

Abstract
To establish a rational molecular design for bisphosphonate (BP)-modified proteins for efficient bone targeting, a pharmacokinetic study was performed using a series of alendronate (ALN), a nitrogen-containing BP, modified proteins with various molecular weights and varying degrees of modification. Four proteins with different molecular weight—yeast glutathione reductase (GR; MW: 112,000 Da), bovine serum albumin (BSA; MW: 67,000 Da), recombinant human superoxide dismutase (SOD; MW: 32,000 Da), and chicken egg white lysozyme (LZM; MW: 14,000 Da)—were modified with ALN to obtain ALN-modified proteins. Pharmacokinetic analysis of the tissue distribution of the ALN-modified and unmodified proteins was performed after radiolabeling them with indium-111 (111In) by using a bifunctional chelating agent. Calculation of tissue uptake clearances revealed that the bone uptake clearances of 111In-ALN-modified proteins were proportional to the degree of ALN modification. 111In-GR-ALN and BSA-ALN, the two high-molecular-weight proteins, efficiently accumulated in bones, regardless of the degree of ALN modification. Approximately 36 and 34% of the dose, respectively, was calculated to be delivered to the bones. In contrast, the maximum amounts taken up by bone were 18 and 13% of the dose for 111In-SOD-ALN(32) and LZM-ALN(9), respectively, because of their high renal clearance. 111In-SOD modified with both polyethylene glycol (PEG) and ALN (111In-PEG-SOD-ALN) was efficiently delivered to the bone. Approximately 36% of the dose was estimated to be delivered to the bones. In an experimental bone metastasis mouse model, treatment with PEG-SOD-ALN significantly reduced the number of tumor cells in the bone of the mice. These results indicate that the combination of PEG and ALN modification is a promising approach for efficient bone targeting of proteins with a high total-body clearance.
Introduction
The prevalence of bone diseases is rapidly increasing as the population ages. Various endogenous proteins, including enzymes and cytokines, represent new candidates as therapeutic agents to treat bone diseases [1–4]. However, most protein drugs intended for bone diseases do not have a high affinity for bone hydroxyapatite under physiological conditions, which limits their therapeutic potential [1]. Therefore, targeting protein drugs to the bone is essential for the treatment of bone diseases. Of the various strategies available, chemical modification is an attractive approach for the targeted delivery of protein drugs, in terms of efficacy and safety, because protein drugs have several functional groups that can be conjugated with ligands or polymers [5].
To date, bone targeting of protein drugs by chemical modification has been studied with several molecules, including tetracycline, acidic oligopeptides, and bisphosphonates (BPs) [6–8]. BPs strongly inhibit the function of osteoclasts that mediate bone resorption, thereby allowing for their use in the treatment of Paget’s disease, bone metastasis, hypercalcemia of malignancy, and osteoporosis [9,10]. In addition, BPs are carbon-substituted pyrophosphate analogs that exhibit a high affinity for hydroxylapatite. Consequently, BPs are rapidly distributed to the bone or excreted in the urine in equal proportion following intravenous injection [11,12]. Accordingly, the modification of protein drugs with BPs could be useful for bone targeting. It was reported that the bone accumulation of osteoprotegerin, a potent receptor activator of nuclear factor κB ligand (RANKL) inhibitor that prevents osteoclastogenesis, was increased following BP modification [13]. Furthermore, bone metastasis was suppressed by intravenous injection of BP-modified bovine catalase in mice [14]. Despite these promising characteristics, there are no systemic studies on the application of BP modification to the bone targeting of protein drugs with different physicochemical and biological characteristics. In fact, the relationship between the physicochemical characteristics of proteins, such as the molecular weight and degree of modification with BP, and their in vivo pharmacokinetic profiles are not fully understood.
The aim of the present study was to establish a rational molecular design of BP-modified proteins for efficient bone targeting in vivo. In this study, we used the nitrogen-containing BP, alendronate (ALN) and conjugated it to various proteins to obtain ALN-modified proteins. The bone targeting efficiency of ALN-modified proteins with various molecular weights and degree of modification was examined after intravenous injection in mice, and pharmacokinetic analyses were performed to obtain quantitative relationships between these two factors.
Materials and Methods
Animals
Male ddY mice (5-week-old) and female C57/BL6 mice (7-week-old) were purchased from Japan SLC Inc. (Shizuoka, Japan), and were maintained under conventional housing conditions. All animal experiments were conducted in accordance with the principles and procedures outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The Animal Experimentation Committee of the Kyoto Pharmaceutical University approved all protocols for animal experiments.
Chemicals
Glutathione reductase from yeast (GR, 1,000 U/mL) was purchased from Oriental Yeast, Tokyo, Japan. Bovine serum albumin (BSA) was purchased from Sigma-Aldrich, St. Louis, MO. Human recombinant superoxide dismutase solution (SOD, 300 or 450 U/μL) was purchased from Wako Pure Chemical Industries, Osaka, Japan. Lysozyme from chicken egg whites (LZM, >9,000 U/mg) was purchased from MP Biomedicals, Illkirch, France. Alendronic acid, monosodium salt, trihydrate (ALN) was purchased from Toronto Research Chemicals, Brisbane, North York, Canada. Methoxypolyethylene glycol N-succinimidyl succinate (activated PEG; average molecular weight 5,200 Da) was purchased from NOF Co, Tokyo, Japan. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDAC), diethylenetriaminepentaacetic acid (DTPA) anhydride and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were purchased from Dojindo Laboratory, Kumamoto, Japan. D-luciferin was purchased from Promega, Madison, WI. Pharmalyte 3–10 and amberlyte IRN-150L were purchased from GE Healthcare UK Ltd.,Buckinghamshire, UK.111Indium chloride ([111In]InCl3) was supplied by Nihon Medi-Physic, Takarazuka, Japan. All other chemicals were obtained commercially as reagent-grade products.
Synthesis and characterization of ALN-modified proteins
The ALN-modified proteins were synthesized by covalent attachment of ALN to the proteins. GR (MW: 112,000 Da), BSA (MW: 67,000 Da), SOD (MW: 32,000 Da) and LZM (MW: 14,000 Da) were used as model proteins in this study. The coupling reaction of a protein with amine of ALN by EDAC was performed according to a previously published method with slight modifications [15]. In brief, protein was added to the ALN dissolved in 0.1 M HEPES (pH 6.5) with EDAC as a carboxyl activating agent. After incubation on ice for 1 h, a second aliquot of EDAC was added to the solution, and the reaction mixture was stirred overnight at 4°C. The pH of the reaction solution was adjusted to 6.5–7.0 during the first 2 h. The number of ALN residues per protein molecule was controlled by the molar ratio of reagents, protein, ALN and EDAC. The reaction mixture was applied to a column (Sephadex G-25 coarse, GE Healthcare, Little Chalfont, UK) and fractions with larger molecular weights were collected. To remove protein aggregates formed during the synthesis, reaction products were ultrafiltered through a membrane (Vivaspin, GE Healthcare, Little Chalfont, UK) that traps and retains large protein aggregates. Each derivative was then washed and concentrated by ultrafiltration against distilled water, and lyophilized. These protein derivatives were subjected to SDS-PAGE analysis, and all were found to produce one band (data not shown). To synthesize the polyethylene glycol-SOD-ALN conjugate (PEG-SOD-ALN), SOD was mixed with activated PEG in borate buffer (pH 9.0) for 24 h at 4°C in the dark [16], and then mixed with ALN as described above. The number of ALN conjugated to each derivative was determined by the molybdenum method [17]. The number of PEG was estimated by the number of unreacted free amino groups, which was determined by the trinitrobenzenesulfonic acid method [18]. The apparent molecular weight of each derivative was estimated in reference to the number of ALN and PEG.
Isoelectric point of native proteins and ALN-modified proteins
The isoelectric point (pI) was measured by isoelectric focusing as reported previously [19]. The gel consisted of acrylamide, bisacrylamide, glycerol, pharmalyte 3–10 for IEF, amberlyte IRN-150L, TEMED, and ammonium persulfate. H2SO4 (0.1 M) and NaOH (0.1 M) were used as the anolyte and catholyte, respectively. Samples were applied on the gel and subjected to electrophoresis at 500 V for 9 h, 600 V for 1 h and 800 V for 2 h. Aspergillus niger amyloglucosidase (pI 3.5), Aspergillus niger glucose oxidase (pI 4.2), soybean trypsin inhibitor (pI 4.5), bovine milk β-lactoglobulin (pI 5.3, 5.2), bovine erythrocytes carbonic anhydrase (pI 6.0), horse muscle myoglobin (pI 6.9, 7.4), Lens culinaris lectin (pI 7.8, 8.0, 8.3), bovine pancreas ribonuclease A (pI 9.5), and pig heart cytochrome C (pI 10.7) were used as pI markers.
In vivo disposition experiment
Native proteins and ALN-modified proteins were radiolabeled with 111In using the bifunctional chelating agent DTPA anhydride according to the method of Hnatowich et al., which was described in a previous report [20,21]. In brief, DTPA-protein derivatives were synthesized in 0.1 M HEPES buffer (pH 7) and reacted with 111InCl3 to obtain 111In-labeled compound in 0.1 M citrate buffer (pH 5.5). The reaction mixture was applied to a PD-10 column (GE Healthcare, Little Chalfont, UK) and eluted with 0.1 M citrate buffer (pH 5.5). The derivative fractions were collected and concentrated by ultrafiltration. Each 111In-labeled compound was dissolved in saline, and the protein concentration was adjusted by the addition of non-radiolabeled protein to the solution. The solution of 111In-protein was injected into the tail vein of mice at a dose of 1 mg protein/kg. At the appropriate time after injection, blood was collected from the vena cava under ether anesthesia, and the mice were then killed. Heparin sulfate was used as an anticoagulant. Plasma was obtained from the blood by centrifugation at 5,000 g for 5 min. The liver, kidneys, spleen, heart, lungs, and bones of the lower extremity, i.e., tibiae and femurs, were collected, rinsed with saline, and weighed; urine in the bladder was also collected. The radioactivity in each sample was counted using an automatic gamma counter (1480 Wizard, Perkin-Elmer, Boston, MA, USA). The total wet bone weight was estimated as 12% of the body weight, and the radioactivity in the bone was based on activity determined in tibiae and femurs extrapolated to whole wet bone as reported previously [22].
Calculation of area under the plasma concentration-time curve (AUC) and clearance
The 111In-radioactivity concentration in plasma was normalized with respect to the percentage of the dose/ml and the time courses were analyzed using the nonlinear least-squares program MULTI [23]. The tissue distribution profiles were evaluated using tissue uptake clearance (CLtissue) according to the integration plot analysis. The amount in a tissue at time t (Xt) and the area under the plasma concentration-time curve (AUC) from time 0 to t (AUC0-t) was normalized with the plasma concentration at time t (Ct), and CLtissue was obtained from the slope of the plot of Xt/Ct versus AUC0-t/Ct [24].
Effect of PEG-SOD-ALN on experimental bone metastasis
Experimental bone metastasis was induced by injecting 2 x 105 murine B16-BL6 cells labeled with firefly luciferase gene (B16-BL6/Luc cells) [25] in 0.1 mL of RPMI into the left ventricle of C57/BL6 mice. One and three days after tumor inoculation, saline, PEG-SOD or PEG-SOD-ALN was injected into the lateral tail vein at a dose of 500 U/mouse. At 14 days after tumor inoculation, mice were injected intraperitoneally with 2.5 mg D-luciferin, anesthetized with an intraperitoneal injection of pentobarbital (50 mg/kg), and then imaging was performed using a LAS-4000 (Fujifilm, Tokyo, Japan). Then, mice were killed, and the bones of lower limbs were excised, and the luciferase activity in the tissue was measured in a luminometer (Lumat LB 9507, EG & G Berthold, Bad Wild-bad, Germany). The luciferase activity was converted to the number of the tumor cells using a regression line, as reported previously [25].
Results
Physicochemical characteristics of ALN-modified proteins
Table 1 summarizes the physicochemical characteristics of the synthesized ALN-modified proteins. The numbers of ALN molecules on the proteins ranged from 3 to 40. The number of ALN-modified carboxyl groups depended on the amount of reacted ALN. PEG-SOD and PEG-SOD-ALN had an average of 10 PEG molecules per SOD molecule. The isoelectric points of GR, BSA, and SOD were increased depending on the number of ALN residues, although the apparent molecular weights were barely altered by ALN modification. Unfortunately, the isoelectric points of LZM-ALN and PEG conjugates could not be determined, because of the out-of-range and neutral isoelectric point of PEG, respectively.
Table 1. Physicochemical characteristics of ALN-modified proteins.
| Compound | Number of ALN Residues (mol/mol) a) | Proportion of Modified Carboxyl Groups (%)a) | Number of PEG (mol/mol) b) | Estimated Molecular Weight c) | Isoelectric Point |
|---|---|---|---|---|---|
| GR | 0 | 0 | - | 112,000 | 4.5 |
| GR-ALN(4) | 3.79 | 1.11 | - | 113,000 | 8 |
| GR-ALN(17) | 17.3 | 5.06 | - | 117,000 | 8.3 |
| BSA | 0 | 0 | - | 67,000 | 4.8 |
| BSA-ALN (3) | 2.47 | 2.52 | - | 67,800 | 4.8 |
| BSA-ALN (28) | 27.7 | 28.3 | - | 74,600 | 9.5–10.7 |
| BSA-ALN (40) | 39.7 | 40.5 | - | 77,900 | >10.7 |
| SOD | 0 | 0 | - | 32,000 | 4.9 |
| SOD-ALN(3) | 2.99 | 7.88 | - | 32,800 | 4.9 |
| SOD-ALN(32) | 31.6 | 83.2 | - | 40,700 | 10.7 |
| PEG(10)-SOD | 0 | 0 | 9.62 | 82,000 | N.D. |
| PEG(10)-SOD-ALN(4) | 3.79 | 10 | 9.62 | 83,100 | N.D. |
| LZM | 0 | 0 | - | 14,000 | 11.2 |
| LZM-ALN(5) | 29.9 | 29.9 | - | 15,400 | N.D. |
| LZM-ALN(9) | 49.4 | 49.4 | - | 16,400 | N.D. |
a) The alendronate (ALN) content was determined by the molybdenum method using ALN as a standard. The number of ALN residues was calculated using the molar ratio of ALN residues attached to the protein.
b)The number of PEG was estimated by the number of unreacted free amino groups, which was determined by the trinitrobenzenesulfonic acid method.
c) The molecular weight of each protein derivative was estimated by considering of the number of ALN residues and PEG modifications.
N.D.; not determined
Distribution of 111In-native proteins after intravenous administration
Figs 1–5 show the time course of the plasma concentrations and the cumulative amounts of 111In-radioactivity in major organs after intravenous injection of 111In-native- and ALN-modified proteins into mice. 111In-LZM and 111In-SOD quickly disappeared from plasma, whereas the other proteins with high-molecular weights remained in the circulation for a longer period of time (Figs 3 and 4). These results are in good agreement with previous studies [26–28]. The amount of 111In-GR, 111In-BSA, 111In-SOD and 111In-LZM accumulated in bone were 9.1, 2.1, 0.3, and 2.2% of the initial dose 3 h after injection, respectively (Figs 1–4). Approximately 77 and 92% of 111In-radioactivity accumulated in the kidney 3 h after injection of 111In- SOD and LZM, respectively (Figs 3 and 4), whereas those of 111In-GR and 111In-BSA were less than 2% (Figs 1 and 2). Other tissues exhibited no significant radioactivity after injection of all 111In-native proteins.
Fig 1. Time-courses of the plasma concentration, and bone, liver, kidney, and lung accumulation of 111In-labeled GR derivatives in mice after intravenous injection at a dose of 1 mg protein/kg.

The total wet bone weight was estimated as 12% of the body weight, and the radioactivity in the bone was based on activity determined in tibiae and femurs extrapolated to whole wet bone as reported previously [22]. Results are expressed as the mean ± S.D. of three mice. ●: GR, ○: GR-ALN (4), ▼: GR-ALN (17).
Fig 5. Time-courses of the plasma concentration, and bone, liver, kidney, and lung accumulation of 111In-labeled PEG-SOD derivatives in mice after intravenous injection at a dose of 1 mg protein/kg.

The total wet bone weight was estimated as 12% of the body weight, and the radioactivity in the bone was based on activity determined in tibiae and femurs extrapolated to whole wet bone as reported previously [22]. Results are expressed as the mean ± S.D. of three mice. ●: SOD, ○: PEG(10)-SOD, ▼: PEG(10)-SOD-ALN (4).
Fig 3. Time-courses of the plasma concentration, and bone, liver, kidney, and lung accumulation of 111In-labeled SOD derivatives in mice after intravenous injection at a dose of 1 mg protein/kg.

The total wet bone weight was estimated as 12% of the body weight, and the radioactivity in the bone was based on activity determined in tibiae and femurs extrapolated to whole wet bone as reported previously [22].Results are expressed as the mean ± S.D. of three mice. ●: SOD, ○: SOD-ALN (3), ▼: SOD-ALN (32).
Fig 4. Time-courses of the plasma concentration, and bone, liver, kidney, and lung accumulation of 111In-labeled LZM derivatives in mice after intravenous injection at a dose of 1 mg protein/kg.

The total wet bone weight was estimated as 12% of the body weight, and the radioactivity in the bone was based on activity determined in tibiae and femurs extrapolated to whole wet bone as reported previously [22].Results are expressed as the mean ± S.D. of three mice. ●: LZM, ○: LZM-ALN (5), ▼: LZM-ALN (9).
Fig 2. Time-courses of the plasma concentration, and bone, liver, kidney, and lung accumulation of 111In-labeled BSA derivatives in mice after intravenous injection at a dose of 1 mg protein/kg.

The total wet bone weight was estimated as 12% of the body weight, and the radioactivity in the bone was based on activity determined in tibiae and femurs extrapolated to whole wet bone as reported previously [22].Results are expressed as the mean ± S.D. of three mice. ●: BSA, ○: BSA-ALN (3), ▼: BSA-ALN (28), ▽: BSA-ALN (40).
Distribution of 111In-ALN-modified proteins after intravenous administration
111In-ALN-modified proteins were taken up by the bone in various amounts, according to the degree of modification (Figs 1–5). Fig 6 summarizes the bone accumulation of 111In-ALN-modified proteins as a function of the number of ALN residues. The maximum amount taken up by the bone was 36, 34, 18, and 13% of the dose for 111In-GR-ALN(4),111In-BSA-ALN(3), 111In-SOD-ALN(32) and 111In-LZM-ALN(9), respectively (Figs 1–6). Although the bone accumulation of 111In-GR-ALN and 111In-BSA-ALN was greatly increased with a low degree of modification, the increase in the number of attached ALN residues accelerated accumulation in the liver, kidney, and lung, and reduced bone accumulation (Figs 1, 2 and 6). 111In-SOD-ALN and 111In-LZM-ALN accumulated in the bone depending on the number of ALN residues. However, 111In-radioactivity after intravenous injection of 111In-SOD-ALN and 111In-LZM-ALN was observed mainly in the kidney or liver (Figs 3 and 4 and 6). In contrast, 111In-PEG-SOD-ALN and 111In-PEG-SOD slowly disappeared from the plasma and accumulated in the bone. Approximately 36 and 8.4% of the dose were recovered in the bone 24 h after injection, respectively (Figs 5 and 6). Little radioactivity was counted in any other tissues for all ALN-modified proteins.
Fig 6. Effect of the number of ALN residues on bone accumulation of 111In-labeled ALN-modified proteins.
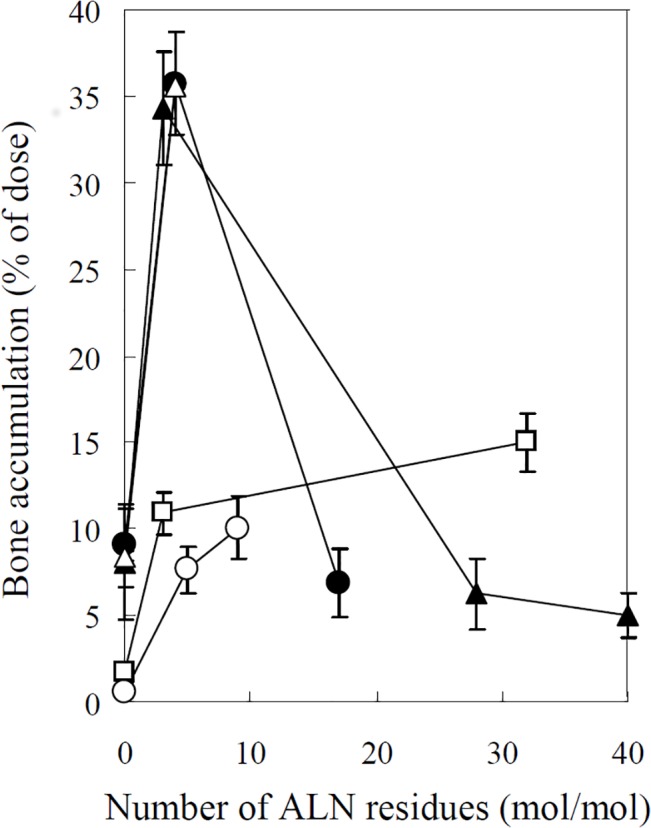
111In-labeled ALN-modified proteins were intravenously administered into mice at a dose of 1 mg protein/kg. The total wet bone weight was estimated as 12% of the body weight, and the radioactivity in the bone was based on activity determined in tibiae and femurs extrapolated to whole wet bone as reported previously [22]. Results are expressed as the mean ± S.D. of three mice. ●: GR-ALN, ▲: BSA-ALN, □: SOD-ALN, ○: LZM-ALN, △: PEG-SOD-ALN.
Calculation of pharmacokinetic parameters
To quantitatively compare the distribution profiles of the 111In-native proteins and 111In-ALN-modified proteins, CLtotal, CLtissue, and AUC were calculated based on the distribution data (Table 2). 111In-GR and BSA had large AUCs and low total clearances, reflecting the high plasma retention after intravenous injection. The AUCs of 111In- SOD and 111In-LZM were much lower than those of 111In-GR and BSA, but increased depending on the molecular weight. The CLtotal of native proteins were almost inversely correspondent with their AUC. The CLbone of 111In-GR, 111In-BSA, 111In-SOD and 111In-LZM were 0.04, 0.02, 0.10, and 1.39 ml/h, respectively, which were quite low compared with their CLtotal.
Table 2. AUC and clearance of 111In-labeled ALN-modified proteins in mice after intravenous injection of 1 mg protein/kg.
| Compound | AUC(h•% of dose/ml) | CLtotal (ml/h) | CLbone (ml/h) | CLliver (ml/h) | CLkidney (ml/h) | CLlung (ml/h) | CLurine (ml/h) |
|---|---|---|---|---|---|---|---|
| GR | 545 | 0.18 | 0.04 | 0.02 | 0.01 | 0.002 | 0.003 |
| GR-ALN(4) | 130 | 0.77 | 0.37 | 0.22 | 0.04 | 0.006 | 0.02 |
| GR-ALN(17) | 1.91 | 52.4 | 1.37 | 35.8 | 1.87 | 1.37 | 0.38 |
| BSA | 259 | 0.39 | 0.02 | 0.03 | 0.01 | 0.003 | 0.004 |
| BSA-ALN (3) | 265 | 0.38 | 0.12 | 0.03 | 0.02 | 0.002 | 0.03 |
| BSA-ALN (28) | 1.86 | 53.8 | 2.21 | 32.6 | 2.17 | 3.19 | 0.41 |
| BSA-ALN (40) | 0.81 | 124 | 6.73 | 62.8 | 2.62 | 18 | 2.14 |
| SOD | 9.64 | 10.4 | 0.1 | 0.02 | 8.43 | 0.02 | 3.28 |
| SOD-ALN(3) | 6.49 | 15.4 | 1.51 | 0.45 | 11.8 | 0.02 | 0.75 |
| SOD-ALN(32) | 2.32 | 43.1 | 6.26 | 14.7 | 7.65 | 0.36 | 6.85 |
| PEG(10)-SOD | 207 | 0.48 | 0.12 | 0.03 | 0.02 | 0.002 | 0.002 |
| PEG(10)-SOD-ALN(4) | 30.8 | 3.25 | 0.94 | 0.11 | 0.38 | 0.01 | 0.06 |
| LZM | 4.18 | 24 | 1.39 | 0.65 | 9.5 | 0.38 | 2.99 |
| LZM-ALN(5) | 3.48 | 28.7 | 2.55 | 3.06 | 12.1 | 0.59 | 2.43 |
| LZM-ALN(9) | 3.5 | 28.6 | 2.35 | 1.76 | 13.1 | 0.35 | 2.54 |
ALN modification increased the CLbone of all the proteins, without significantly changing the tissue uptake clearance in any other organ, except for the liver, kidneys, and lungs. The maximum CLbone of 111In-GR-ALN, 111In-BSA-ALN, 111In-SOD-ALN, and 111In-LZM-ALN were 1.37, 6.73, 6.26, and 2.55 ml/hr, respectively, which were much higher than those of native protein. The CLtotal of 111In-ALN-modified proteins were proportional to the degree of modification, and CLliver, CLrenal, or CLlung accounted for most of the CLtotal in proteins with a high degree of modification.
Among the series of SOD derivatives, PEG-SOD had the highest AUC. The AUC of 111In-PEG-SOD-ALN was much higher than that of 111In-SOD-ALN and 111In-SOD. Additionally, the CLliver and CLkidney of 111In-PEG-SOD-ALN were much lower than those of 111In-SOD-ALN and 111In-SOD. In addition, 111In-PEG-SOD-ALN showed the highest CLbone among the SOD derivatives. The uptake rate in the bone of 111In-PEG-SOD-ALN was 94-fold greater than that of 111In-SOD.
Inhibition of bone metastasis of melanoma cells by PEG-SOD-ALN
Fig 7A shows the number of B16–BL6/Luc cells in the tibiae and femurs of the bone metastasis mouse model. Treatment with PEG-SOD-ALN significantly reduced the number of tumor cells in the bone of the mice. In marked contrast, PEG-SOD had no statistically significant effects on the number of tumor cells in the bones.
Fig 7. Effect of SOD derivatives on the growth of metastatic tumor cells in the bones of lower limbs after its administrations 1 and 3 days after tumor injection.

(A) Number of B16BL6/Luc cells in the bones of lower limb of mice administered with PEG-SOD or PEG-SOD-ALN. Mice were sacrificed at 14 days after tumor inoculation. Results are expressed as the mean ± SD of 5–6 mice. *, a statistically significant difference compared with the saline group (P < 0.05). (B) Macroscopic observation of bone metastasis in the mouse lower limb at 14 days after inoculation of B16BL6/Luc cells (2×105 cells/mouse) into the mouse left ventricle. (a) Saline, (b) PEG-SOD, (c) PEG-SOD-ALN.
Fig 7B shows the macroscopic observation of B16–BL6/Luc cells in the tibiae and femurs of the bone metastasis mouse model. PEG-SOD-ALN effectively suppressed the metastatic colonies in the tibiae and femurs of the bone metastasis mouse model. S1 Fig shows the in vivo imaging on the distribution of the tumor cells in live mice monitored by the luciferase activity. Similarly, the growth of metastatic tumor cells was effectively inhibited by PEG-SOD-ALN. These data strongly supported the quantitative results.
Statistical analysis
Statistical comparisons with the vehicle control group were performed using Dunnett's test at a significance level of p < 0.05.
Discussion
To establish a rational molecular design for BP-modified proteins to efficiently target the bone in vivo, we performed a systemic pharmacokinetic study using a series of ALN-modified proteins with various molecular weights and degrees of modification. The pharmacokinetic study showed that the bone uptake clearance of ALN-modified proteins was closely related to the degree of ALN modification. In a preliminary experiment, we examined the stability of BSA-ALN(3) in mouse plasma by measuring free ALN using HPLC. No free ALN was detected in the sample 3 h after incubation at 37°C (data not shown), indicating that the linkage of ALN with proteins was stable in plasma. Therefore, the high bone uptake clearance of ALN-modified proteins could be explained by the high affinity of ALN to hydroxylapatite, the major component of bone [6–8,11,12]. However, ALN modification did not change the high renal clearance of SOD and LZM, the two low-molecular weight proteins, and hardly increased the bone accumulation compared with GR and BSA, the high-molecular weight proteins with long blood circulation. These results suggest that the affinity of ALN to bone is not sufficient for the effective bone targeting of proteins with high total-body clearance. The greater bone distribution of PEG-SOD-ALN could be the consequence of reduced renal clearance, affording a greater opportunity to interact with bone, because it was reported that PEG modification increases the plasma retention of proteins in vivo [16, 29, 30]. Although there was a concern that the interaction of ALN of the ALN-modified proteins with bone could be sterically prevented by the bulky PEG molecules [13], the pharmacokinetic analysis in the present study indicates that PEG modification barely reduces the affinity of SOD-ALN to bone. Thus, the combination of PEG and ALN modification is a promising approach for efficient bone targeting of proteins with high total-body clearance.
The bone accumulation of GR-ALN and BSA-ALN, the two high-molecular weight proteins, was almost inversely correlated with the number of ALN residues. In general, the electric charge of proteins also plays an important role in the tissue distribution of protein derivatives [5, 31–33]. It was reported that cationic proteins, such as amidated proteins, are rapidly accumulated in the liver after intravenous injection [32,33]. In the present study, ALN modification increased the positive charge depending on the number of ALN residues, likely due to the fact that the carboxyl residues of the proteins are modified. These results suggest that the greater hepatic distribution of GR-ALN and BSA-ALN with large number of ALN moieties is due to the electrostatic interaction between the negatively charged cell surface and the increased positive charge of GR and BSA [32, 33]. Furthermore, it was also reported that high concentrations of BPs form insoluble complexes with calcium or insoluble aggregates in the blood [34]. Therefore, the large number of ALN residues on GR-ALN and BSA-ALN may also enhance the aggregation of GR-ALN or BSA-ALN in the blood through interactions with calcium in the blood, thereby enhancing uptake by the reticuloendothelial system in the liver and accumulation in the pulmonary capillary. These two effects could be interrelated in the tissue distribution, affecting the fate of GR-ALN and BSA-ALN with a large number of ALN moieties. Although the calculation of bone uptake clearance revealed that the affinity of ALN-modified proteins to bone was proportional to the degree of modification, the results of the tissue distribution suggest that approximately 3–4 ALN/protein is the optimal degree of modification for bone targeting of these proteins.
It was reported that bones possess a membrane consisting of lining cells, which function as a marrow-blood barrier. Thus, the accessibility of exogenous large substances to the bone surface is extremely limited [7, 35]. In contrast, it was also reported that exogenous macromolecules, which are approximately 60 nm in diameter, accumulated within myeloid bone marrow interstitial space [36]. Thus, there have been several controversies concerning the vascular permeability of bone marrow. In the present study, ALN-modified proteins showed high bone uptake clearance (0.12–6.73 ml/h) compared with unmodified proteins (0.02–1.39 ml/h). Previous studies reported that the bone plasma flow rate was approximately 10.2 ml/h for a 22 g mouse [37]. These results indicate that the bone uptake of proteins is not limited by the marrow-blood barrier. In bone metastasis, where tumor cells cause vascular endothelial cell junctions to loosen and increase vascular permeability [38], the transport of ALN-modified proteins through the marrow-blood barrier would be considered relatively easy. In addition, it was reported that BPs have a high affinity to hydroxyapatite at areas of increased bone turnover, such as bone metastasis [12].
The inhibitory effects on bone metastasis treated with PEG-SOD-ALN may be associated with these results, together with those on pharmacokinetics studies in the present study. Because reactive oxygen species (ROS) play important roles in a series of steps of tumor metastasis and the tumor metastasis is effectively prevented by targeting of ROS scavenging SOD and catalase [39], the inhibitory effect on bone metastasis treated with PEG-SOD-ALN could be explained by efficient ROS elimination in the bone. Although further studies are needed to elucidate the detailed mechanism underlying this inhibition, these results suggest that our bone targeting strategy using BP is a promising approach for the treatment of bone metastasis.
Conclusions
The efficient bone targeting of GR and BSA, two high-molecular weight proteins with long blood circulation, was successfully achieved by ALN modification; however, decreased bone distribution was observed with a high degree of modification. These results indicate that approximately 3–4 ALN/protein is an optimal degree of modification for bone targeting of proteins. In contrast, low-molecular-weight proteins, such as SOD and LZM with a high total-body clearance, need both modification with 3–4 ALN/protein and PEG for efficient delivery to bone. Thus, the present study has provided useful basic information for therapeutic strategies and the molecular design of BP-modified proteins for use as therapeutic agents and drug carriers for bone targeting in vivo.
Supporting Information
Visualization of firefly luciferase gene expression in the lower limb at 14 days after inoculation. (a) Saline, (b) PEG-SOD, (c) PEG-SOD-ALN.
(TIF)
Acknowledgments
This work was supported in part by the Kyoto Pharmaceutical University Fund for the Promotion of Scientific Research.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the Kyoto Pharmaceutical University Fund for the Promotion of Scientific Research. HK recieved the funding from Kyoto Pharmaceutical University (internal to our organization). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Gittens SA, Bansal G, Zernicke RF, Uludağ H. Designing proteins for bone targeting. Adv Drug Deliv Rev. 2005; 57: 1011–1036. [DOI] [PubMed] [Google Scholar]
- 2. Uludag H, Yang J. Targeting systemically administered proteins to bone by bisphosphonate conjugation. Biotechnol Prog. 2002; 18: 604–611. [DOI] [PubMed] [Google Scholar]
- 3. Tu SM, Lin SH. 2008. Current trials using bone-targeting agents in prostate cancer. Cancer J. 2008; 14: 35–39. 10.1097/PPO.0b013e318161d32d [DOI] [PubMed] [Google Scholar]
- 4. Azim H, Azim H.A. Jr. Targeting RANKL in breast cancer: bone metastasis and beyond. Expert Rev Anticancer Ther. 2013; 13: 195–201. 10.1586/era.12.177 [DOI] [PubMed] [Google Scholar]
- 5. Takakura Y, Hashida M. Macromolecular carrier systems for targeted drug delivery: pharmacokinetic considerations on biodistribution. Pharm Res. 1996; 13: 820–831. [DOI] [PubMed] [Google Scholar]
- 6. Wang D, Miller SC, Kopecková P, Kopecek J. Bone-targeting macromolecular therapeutics. Adv Drug Deliv Rev. 2005; 57: 1049–1076. [DOI] [PubMed] [Google Scholar]
- 7. Hirabayashi H, Fujisaki J. Bone-specific Drug Delivery Systems Approaches via Chemical Modification of Bone-Seeking Agents. Clin Pharmacokinet. 2003; 42: 1319–1330. [DOI] [PubMed] [Google Scholar]
- 8. Low SA, Kopeček J. Targeting polymer therapeutics to bone. Adv Drug Deliv Rev. 2012; 64: 1189–1204. 10.1016/j.addr.2012.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fleisch H. Experimental basis for the use of bisphosphonates in Paget's disease of bone. Clin Orthop Relat Res. 1987; 217: 72–78. [PubMed] [Google Scholar]
- 10. Lambrinoudaki I, Christodoulakos G, Botsis D. Bisphosphonates. Ann N Y Acad Sci. 2006; 1092: 397–402. [DOI] [PubMed] [Google Scholar]
- 11. Lin JH. Bisphosphonates: a review of their pharmacokinetic properties. Bone. 1996; 18: 75–85. [DOI] [PubMed] [Google Scholar]
- 12. Porras AG, Holland SD, Gertz BJ. Pharmacokinetics of alendronate. Clin Pharmacokinet. 1999; 36: 315–328. [DOI] [PubMed] [Google Scholar]
- 13. Doschak MR, Kucharski CM, Wright JE, Zernicke RF, Uludağ H. Improved bone delivery of osteoprotegerin by bisphosphonate conjugation in a rat model of osteoarthritis. Mol. Pharmaceutics. 2009; 6: 634–640. [DOI] [PubMed] [Google Scholar]
- 14. Zheng Y, Nishikawa M, Ikemura M, Yamashita F, Hashida M. Development of bone-targeted catalase derivatives for inhibition of bone metastasis of tumor cells in mice. J Pharm Sci. 2012; 101: 552–557. 10.1002/jps.22773 [DOI] [PubMed] [Google Scholar]
- 15. Futami J, Maeda T, Kitazoe M, Nukui E, Tada H, Seno M et al. , Preparation of potent cytotoxic ribonucleases by cationization: enhanced cellular uptake and decreased interaction with ribonuclease inhibitor by chemical modification of carboxyl groups. Biochemistry. 2001; 40:7518–7524. [DOI] [PubMed] [Google Scholar]
- 16. Abuchowski A, van Es T, Palczuk NC, Davis FF. Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J Biol Chem. 1977; 252:3578–3581. [PubMed] [Google Scholar]
- 17. Ames BN. Assay of inorganic phosphate, total phosphate and phosphatases. Methods Enzymol. 1996; 8: 115–117. [Google Scholar]
- 18. Habeeb AF. Determination of free amino groups in proteins by trinitrobenzenesulfonic acid. Anal. Biochem 1966; 14: 328–336. [DOI] [PubMed] [Google Scholar]
- 19. Yamasaki Y, Hisazumi J, Yamaoka K, Takakura Y. Efficient scavenger receptor-mediated hepatic targeting of proteins by introduction of negative charges on the proteins by aconitylation: the influence of charge density and size of the proteins molecules. Eur J Pharm Sci. 2003; 18: 305–312. [DOI] [PubMed] [Google Scholar]
- 20. Hnatowich DJ, Layne WW, Childs RL. The preparation and labeling of DTPA-coupled albumin. Int J Appl Radiat Isot. 1982; 33: 327–332. [DOI] [PubMed] [Google Scholar]
- 21. Katsumi H, Nishikawa M, Ma SF, Yamashita F, Hashida M. Physicochemical, tissue distribution, and vasodilation characteristics of nitrosated serum albumin: delivery of nitric oxide in vivo. J Pharm Sci. 2004; 93: 2343–2352. [DOI] [PubMed] [Google Scholar]
- 22. El-Mabhouh A, Angelov C, McEwan A, Jia G, Mercer J. Preclinical investigations of drug and radionuclide conjugates of bisphosphonates for the treatment of metastatic bone cancer. Cancer Biother Radiopharm. 2004; 19: 627–640. [DOI] [PubMed] [Google Scholar]
- 23. Yamaoka K, Tanigawara Y, Nakagawa T, Uno T. A pharmacokinetic analysis program (multi) for microcomputer. J Pharmacobiodyn. 1981; 4: 879–885. [DOI] [PubMed] [Google Scholar]
- 24. Takakura Y, Takagi A, Hashida M, Sezaki H. Disposition and tumor localization of mitomycin C-dextran conjugates in mice. Pharm Res. 1987; 4: 293–300. [DOI] [PubMed] [Google Scholar]
- 25. Hyoudou K, Nishikawa M, Umeyama Y, Kobayashi Y, Yamashita F, Hashida M. Inhibition of metastatic tumor growth in mouse lung by repeated administration of polyethylene glycol-conjugated catalase: Quantitative analysis with firefly luciferase-expressing melanoma cells. Clin Cancer Res. 2004; 10: 7685–7691. [DOI] [PubMed] [Google Scholar]
- 26. Nishikawa M, Hirabayashi H, Takakura Y, Hashida M. Design for cell-specific targeting of proteins utilizing sugar-recognition mechanism: effect of molecular weight of proteins on targeting efficiency. Pharm Res. 1995; 12: 209–214. [DOI] [PubMed] [Google Scholar]
- 27. Takakura Y, Fujita T, Hashida M, Sezaki H. Disposition characteristics of macromolecules in tumor-bearing mice. Pharm Res. 1990; 7: 339–346. [DOI] [PubMed] [Google Scholar]
- 28. Mihara K, Hojo T, Fujikawa M, Takakura Y, Sezaki H, Hashida M. Disposition characteristics of protein drugs in the perfused rat kidney. Pharm Res. 1993; 10: 23–27. [DOI] [PubMed] [Google Scholar]
- 29. Katsumi H, Nishikawa M, Nishiyama K, Hirosaki R, Nagamine N, Okamoto H et al. , Development of PEGylated serum albumin with multiple reduced thiols as a long-circulating scavenger of reactive oxygen species for the treatment of fulminant hepatic failure in mice. Free Radic Biol Med. 2014; 69: 318–323 10.1016/j.freeradbiomed.2014.01.036 [DOI] [PubMed] [Google Scholar]
- 30. Katsumi H, Nishikawa M, Yamashita F, Hashida M. Development of polyethylene glycol-conjugated poly-S-nitrosated serum albumin, a novel S-Nitrosothiol for prolonged delivery of nitric oxide in the blood circulation in vivo. J Pharmacol Exp Ther. 2005; 314: 1117–1124. [DOI] [PubMed] [Google Scholar]
- 31. Yamasaki Y, Sumimoto K, Nishikawa M, Yamashita F, Yamaoka K, Hashida M et al. , Pharmacokinetic analysis of in vivo disposition of succinylated proteins targeted to liver nonparenchymal cells via scavenger receptors: importance of molecular size and negative charge density for in vivo recognition by receptors. J Pharmacol Exp Ther. 2002; 301: 467–477. [DOI] [PubMed] [Google Scholar]
- 32. Ma SF, Nishikawa M, Katsumi H, Yamashita F, Hashida M. Cationic charge-dependent hepatic delivery of amidated serum albumin. J Control Release. 2015; 102: 583–594. [DOI] [PubMed] [Google Scholar]
- 33. Nishida K, Mihara K, Takino T, Nakane S, Takakura Y, Hashida M et al. , Hepatic disposition characteristics of electrically charged macromolecules in rat in vivo and in the perfused liver, Pharm Res. 1991; 8: 437–444. [DOI] [PubMed] [Google Scholar]
- 34. Zojer N, Keck AV, Pecherstorfer M. Comparative tolerability of drug therapies for hypercalcaemia of malignancy. Drug Saf. 1999; 21: 389–406. [DOI] [PubMed] [Google Scholar]
- 35. Talmage RV. Morphological and physiological consideration in a new concept of calcium transport in bone. Am J Anat. 1970; 129: 467–476. [DOI] [PubMed] [Google Scholar]
- 36. Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res. 2010; 2: 14 10.1186/2040-2384-2-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gerlowski LE, Jain RK. Physiologically based pharmacokinetic modeling: principles and applications. J Pharm Sci. 1983; 72: 1103–1127. [DOI] [PubMed] [Google Scholar]
- 38. Mundy GR. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002; 2: 584–593. [DOI] [PubMed] [Google Scholar]
- 39. Nishikawa M, Hashida M. Inhibition of tumour metastasis by targeted delivery of antioxidant enzymes. Expert Opin Drug Deliv. 2006; 3:355–369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Visualization of firefly luciferase gene expression in the lower limb at 14 days after inoculation. (a) Saline, (b) PEG-SOD, (c) PEG-SOD-ALN.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.