scholar]
- 40.High FA, Epstein JA. The multifaceted role of Notch in cardiac development and disease. Nat Rev Genet. 2008;9:49–61. doi: 10.1038/nrg2279. [DOI] [PubMed] [Google Scholar]
- 41.Gridley T. Notch signaling in vascular development and physiology. Development. 2007;134:2709–2718. doi: 10.1242/dev.004184. [DOI] [PubMed] [Google Scholar]
- 42.Hofmann JJ, Iruela-Arispe ML. Notch signaling in blood vessels: who is talking to whom about what? Circ Res. 2007;100:1556–1568. doi: 10.1161/01.RES.0000266408.42939.e4. [DOI] [PubMed] [Google Scholar]
- 43.Kohler C, Bell AW, Bowen WC, Monga SP, Fleig W, Michalopoulos GK. Expression of Notch-1 and its ligand Jagged-1 in rat liver during liver regeneration. Hepatology. 2004;39:1056–1065. doi: 10.1002/hep.20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leker RR. Manipulation of endogenous neural stem cells following ischemic brain injury. Pathophysiol Haemost Thromb. 2006;35:58–62. doi: 10.1159/000093545. [DOI] [PubMed] [Google Scholar]
- 45.Williams CK, Li JL, Murga M, Harris AL, Tosato G. Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood. 2006;107:931–939. doi: 10.1182/blood-2005-03-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444:1032–1037. doi: 10.1038/nature05355. [DOI] [PubMed] [Google Scholar]
- 47.Thurston G, Noguera-Troise I, Yancopoulos GD. The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer. 2007;7:327–331. doi: 10.1038/nrc2130. [DOI] [PubMed] [Google Scholar]
- 48.Pajvani UB, Shawber CJ, Samuel VT, Birkenfeld AL, Shulman GI, Kitajewski J, et al. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat Med. 2011;17:961–967. doi: 10.1038/nm.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rubio-Aliaga I, Przemeck GK, Fuchs H, Gailus-Durner V, Adler T, Hans W, et al. Dll1 haploinsufficiency in adult mice leads to a complex phenotype affecting metabolic and immunological processes. PLoS One. 2009;4:e6054. doi: 10.1371/journal.pone.0006054. doi:10.1371/journal.pone.0006054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shimizu T, Tanaka T, Iso T, Doi H, Sato H, Kawai-Kowase K, et al. Notch signaling induces osteogenic differentiation and mineralization of vascular smooth muscle cells: role of Msx2 gene induction via Notch-RBP-Jk signaling. Arterioscler Thromb Vasc Biol. 2009;29:1104–1111. doi: 10.1161/ATVBAHA.109.187856. [DOI] [PubMed] [Google Scholar]
- 51.Li Y, Takeshita K, Liu PY, Satoh M, Oyama N, Mukai Y, et al. Smooth muscle Notch1 mediates neointimal formation after vascular injury. Circulation. 2009;119:2686–2692. doi: 10.1161/CIRCULATIONAHA.108.790485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doi H, Iso T, Shiba Y, Sato H, Yamazaki M, Oyama Y, et al. Notch signaling regulates the differentiation of bone marrow-derived cells into smooth muscle-like cells during arterial lesion formation. Biochem Biophys Res Commun. 2009;381:654–659. doi: 10.1016/j.bbrc.2009.02.116. [DOI] [PubMed] [Google Scholar]
- 53.Liu ZJ, Tan Y, Beecham GW, Seo DM, Tian R, Li Y, et al. Notch activation induces endothelial cell senescence and pro-inflammatory response: implication of Notch signaling in atherosclerosis. Atherosclerosis. 2012;225:296–303. doi: 10.1016/j.atherosclerosis.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee SU, Maeda M, Ishikawa Y, Li SM, Wilson A, Jubb AM, et al. LRF-mediated Dll4 repression in erythroblasts is necessary for hematopoietic stem cell maintenance. Blood. 2013;121:918–929. doi: 10.1182/blood-2012-03-418103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van der Wal AC, Dingemans KP, van den Bergh Weerman M, Das PK, Becker AE. Specialized membrane contacts between immunocompetent cells in human atherosclerotic plaques. Cardiovasc Pathol. 1994;3:81–85. doi: 10.1016/1054-8807(94)90037-X. [DOI] [PubMed] [Google Scholar]
- 56.Outtz HH, Wu JK, Wang X, Kitajewski J. Notch1 deficiency results in decreased inflammation during wound healing and regulates vascular endothelial growth factor receptor-1 and inflammatory cytokine expression in macrophages. J Immunol. 2010;185:4363–4373. doi: 10.4049/jimmunol.1000720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsao PN, Wei SC, Huang MT, Lee MC, Chou HC, Chen CY, et al. Lipopolysaccharide-induced Notch signaling activation through JNK-dependent pathway regulates inflammatory response. J Biomed Sci. 2011;18:56. doi: 10.1186/1423-0127-18-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fukushima A, Sumi T, Ishida W, Ojima A, Kajisako M, Koyanagi A, et al. Notch ligand Delta-like4 inhibits the development of murine experimental allergic conjunctivitis. Immunol Lett. 2008;121:140–147. doi: 10.1016/j.imlet.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 59.Kassner N, Krueger M, Yagita H, Dzionek A, Hutloff A, Kroczek R, et al. Cutting edge: Plasmacytoid dendritic cells induce IL-10 production in T cells via the Delta-like-4/Notch axis. J Immunol. 2010;184:550–554. doi: 10.4049/jimmunol.0903152. [DOI] [PubMed] [Google Scholar]
- 60.Koyanagi A, Sekine C, Yagita H. Expression of Notch receptors and ligands on immature and mature T cells. Biochem Biophys Res Commun. 2012;418:799–805. doi: 10.1016/j.bbrc.2012.01.106. [DOI] [PubMed] [Google Scholar]
- 61.Moriyama Y, Sekine C, Koyanagi A, Koyama N, Ogata H, Chiba S, et al. Delta-like 1 is essential for the maintenance of marginal zone B cells in normal mice but not in autoimmune mice. Int Immunol. 2008;20:763–773. doi: 10.1093/intimm/dxn034. [DOI] [PubMed] [Google Scholar]
- 62.Oishi H, Sunamura M, Egawa S, Motoi F, Unno M, Furukawa T, et al. Blockade of delta-like ligand 4 signaling inhibits both growth and angiogenesis of pancreatic cancer. Pancreas. 2010;39:897–903. doi: 10.1097/MPA.0b013e3181ce7185. [DOI] [PubMed] [Google Scholar]
- 63.Sekine C, Moriyama Y, Koyanagi A, Koyama N, Ogata H, Okumura K, et al. Differential regulation of splenic CD8- dendritic cells and marginal zone B cells by Notch ligands. Int Immunol. 2009;21:295–301. doi: 10.1093/intimm/dxn148. [DOI] [PubMed] [Google Scholar]
- 64.Yamanda S, Ebihara S, Asada M, Okazaki T, Niu K, Ebihara T, et al. Role of ephrinB2 in nonproductive angiogenesis induced by Delta-like 4 blockade. Blood. 2009;113:3631–3639. doi: 10.1182/blood-2008-07-170381. [DOI] [PubMed] [Google Scholar]
- 65.Subramanian S, Han CY, Chiba T, McMillen TS, Wang SA, Haw A, 3rd, et al. Dietary cholesterol worsens adipose tissue macrophage accumulation and atherosclerosis in obese LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:685–691. doi: 10.1161/ATVBAHA.107.157685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005;435:964–968. doi: 10.1038/nature03589. [DOI] [PubMed] [Google Scholar]
- 67.Gale NW, Dominguez MG, Noguera I, Pan L, Hughes V, Valenzuela DM, et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci U S A. 2004;101:15949–15954. doi: 10.1073/pnas.0407290101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Libby P, Aikawa M. Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nat Med. 2002;8:1257–1262. doi: 10.1038/nm1102-1257. [DOI] [PubMed] [Google Scholar]
- 69.Kobayashi N, Hata N, Kume N, Yokoyama S, Shinada T, Tomita K, et al. Matrix metalloproteinase-9 for the earliest stage acute coronary syndrome. Circ J. 2011;75:2853–2861. doi: 10.1253/circj.cj-11-0640. [DOI] [PubMed] [Google Scholar]
- 70.Aikawa E, Otto CM. Look more closely at the valve: imaging calcific aortic valve disease. Circulation. 2011;125:9–11. doi: 10.1161/CIRCULATIONAHA.111.073452. [DOI] [PubMed] [Google Scholar]
- 71.New SE, Aikawa E. Cardiovascular calcification: an inflammatory disease. Circ J. 2011;75:1305–1313. doi: 10.1253/circj.cj-11-0395. [DOI] [PubMed] [Google Scholar]
- 72.Shao JS, Cheng SL, Sadhu J, Towler DA. Inflammation and the osteogenic regulation of vascular calcification: a review and perspective. Hypertension. 2010;55:579–592. doi: 10.1161/HYPERTENSIONAHA.109.134205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Osipo C, Golde TE, Osborne BA, Miele LA. Off the beaten pathway: the complex cross talk between Notch and NF-kappaB. Lab Invest. 2008;88:11–17. doi: 10.1038/labinvest.3700700. [DOI] [PubMed] [Google Scholar]
- 74.Shanahan CM. Inflammation ushers in calcification: a cycle of damage and protection? Circulation. 2007;116:2782–2785. doi: 10.1161/CIRCULATIONAHA.107.749655. [DOI] [PubMed] [Google Scholar]
- 75.Yuan JS, Kousis PC, Suliman S, Visan I, Guidos CJ. Functions of notch signaling in the immune system: consensus and controversies. Annu Rev Immunol. 2010;28:343–365. doi: 10.1146/annurev.immunol.021908.132719. [DOI] [PubMed] [Google Scholar]
- 76.Williams KJ, Feig JE, Fisher EA. Rapid regression of atherosclerosis: insights from the clinical and experimental literature. Nat Clin Pract Cardiovasc Med. 2008;5:91–102. doi: 10.1038/ncpcardio1086. [DOI] [PubMed] [Google Scholar]