

Abstract
The advent of polymeric materials has significantly promoted the development and rapid growth of various technologies in biomedical applications, such as tissue engineering and controlled drug and gene delivery. Water-soluble polypeptides bearing functional side chains and adopting stable secondary structures are a new class of functional polymeric materials of potentially broad applications in medicine and biotechnology. In this article, we summarize our recent effort on the design and synthesis of the water-soluble α-helical ionic polypeptides originally developed in our laboratory and highlight their applications in cell membrane penetration and non-viral gene/siRNA delivery.
Keywords: ionic polypeptide, amino acid, N-carboxyanhydride (NCA), helical conformation, cell penetration, non-viral gene delivery, membrane active peptide, polyarginine analogue, non-viral siRNA delivery, functional polypeptide, ring-opening polymerization, biomaterials, TNF-α, mannose targeting, oral siRNA delivery
Introduction
Polypeptides are widely used as biocompatible materials,[1] and a unique characteristic of polypeptides is their innate ability to adopt ordered conformations such as α-helices and β-sheets through cooperative hydrogen-bonding. These conformations impart polypeptides with desirable properties and functionalities in various biomedical applications, such as tissue engineering and drug delivery.[2]
Cationic polypeptides were also the first class of biomaterials used as non-viral gene delivery vectors.[3] However, conventional cationic polypeptides such as poly-L-lysine (PLL) and its modified derivatives often suffer from low transfection efficiencies despite their abilities to condense anionic plasmid DNA.[4] Polymers with better transfection efficiency, such as polyethyleneimine (PEI), have progressively replaced polypeptides as polymeric transfection reagents,[5] while PLL has assumed diminished roles in other functions relevant to transfection and sometimes is even used as negative control in gene transfection. However, specialty peptides, such as cell penetrating peptides (CPPs) exemplified by penetratin[6], transportan[7] and TAT (HIV Tat-derived peptide with the sequence of RKKRRQRRR),[8] have been explored as component materials incorporated into existing delivery vectors to promote cell internalization, endosomal escape, and overall transfection efficiency.[9] Helical conformation is often observed in CPPs or formed in CPPs during membrane transduction, and has been closely connected to their membrane activity.[10] However, due to their short length and lack of adequate cationic charge density, CPPs are often incompetent as gene delivery vectors on their own. Therefore, it is of great interest in the design and synthesis of polypeptide-based vectors that possess the structural characteristics of CPPs (i.e. helical secondary structure) yet can also function as stand-alone gene delivery vectors.
Synthesis of Polypeptides Using Ring-Opening Polymerization of N-Carboxyanhydrides (NCAs)
Polypeptides are usually prepared through amine-initiated ring-opening polymerization (ROP) of α-amino acid N-carboxyanhydrides (NCAs) and the resulting polypeptides typically have uncontrolled molecular weights (MWs) and broad molecular weight distributions (MWDs).[11] In the past decade, a few controlled NCA polymerizations were reported using ammonium[12], transition metal complexes as initiators[13], or conventional amine initiators under high vacuum[14]. Mechanistic studies were also conducted on how temperature, pressure, and impurities affect the polymerization.[15] These achievements collectively contributed to the improvement of traditional amine-initiated NCA polymerization. We recently reported hexamethyldisilazane (HMDS)-mediated, controlled NCA polymerization and identified trimethylsilyl carbamate (TMS-CBM) moiety as an unusual chain-propagation group (Figure 1A).[16] This methodology allows better control over MW and MWD, and thus enables the preparation of well-defined block co-polypeptides. In addition, this metal-free living polymerization can be expanded to various N-trimethylsilyl (N-TMS) amines and allows facile functionalization of C-termini of polypeptides (Figure 1B)[17] by a variety of functional groups including alkene, alkyne, and norbornene. For instance, this system can be integrated with ring-opening metathesis polymerization (ROMP) by attaching TMS group on ROMP monomers or chain transfer agents (CTAs), such that well-defined polypeptides containing brush-like[18] or block polymers[19] can be synthesized.
Figure 1.
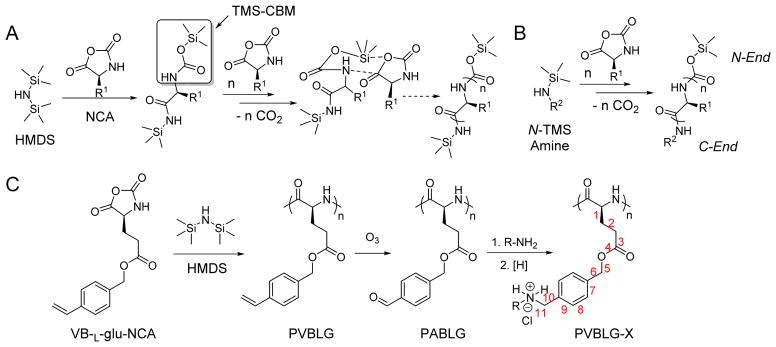
A) Proposed mechanism of hexamethyldisilazane (HMDS)-initiated controlled ROP of NCA.[16] Copyright 2007 American Chemical Society. Reproduced with permission. B) Synthesis of C-termini functionalized polypeptides initiated by N-TMS amines.[17] Copyright 2008 American Chemical Society. Reproduced with permission. C) Synthesis of water-soluble α-helical polypeptides PVBLG-X.[29] Copyright 2011 Nature Publishing Group. Reproduced with permission.
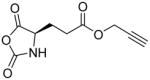
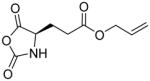
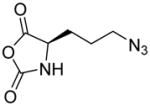
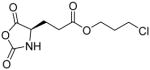
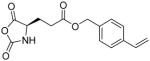
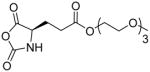
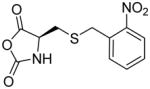
In addition, new NCA monomers derived from non-canonical amino acids have emerged as attractive motifs, and substantial efforts have thus been made in order to integrate more functionalities into polypeptides. Some recently developed NCAs are listed in Table 1. These NCA monomers bear functional groups that are amenable either to post-modifications (e.g., alkynyl[20], alkenyl[21], azide[22] and chloro[23] groups) or stimuli-responsive[24]. In conjunction with these efforts, we also developed novel NCA monomers based on glutamate, [25] such as the γ-(4-vinylbenzyl)-L-glutamate NCA (VB-L-Glu-NCA) bearing vinyl benzyl groups that can be converted into various functional groups including alcohol, aldehyde, carboxylic acid, vicinal diol, chloride, and aromatic ring via highly efficient post-polymerization modifications.[25a] The versatility of this chemistry presents great potential in generating polypeptides with diverse side chain functionalities and properties, which greatly potentiated the applications of polypeptide-based materials.
Table 1.
The structures of recently developed NCAs.
Water-Soluble Ionic Polypeptides with Unusual Helical Stability
Helical secondary structure is one of the most common characteristics of polypeptides and plays important roles in their biological activities. However, the usefulness of α-helical polypeptides comprised of natural amino acids, such as poly-L-alanine, is limited because of their poor aqueous solubility and processability.[26] Therefore, tremendous efforts have been devoted to the design and synthesis of water soluble polypeptides with stable helical conformation. Incorporation of charged groups into polypeptides significantly improves water solubility, which however, also leads to reduced helical stability because of increased side-chain charge repulsion.[27] Comparatively, introducing neutral hydrophilic groups (e.g., hydroxyl group, sugar moieties, or oligo ethylene glycol) onto the side chains can yield water-soluble polypeptides with stable helical structures.[28] However, these neutral polypeptides may not be easily used for gene delivery due to their lack of cationic charges to condense nucleic acids. Charged, water-soluble polypeptides that adopt stable α-helical conformations would be an interesting alternative to these water-soluble, neutral, helical polypeptides in gene delivery.
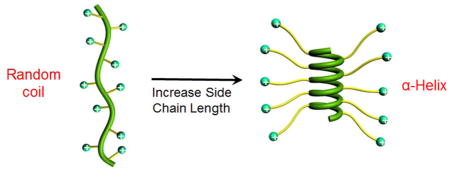
We designed and synthesized ionic α-helical polypeptides by elongating the separation distance between the polypeptide backbone and the distally positioned side charged groups.[29] We reason that when the charge is moved further away from the backbone, the helix-disrupting, side-chain charge repulsion would be reduced. Additionally, the enhanced hydrophobic interaction due to the elongated side chains would also serve to stabilize the helical conformation (Figure 2A). We thereafter unraveled that when the charged residue was placed at a minimum of 11 σ-bond away from the backbone (Figure 1C), the resulting polypeptide (PAHG and PVBLG-1, Figure 2B) showed good water solubility and stable helical structures (Figure 2C). The helical structure was also found to be highly stable under various physiochemical conditions. The helicity was stable over a wide range of pH (pH 1~10) and temperature (5~60 °C) (Figure 2D and 2E), both of which are important parameters for biomedical applications (such as the pH variations among different extracellular/intracellular compartments). In addition, the helicity was also stable against high salt concentration (up to 3 M) and the denaturing reagent (urea, up to 2 M).[29] Such unique helical stability would allow the polypeptides to maintain integral secondary structures under a variety of complex physiological conditions, which largely broadens their utility in biomedical applications.
Figure 2.

A) Schematic illustration showing the random coil to helix transformation of cationic polypeptides upon elongation of side chain length.[29] Copyright 2012 Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. B) Chemical structures of PAHG, PLL, and PVBLG-1. C) Circular dichroism (CD) spectra of PAHG, PVBLG-1, and PLL in aqueous solution at pH 3. Both PAHG and PVBLG-1 adopted helical conformation while PLL adopted random coil conformation.[29] Copyright 2011 Nature Publishing Group. Reproduced with permission. D) The pH-dependence of the residue molar ellipticity at 222 nm for PAHG, PVBLG-1, and PLL.[29] Copyright 2011 Nature Publishing Group. Reproduced with permission. E) Temperature-dependence of residue molar ellipticity at 222 nm for PAHG and PVBLG-1 at pH 3.[29] Copyright 2011 Nature Publishing Group. Reproduced with permission.
Bioactive Cationic α-Helical Polypeptide Template for Cell Penetration and Non-Viral Gene Delivery
Based on our aforementioned efforts in controlling NCA polymerization and synthesizing α-helical ionic polypeptides, we are able to generate novel non-viral gene delivery vectors based on cationic helical polypeptides that are able to simultaneously condense genes and mediate membrane transduction.[30] As such, a library of cationic α-helical polypeptides with various amine side groups (PVBLG-X) was synthesized via ROP of VB-L-Glu-NCA and subsequent side-chain amination (Figure 1C). These polypeptides were screened in terms of gene transfection efficiencies in attempts to identify particular candidates with appropriate balance between hydrophilicity (i.e., DNA binding strength) and hydrophobicity (i.e., membrane activity). The top-performing polypeptide, named PVBLG-8 (Figure 3A), was identified which demonstrated 12-fold higher transfection efficiency than the commonly used transfection reagent polyethylenimine (PEI) (Figure 3B). By observing the cellular internalization pattern of a membrane impermeable fluorescent dye (calcein) in COS-7 cells, we discovered that treatment with PVBLG-8 led to permeated intracellular distribution of calcein compared to the punctated spots of free calcein, which suggested that PVBLG-8 was able to promote membrane permeation by destabilizing the cell membrane and generating pores (Figure 4A). In addition, PVBLG-8 with a concentration of 10 μg/mL incubated with murine erythrocytes resulted in 80% hemoglobin leakage, further indicating the membrane disruptive ability of the polymer.[31] Transfection studies were also performed in the presence of inhibitors such as bafilomycin that prevents proton transport and acidification of endosomes, or nocodazole that leads to the accumulation of endocytosed material in early endosomes via the disruption of active cellular transport processes. Bafilomycin showed negligible effect on PVBLG-8-mediated transfection.[30] Meanwhile, nocodazole induced a two-fold increase in the transfection efficiency of PVBLG-8, which suggested that as more complexes accumulated the effective polymer concentration became sufficient enough to cause enhanced vesicle lysis.[30] These results collectively indicated that PVBLG-8 was able to mediate endosomal escape towards effective gene transfection mainly by disrupting the endosomal membranes rather than via the “proton sponge” effect. As part of the above assessments, PVBDLG-8, an analogue of PVBLG-8 with non-helical, distorted conformation prepared from the racemic D,L-NCA, was unable to mediate efficient pore formation on the cell membranes indicated by a lower percentage of hemolysis compared to that of the helical polypeptide at the same concentration,[31] and showed negligible transfection efficiency (Figure 3B). Such observation thus highlighted the essential roles of helical structure in mediating membrane penetration and gene delivery which can present a rigid amphiphilic structure to promote the membrane interactions.
Figure 3.
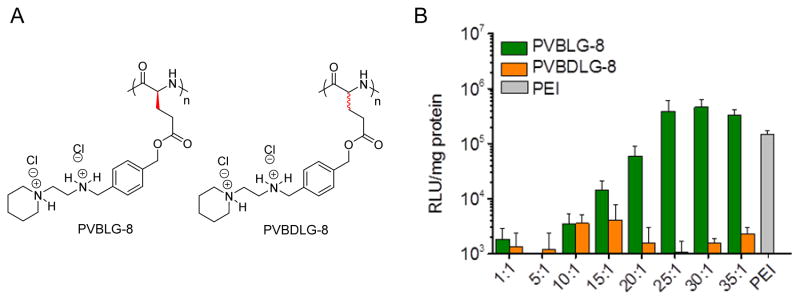
A) Chemical structure of helical PVBLG-8 and non-helical PVBDLG-8. B) In vitro transfection efficiency of PVBLG-8/pCMV-Luc and PVBDLG-8/pCMV-Luc polyplexes in COS-7 cells at various polymer/DNA weight ratios. PEI (25 kDa) at a 7.5:1 polymer/DNA weight ratio was included as a control.[30] Copyright 2012 Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Figure 4.

A) Calcein uptake in COS-7 cells treated with PVBLG-8 at various concentrations. In the absence of an agent capable of pore formation, calcein, a fluorescent dye, is unable to permeate intact cell membranes and thus taken up by cells in a pinocytic fashion, resulting in the appearance of small punctate intracellular fluorescent spots. As the concentration of PVBLG-8 in the extracellular medium was increased, the intracellular fluorescence of calcein became more diffuse, which indicated membrane permeation and non-endosomal uptake.[30] Copyright 2012 Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. B) Schematic presentation of a proposed model for the cellular internalization of PVBLG-8/siRNA complexes.[31] Copyright 2012 The American Society of Gene & Cell Therapy. Reproduced with permission.
Apart from DNA, PVBLG-8 was also able to mediate effective cellular delivery of small interference RNA (siRNA).[31] Because of the rigid structure of both PVBLG-8 and siRNA and the relatively short length of the siRNA molecule, the condensation capacity of PVBLG-8 towards siRNA was lower than that towards plasmid DNA.[30,31] Nevertheless, we noted that PVBLG-8-mediated intracellular delivery of siRNA did not necessarily require condensation of siRNA. Namely, pre-treatment of cells with free PVBLG-8 prior to addition of free siRNA led to comparable cellular uptake of siRNA as compared to co-delivered materials (PVBLG-8 and siRNA were added to the cell media concurrently).[31] Such observation further substantiated that the helical PVBLG-8 was able to induce pore formation on cell membranes to promote the direct trans-membrane diffusion of small nucleic acids, such as siRNA that contains only 21–27 nucleotides (Figure 4B). Traditional CPPs often require the formation of acidic environment when disrupting biological membranes and mediating cellular uptake as well as endosomal escape, and they will be ineffective if the internalization route avoids rapid acidification, such as caveolae endocytosis.[32] In this regard, PVBLG-8 with stable and pH-independent helical conformation showed advantages as siRNA delivery vectors.
Cationic Helical Polypeptides Enhance Delivery Efficiencies of Other Systems
The membrane activities of PVBLG-8 not only endow it with potent gene/siRNA delivery capabilities, but also allow it to enhance the delivery efficiencies of existing systems. For instance, the incorporation of PVBLG-8 into self-assembled nanocomplexes (SSANs) consisting of oleyl trimethyl chitosan, oleyl-PEG-mannose, and DNA notably altered the intracellular kinetics of SSANs.[33] SSANs without PVBLG-8 were mainly internalized via clathrin-mediated endocytosis, an acidic and digestive route that often involves endosomal entrapment, as evidenced by the inhibited cell uptake level by 70~80% at 4 °C or following treatment with chlorpromazine, an inhibitor of clathrin-mediated endocytosis. Comparatively, the cellular uptake level of SSANs with PVBLG-8 was only reduced by 30% at 4 °C and endocytic inhibitors exerted slight or negligible inhibitory effect, which implied that PVBLG-8 allowed majority of the SSANs to enter the cells via energy-independent direct transduction after puncturing pores on cell membranes (Figure 5). In addition to the internalization pathway, PVBLG-8 also altered the mechanisms underlying endosomal/lysosomal escape. The transfection efficiency of SSANs without PVBLG-8 was greatly enhanced by chloroquine that buffers the pH of late endosomes/lysosomes while was decreased upon treatment with bafilomycin, indicating that they partially escaped the endosomes/lysosomes via the “proton sponge” effect. Nevertheless, neither chloroquine nor bafilomycin had appreciable effect on SSANs with PVBLG-8, which suggested that PVBLG-8 mediated endosomal escape by destabilizing the endosomal membranes rather than by the “proton sponge” effect. In consistence with such huge discrepancy in the intracellular fate, incorporation of PVBLG-8 remarkably potentiated the transfection efficiency of SSANs by two orders of magnitude.[33]
Figure 5.
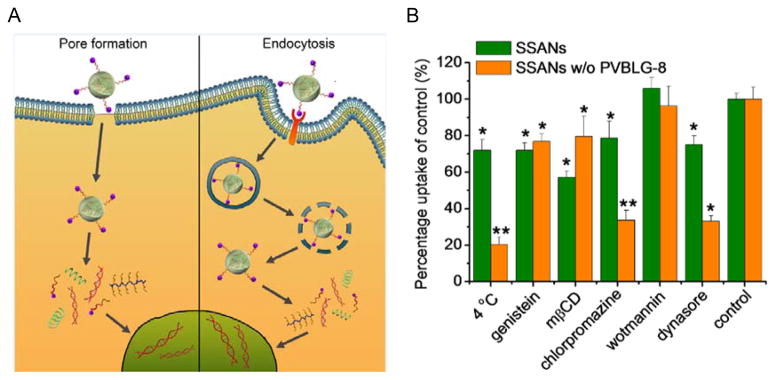
A) Schematic illustration of the internalization pathways of supramolecular self-assembled nanocomplexes (SSANs) containing PVBLG-8.[33] Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. B) Mechanistic probes of the intracellular kinetics of SSANs w/and w/o PVBLG-8 (genistein, methyl-β-cyclodextrin (mβCD) = caveolae inhibitor; chlorpromazine = clathrin-mediated endocytosis inhibitor; dynasore = clathrin-mediated endocytosis and caveolae inhibitor; wortmannin = macropinocytotic inhibitor).[33] Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
In a similar approach, PVBLG-8 was also incorporated to supramolecular self-assembled nanoparticles (SSNPs) comprising oleyl trimethyl chitosan, oleyl-PEG-mannose, oleyl-PEG-cysteine, sodium tripolyphosphate, and siRNA against tumor necrosis factor α (TNF-α), to strengthen their capabilities in oral siRNA delivery (Figure 6A).[34] Particularly, the potent membrane activities of PVBLG-8 notably enhanced the internalization of SSNPs by normal enterocytes as well as M cells in the small intestine, thereby resulting in augmented intestinal absorption level of the siRNA cargo via the intracellular pathway (Figure 6B). Nevertheless, PVBLG-8 had negligible effect on the paracellular transport via the tight junctions, as evidenced by the unappreciable alteration in the transepithelial electrical resistance values after incorporation of PVBLG-8 into the SSNPs. [34] Similar trend was also noted in gut-associated macrophages, the target cells of TNF-α siRNA-mediated gene silencing. In the in vitro studies in RAW 264.7 cells (murine macrophages), incorporation of PVBLG-8 greatly enhanced the cellular uptake level of SSNPs due to its potent membrane disruption/destabilization capabilities, and thus triggered higher TNF-α knockdown levels than SSNPs without PVBLG-8. The unique helical stability of PVBLG-8 between pH 2 and 9 allows it to maintain helix-dependent membrane activities after passing the gastric (acidic) and intestinal (weak basic) environments, such that their capabilities to promote intestinal absorption and gene silencing in gut-associated macrophages can be guaranteed. As a consequence of the facilitated intestinal absorption as well as the translocation of transfected gut-associated macrophages to systemic reticuloendothelial tissues, PVBLG-8 finally enhanced the capacity of orally delivered SSNPs to alleviate lipopolysaccharide (LPS)-induced systemic TNF-α production (Figure 6C), and thus demonstrated desired anti-inflammatory efficacy against hepatic injury at a low dose of 50 μg/kg. This is the first example of a polymeric nanocarrier that can simultaneously address the material requirement for oral siRNA delivery, and the essential roles of PVBLG-8 render it great potentials for in vivo applications.
Figure 6.

A) Schematic illustration of supramolecular self-assembled nanoparticles (SSNPs) developed for oral delivery of TNF-α siRNA (oleyl trimethyl chitosan = OTMC; oleyl-PEG-mannose = OPM; oleyl-PEG-cysteine = OPC; tripolyphosphate = TPP). SSNPs were constructed through electrostatic and hydrophobic self-assembly of each component, which had a particle size of about 120 nm.[34] Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. B) Intestinal absorption of Cy3-siRNA-containing SSNPs in the in vitro follicle-associated epithelial (FAE) and non-FAE models. Results were represented as apparent permeability coefficient (Papp).[34] Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. C) Relative TNF-α mRNA levels in mouse liver, spleen, and lung 24 h after oral gavage of SSNPs at 200 μg/kg.[34] Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Functionalization and Reconfiguration of Polypeptides toward Maximized Gene Delivery Capabilities
The potent membrane activity of PVBLG-8 that stems from both of its cationic density and helicity greatly contributes to its gene delivery efficiency, while in the meantime, excessive membrane activity will also cause irreversible damage to cell membranes that ultimately leads to undesired cytotoxicity. We thus sought additional strategies to reduce the material toxicity and simultaneously maximize its delivery efficiencies. Incorporation of poly(ethylene glycol) (PEG) moieties has been proven as an effective strategy to reduce material cytotoxicity and improve the polyplex stability.[35] However, PEGylation might also reduce the membrane activity and the overall efficacy because the hydrophilic PEG corona prevents the interactions between polyplexes and cell membrane.[36] Motivated by these understandings, we designed and synthesized various compositionally equivalent yet topologically different PEG-PVBLG-8 copolymers (Figure 7A), attempting to elucidate the effect of polymer topology on the gene delivery efficiency and identify the optimal architecture toward maximized transfection efficiency and minimized cytotoxicity.[37] Di-block, ABA tri-block, graft, and star (8-arm) PEG-PVBLG-8 copolymers were prepared by using different amine-terminated PEG as initiators or grafting moieties. Di-block and tri-block copolymers displayed lower membrane activities and cytotoxicities, and a possible balance thereof led to uncompromised gene transfection efficiency compared to the non-PEGylated homopolymer, PVBLG-8. The graft copolymer exhibited lower DNA binding affinity and membrane activity and overall reduced transfection efficiency, which was attributed to the steric effect of the PEG segments that prevented the polymer from accessing plasmid DNA as well as cell membranes. The star copolymer possessed the highest membrane activity yet relatively low cytotoxicity, thus resulting in the highest transfection efficiency that outperformed the non-PEGylated PVBLG-8 and Lipofectamine™ 2000 (LPF2000) (Figure 7B and 7C). The extraordinary permeability of the star copolymer was believed to stem from its densely charged architecture as a “multivalent cationic sphere”, which favored interactions with negatively charged cell membranes.
Figure 7.

A) Schematic representation of PEG-PVBLG-8 copolymers with different architectures.[37] Copyright 2013 Elsevier B.V. Reproduced with permission. B) Cytotoxicity of polyplexes (N/P ratio = 20) towards HeLa cells as determined by the MTT assay.[37] Copyright 2013 Elsevier B.V. Reproduced with permission. C) Transfection efficiencies of polyplexes in HeLa cells at various N/P ratios.[37] Copyright 2013 Elsevier B.V. Reproduced with permission.
In another approach, we designed a light-responsive PVBLG-8 derivative (PDMNBLG-r-PVBLG-8) that maintains high membrane activity pre-transfection while transforms to a helix-distorted and less charged state post-transfection to facilitate intracellular DNA release as well as eliminate long-term material toxicity (Figure 8).[38] Upon external light triggers (UV or NIR), the poly(4,5-dimethoxy-2-nitrobenzyl glutamate) (PDMNBLG) domain will yield pendant carboxylate groups via light-triggered de-esterfication (Figure 8A). The intramolecular electrostatic attraction between negatively charged carboxylate groups and positively charged amine groups would thus reduce the net charge density and disrupt the helical structures, collectively leading to decreased membrane activity and cytotoxicity of the polypeptides. Additionally, the reduced cationic charge density also weakened the DNA binding affinity to promote intracellular DNA unpacking post-transfection, as evidence by the notably promoted separation between red (rhodamine labeled PDMNBLG-r-PVBLG-8) and green (YOYO-1-DNA) fluorescence in the confocal laser scanning microscopy (CLSM) images of transfected HeLa cells pre- and post-UV light exposure (Figure 8B). With the combined effect of reduced toxicity and facilitated DNA release, the gene transfection efficiency was greatly enhanced by one order of magnitude following light irradiation post-transfection.[38] This class of novel cationic helical polypeptides with built-in trigger-responsive domains allows us to precisely control the charge and conformational change of the polypeptides using external stimuli, and ultimately manipulate the intracellular responses to overcome multiple barriers against non-viral gene transfer.
Figure 8.

A) Schematic representation showing the charge and conformational transformation of PDMNBLG-r-PVBLG-8 upon external light triggers.[38] Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. B) CLSM images of HeLa cells incubated with RhB-PDMNBLG-r-PVBLG-8 (red)/YOYO-1-DNA (green) complexes with/without UV irradiation (bar = 20 μm). The orange color indicated colocalization of polymer and DNA, while the separation of green and red fluorescence signals suggested efficient DNA unpacking.[38] Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Arginine residues have been found rich in many CPPs and their guanidinium head groups have been closely tied to the penetrating capabilities due to their interactions with the sulfate groups of glycosaminoglycans localized on cell membranes.[39] Since all the aforementioned helical polypeptides contain amine groups on the side chain terminals, we were thus motivated to develop guanidinium-rich helical polypeptides to further potentiate the membrane activities as well as the gene delivery efficiencies. By taking advantage of the controlled ROP of NCA and highly efficient “click” chemistry, a library of cationic, helical, poly(arginine) mimics were developed where the guanidinium side charged groups were placed distally at a minimum separation of 13 σ-bond away from the backbone.[40] The resulting polypeptides adopted stable helical structures in comparison to poly(L-arginine) (PLR) that adopts random coil conformation under physiological conditions, and they exhibited higher cell penetration capabilities than commercial CPPs such as Arg9 and TAT, mainly via the pore formation mechanism. Further increment of the helicity by elongating the side chain length up to 18 σ-bond yielded significantly improved membrane permeability, which again substantiated the importance of helical structure in mediating membrane interactions. Considering that hydrophobic domains would “self-activate” the guanidinium groups to potentiate the penetrating properties,[41] we also integrated hydrophobicity into the polypeptide design by developing a copolypeptide containing alkyl side chains. Particularly, an increase in the alkyl length that correlated to higher hydrophobicity resulted in greatly improved penetration level, while a decrease in the alkyl content from 50% to 10% further potentiated the penetration level, presumably due to increased cationic charge density of the guanidinium groups. Upon our efforts in the structural optimization, P14 with a proper balance among cationic charge density, helicity, and hydrophobicity was identified to be the top-performing material which outperformed commercial CPPs such as Arg9 and TAT by 1–2 orders of magnitude in terms of cell penetration potency (Figure 9A and 9B). With its strong membrane permeability to mediate effective cellular internalization as well as endosomal escape, P14 also exhibited desired efficiency as a molecular transporter for DNA and siRNA, outperforming commercial transfection reagent LPF2000 by up to two orders of magnitude in terms of transfection/gene knockdown efficiencies (Figure 9C).
Figure 9.

A) Structures of P14, a helical poly(arginine) mimic.[40] Copyright 2013 The Royal Society of Chemistry. Reproduced with permission. B) Uptake level of RhB-P14 (20 μg/mL) in HeLa, 3T3-L1 and Raw 264.7 cells following incubation at 37 °C for 2 h. Commercial CPPs, Arg9 and TAT were included as controls.[40] Copyright 2013 The Royal Society of Chemistry. Reproduced with permission. C) Transfection efficiencies of P14/DNA polyplexes (N/P ratio = 15) in HeLa, 3T3-L1, and Raw 264.7 cells. Arg9, TAT, poly(L-arginine) (PLR), and LPF 2000 served as controls.[40] Copyright 2013 The Royal Society of Chemistry. Reproduced with permission.
Conclusions
We summarized the progress of ionic helical polypeptides and highlighted our recent efforts in their biomedical applications for non-viral gene delivery. By taking advantage of organosilicon-initiated controlled NCA polymerization and side-chain post-modification, a series of cationic helical polypeptides with different side-chain functionalities and architectures were developed. These materials possessed desired membrane activities mainly via the pore formation mechanism, and helical secondary structure was demonstrated to be essential toward their membrane permeabilities. As such, by facilitating direct transduction of the gene cargo across cellular as well as endosomal membranes, the helical polypeptides mediated effective gene/siRNA delivery by themselves or greatly strengthened the delivery efficiencies of existing systems. By incorporating multi-functionalities or reconfiguring the polypeptide architectures, we were able to modulate the membrane activities and material toxicities of the helical polypeptides, thus maximizing their gene delivery capabilities. Despite all these success, the detailed mechanism underlying the polypeptide-mediated membrane penetration remains to be unraveled using techniques such as small-angle X-ray scattering. A proper structural reconfiguration as well as formulation is also necessitated to maximize the in vivo gene delivery efficiency towards therapeutic attempts. Therefore, developing new ionic helical polypeptides with trigger-responsive, degradable, and targeting moieties will be a promising subject in the field.
Acknowledgments
We acknowledge the financial support from the NSF (CHE-1153122) and the NIH (NIH Director’s New Innovator Award 1DP2OD007246 and 1R21EB013379).
References
- 1.a) Obeid R, Scholz C. Biomacromolecules. 2011;12:3797. doi: 10.1021/bm201048x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang S, Bai M, Wang L. Scientific reports. 2013;3:2023. doi: 10.1038/srep02023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Deming TJ. Adv Drug Deliv Rev. 2002;54:1145. doi: 10.1016/s0169-409x(02)00062-5. [DOI] [PubMed] [Google Scholar]; b) Deming TJ. Prog Polym Sci. 2007;32:858. [Google Scholar]
- 3.Monsigny M, Roche AC, Midoux P, Mayer R. Adv Drug Deliv Rev. 1994;14:1. [Google Scholar]
- 4.a) Ferkol T, Perales JC, Mularo F, Hanson RW. Proc Natl Acad Sci USA. 1996;93:101. doi: 10.1073/pnas.93.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Putnam D, Gentry CA, Pack DW, Langer R. Proc Natl Acad Sci USA. 2001;98:1200. doi: 10.1073/pnas.031577698. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Okuda T, Sugiyama A, Niidome T, Aoyagi H. Biomaterials. 2004;25:537. doi: 10.1016/s0142-9612(03)00542-8. [DOI] [PubMed] [Google Scholar]
- 5.Hsu CYM, Uludag H. Nat Protoc. 2012;7:935. doi: 10.1038/nprot.2012.038. [DOI] [PubMed] [Google Scholar]
- 6.Terrone D, Sang SLW, Roudaia L, Silvius JR. Biochemistry. 2003;42:13787. doi: 10.1021/bi035293y. [DOI] [PubMed] [Google Scholar]
- 7.Pooga M, Hallbrink M, Zorko M, Langel U. Faseb J. 1998;12:67. doi: 10.1096/fasebj.12.1.67. [DOI] [PubMed] [Google Scholar]
- 8.Brooks H, Lebleu B, Vives E. Adv Drug Deliv Rev. 2005;57:559. doi: 10.1016/j.addr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Martin ME, Rice KG. The AAPS journal. 2007;9:E18. doi: 10.1208/aapsj0901003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Derossi D, Calvet S, Trembleau A, Brunissen A, Chassaing G, Prochiantz A. The Journal of biological chemistry. 1996;271:18188. doi: 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- 11.Deming TJ. Adv Polym Sci. 2006;202:1. [Google Scholar]
- 12.Dimitrov I, Schlaad H. Chem Commun. 2003:2944. doi: 10.1039/b308990h. [DOI] [PubMed] [Google Scholar]
- 13.a) Deming TJ. Nature. 1997;390:386. doi: 10.1038/37084. [DOI] [PubMed] [Google Scholar]; b) Deming TJ. J Am Chem Soc. 1998;120:4240. [Google Scholar]
- 14.Aliferis T, Iatrou H, Hadjichristidis N. Biomacromolecules. 2004;5:1653. doi: 10.1021/bm0497217. [DOI] [PubMed] [Google Scholar]
- 15.a) Habraken GJM, Peeters M, Dietz CHJT, Koning CE, Heise A. Polym Chem. 2010;1:514. [Google Scholar]; b) Pickel DL, Politakos N, Avgeropoulos A, Messman JM. Macromolecules. 2009;42:7781. [Google Scholar]
- 16.Lu H, Cheng J. J Am Chem Soc. 2007;129:14114. doi: 10.1021/ja074961q. [DOI] [PubMed] [Google Scholar]
- 17.Lu H, Cheng J. J Am Chem Soc. 2008;130:12562. doi: 10.1021/ja803304x. [DOI] [PubMed] [Google Scholar]
- 18.Lu H, Wang J, Lin Y, Cheng J. J Am Chem Soc. 2009;131:13582. doi: 10.1021/ja903425x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai Y, Lu H, Ponnusamy E, Cheng J. Chem Commun. 2011;47:10830. doi: 10.1039/c1cc13531g. [DOI] [PubMed] [Google Scholar]
- 20.a) Engler AC, Lee HI, Hammond PT. Angew Chem Int Ed. 2009;48:9334. doi: 10.1002/anie.200904070. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang YG, Zeng YH, Yang JW, Zeng ZH, Zhu FM, Chen XD. Chem Commun. 2011;47:7509. doi: 10.1039/c1cc12177d. [DOI] [PubMed] [Google Scholar]
- 21.a) Sun J, Schlaad H. Macromolecules. 2010;43:4445. [Google Scholar]; b) Tang H, Zhang D. Polym Chem. 2011;2:1542. [Google Scholar]
- 22.Rhodes AJ, Deming TJ. ACS Macro Lett. 2013;2:351. doi: 10.1021/mz4001089. [DOI] [PubMed] [Google Scholar]
- 23.a) Tang H, Zhang D. Biomacromolecules. 2010;11:1585. doi: 10.1021/bm1002174. [DOI] [PubMed] [Google Scholar]; b) Tang H, Zhang D. Polym Chem. 2011;2:1542. [Google Scholar]; c) Song Z, Zheng N, Ba X, Yin L, Zhang R, Ma L, Cheng J. Biomacromolecules. 2014;15:1491. doi: 10.1021/bm5001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Chen C, Wang Z, Li Z. Biomacromolecules. 2011;12:2859. doi: 10.1021/bm200849m. [DOI] [PubMed] [Google Scholar]; b) Fu XH, Shen Y, Fu WX, Li ZB. Macromolecules. 2013;46:3753. [Google Scholar]; c) Liu G, Dong CM. Biomacromolecules. 2012;13:1573. doi: 10.1021/bm300304t. [DOI] [PubMed] [Google Scholar]
- 25.a) Lu H, Bai Y, Wang J, Gabrielson NP, Wang F, Lin Y, Cheng J. Macromolecules. 2011;44:6237. doi: 10.1021/ma201164n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang Y, Lu H, Lin Y, Cheng J. Macromolecules. 2011;44:6641. doi: 10.1021/ma201678r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tang H, Yin L, Lu H, Cheng J. Biomacromolecules. 2012;13:2609. doi: 10.1021/bm3009445. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zheng N, Yin L, Song Z, Ma L, Tang H, Gabrielson NP, Lu H, Cheng J. Biomaterials. 2014;35:1302. doi: 10.1016/j.biomaterials.2013.09.090. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhang R, Zheng N, Song Z, Yin L, Cheng J. Biomaterials. 2014;35:3443. doi: 10.1016/j.biomaterials.2013.12.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Levy Y, Jortner J, Becker OM. Proc Natl Acad Sci USA. 2001;98:2188. doi: 10.1073/pnas.041611998. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dobson CM, Sali A, Karplus M. Angew Chem Int Ed. 1998;37:868. doi: 10.1002/(SICI)1521-3773(19980420)37:7<868::AID-ANIE868>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 27.a) Dobson CM. Nature. 2003;426:884. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]; b) Dill KA. Biochemistry. 1990;29:7133. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 28.a) Lotan N, Yaron A, Berger A. Biopolymers. 1966;4:365. [Google Scholar]; b) Yu M, Nowak AP, Deming TJ, Pochan DJ. J Am Chem Soc. 1999;121:12210. [Google Scholar]; c) Kramer JR, Deming TJ. J Am Chem Soc. 2012;134:4112. doi: 10.1021/ja3007484. [DOI] [PubMed] [Google Scholar]
- 29.Lu H, Wang J, Bai Y, Lang JW, Liu SY, Lin Y, Cheng J. Nat Commun. 2011;2 doi: 10.1038/ncomms1209. [DOI] [PubMed] [Google Scholar]
- 30.Gabrielson NP, Lu H, Yin L, Li D, Wang F, Cheng J. Angew Chem Int Ed. 2012;51:1143. doi: 10.1002/anie.201104262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gabrielson NP, Lu H, Yin L, Kim KH, Cheng J. Mol Ther. 2012;20:1599. doi: 10.1038/mt.2012.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.a) Davidson TJ, Harel S, Arboleda VA, Prunell GF, Shelanski ML, Greene LA, Troy CM. J Neurosci. 2004;24:10040. doi: 10.1523/JNEUROSCI.3643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bjorklund J, Biverstahl H, Graslund A, Maler L, Brzezinski P. Biophys J. 2006;91:L29. doi: 10.1529/biophysj.106.083881. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rejman J, Bragonzi A, Conese M. Mol Ther. 2005;12:468. doi: 10.1016/j.ymthe.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 33.Yin L, Song Z, Kim KH, Zheng N, Gabrielson NP, Cheng J. Adv Mater. 2013;25:3063. doi: 10.1002/adma.201205088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yin L, Song Z, Qu Q, Kim KH, Zheng N, Yao C, Chaudhury I, Tang H, Gabrielson NP, Uckun FM, Cheng J. Angew Chem Int Ed. 2013;52:5757. doi: 10.1002/anie.201209991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a) Deshpande MC, Davies MC, Garnett MC, Williams PM, Armitage D, Bailey L, Vamvakaki M, Armes SP, Stolnik S. J Control Release. 2004;97:143. doi: 10.1016/j.jconrel.2004.02.019. [DOI] [PubMed] [Google Scholar]; b) Venkataraman S, Ong WL, Ong ZY, Loo SCJ, Ee PLR, Yang YY. Biomaterials. 2011;32:2369. doi: 10.1016/j.biomaterials.2010.11.070. [DOI] [PubMed] [Google Scholar]
- 36.Liu TQ, Thierry B. Langmuir. 2012;28:15634. doi: 10.1021/la301390u. [DOI] [PubMed] [Google Scholar]
- 37.Yin L, Song Z, Kim KH, Zheng N, Tang H, Lu H, Gabrielson N, Cheng J. Biomaterials. 2013;34:2340. doi: 10.1016/j.biomaterials.2012.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin L, Tang H, Kim KH, Zheng N, Song Z, Gabrielson NP, Lu H, Cheng J. Angew Chem Int Ed. 2013;52:9182. doi: 10.1002/anie.201302820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wender PA, Galliher WC, Goun EA, Jones LR, Pillow TH. Adv Drug Deliv Rev. 2008;60:452. doi: 10.1016/j.addr.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang H, Yin L, Kim KH, Cheng J. Chemical Science. 2013;4:3839. doi: 10.1039/C3SC51328A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.a) Som A, Reuter A, Tew GN. Angew Chem Int Ed. 2012;51:980. doi: 10.1002/anie.201104624. [DOI] [PubMed] [Google Scholar]; b) Som A, Tezgel AO, Gabriel GJ, Tew GN. Angew Chem Int Ed. 2011;50:6147. doi: 10.1002/anie.201101535. [DOI] [PubMed] [Google Scholar]