

Abstract
Background
Safflower (Carthamus tinctorius L.), an Asteraceae member, yields high quality edible oil rich in unsaturated fatty acids and is resilient to dry conditions. The crop holds tremendous potential for improvement through concerted molecular breeding programs due to the availability of significant genetic and phenotypic diversity. Genomic resources that could facilitate such breeding programs remain largely underdeveloped in the crop. The present study was initiated to develop a large set of novel microsatellite markers for safflower using next generation sequencing.
Principal Findings
Low throughput genome sequencing of safflower was performed using Illumina paired end technology providing ~3.5X coverage of the genome. Analysis of sequencing data allowed identification of 23,067 regions harboring perfect microsatellite loci. The safflower genome was found to be rich in dinucleotide repeats followed by tri-, tetra-, penta- and hexa-nucleotides. Primer pairs were designed for 5,716 novel microsatellite sequences with repeat length ≥ 20 bases and optimal flanking regions. A subset of 325 microsatellite loci was tested for amplification, of which 294 loci produced robust amplification. The validated primers were used for assessment of 23 safflower accessions belonging to diverse agro-climatic zones of the world leading to identification of 93 polymorphic primers (31.6%). The numbers of observed alleles at each locus ranged from two to four and mean polymorphism information content was found to be 0.3075. The polymorphic primers were tested for cross-species transferability on nine wild relatives of cultivated safflower. All primers except one showed amplification in at least two wild species while 25 primers amplified across all the nine species. The UPGMA dendrogram clustered C. tinctorius accessions and wild species separately into two major groups. The proposed progenitor species of safflower, C. oxyacantha and C. palaestinus were genetically closer to cultivated safflower and formed a distinct cluster. The cluster analysis also distinguished diploid and tetraploid wild species of safflower.
Conclusion
Next generation sequencing of safflower genome generated a large set of microsatellite markers. The novel markers developed in this study will add to the existing repertoire of markers and can be used for diversity analysis, synteny studies, construction of linkage maps and marker-assisted selection.
Introduction
A member of the family Asteraceae, Safflower (Carthamus tinctorius L.) is a diploid (2n = 24), mostly self-pollinating dicot with an estimated haploid genome size of 1.4 GB [1]. The crop is grown in wide geographical zones across the world [2] with Kazakhstan and India currently dominating safflower production [3]. It is a multi-purpose crop employed for diverse uses such as dye production, edible oil extraction and for medicinal applications [4]. It has also been exploited for production of biofuel and industrial oil [5,6]. Recently, transgenic safflower has been employed as a plant factory for production of important pharmaceuticals of human interest such as insulin and apo lipoprotein [7–9]. Considering the desirable oil composition of safflower and its resilience to dry conditions, it can serve as an important source of edible oil especially in arid regions of the world. However, undesirable features such as low yield, spiny nature and susceptibility to several biotic stresses have reduced its cultivation in several regions including India [10].
Conventional breeding programs in several crop species have resulted in the development of cultivars with improved yield and increased resistance to several diseases. Improvements can be achieved more efficiently and faster through analysis of global genetic diversity existing in the crop for selection of elite genotypes and by molecular breeding approaches [11]. Application of molecular markers in crop breeding has proven to be a powerful method for improvement of several crop species [12]. A prerequisite for successful implementation of molecular breeding in crops is the availability of strong molecular marker-trait association [11]. A comprehensive program to increase yield is essential for safflower improvement [13]. However, safflower genetics and genomics are largely unexplored and scarcity of reliable molecular markers in safflower is a major limitation for development of effective molecular breeding programs in the crop [14, 15].
A wide range of dominant markers such as random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), inter-simple sequence repeat (ISSR) and sequence-related amplified polymorphism (SRAP) have been used for assessing the genetic diversity of safflower [16–20]. However, the dominant inheritance pattern of these markers does not allow detection of allelic information, which is important for crop breeding. Conversely, co-dominant markers allow detection of allelic diversity but in safflower, the repertoire of co-dominant markers is limited. Since their discovery in early 1980’s, microsatellite markers or SSRs (simple sequence repeats) have gained importance owing to their co-dominant inheritance, multi-allelic nature, wide genome coverage, high reproducibility, high polymorphic index, adaptability to automation, high throughput genotyping as well as efficient transfer to closely related species making them valuable tools for genetics and breeding [21–23]. In safflower, earlier studies used conventional methods of library enrichment or EST databases for development of microsatellite markers. Chapman et al. [24] generated 104 polymorphic EST-SSRs for linkage mapping in safflower. Naresh et al. [25] reported five EST-SSRs used for testing the purity of safflower hybrids. Hamdan et al. [26] isolated 64 polymorphic genomic SSRs from an enriched genomic library of safflower. However, these methods have high development cost and low throughput restricting the use of microsatellite markers [27]. Next generation sequencing (NGS) provide resources for high-throughput SSR development at a lower cost [28, 29]. Mining of NGS data for development of microsatellite markers has been exploited in a variety of plant species viz., pigeon pea, chrysanthemum, chokecherry, grass pea [30–33]. In safflower, Lee et al. [34] reported thirty polymorphic microsatellite markers derived from pyro-sequencing data while Pearl et al. [35] reported first set of 244 single nucleotide polymorphism (SNP) markers. Nonetheless, till date, only 203 polymorphic SSR markers have been reported indicating an urgent need for enrichment of robust co-dominant markers in safflower.
The genus Carthamus includes 18 species of which, C. tinctorius L. is the only cultivated species [1]. The wild species of Carthamus are known to harbor several agronomically desirable traits, which were lost during the course of safflower domestication [36–38]. Transferability of microsatellite markers to closely related species and genera would assist in the identification of marker-trait associations, which could be used for introgression of desirable loci from the wild species to cultivated safflower thus broadening its gene pool. Such markers would also be useful for synteny studies, identification of progenitor species and the study of genome evolution in the crop.
The current study exploited the efficiency of next generation sequencing data for analysis of microsatellite fraction present in safflower genome and derivation of a large set (5,716) of novel microsatellite markers for safflower. A subset of 325 microsatellite markers was experimentally validated using twenty-three geographically diverse safflower accessions. In addition, cross species transferability of polymorphic markers was assessed. Markers generated in this study would serve as important resources for population genetics, construction of linkage maps and marker-assisted selection in the crop.
Materials and Methods
Plant material and genomic DNA extraction
An accession of C. tinctorius L. (PI No: 560175) with high oil content (44%) was used for Illumina paired-end sequencing. A geographically diverse set of 23 safflower accessions belonging to seventeen countries was used to test the developed microsatellite markers. Cross species transferability of polymorphic microsatellite markers was tested using nine wild relatives of C. tinctorius L., including the probable progenitor species (C. oxyacantha and C. palaestinus). Seed samples were obtained from USDA-ARS, WRPIS, Pullman, WA, USA and IPK Gene Bank, Germany. Detailed information on plant material used in this study is given in Table 1.
Table 1. Details of plant material used in the study.
| Species | PI number # | Origin | Ploidy level | Somatic chromosome number |
|---|---|---|---|---|
| Variability study | ||||
| C tinctorius L. | 613514 | Australia | 2X | 24 |
| C tinctorius L. | 401477 | Bangladesh | 2X | 24 |
| C tinctorius L. | 305188 | India | 2X | 24 |
| C tinctorius L. | 401583 | India | 2X | 24 |
| C tinctorius L. | 544007 | China | 2X | 24 |
| C tinctorius L. | 262447 | Kazakhstan | 2X | 24 |
| C tinctorius L. | 304408 (a) | Pakistan | 2X | 24 |
| C tinctorius L. | 304408 (b) | Pakistan | 2X | 24 |
| C tinctorius L. | 388905 | Iran | 2X | 24 |
| C tinctorius L. | 388907 | Iran | 2X | 24 |
| C tinctorius L. | 306687 | Israel | 2X | 24 |
| C tinctorius L. | 340096 | Turkey | 2X | 24 |
| C tinctorius L. | 576991 | Germany | 2X | 24 |
| C tinctorius L. | 613465 | Spain | 2X | 24 |
| C tinctorius L. | 306599 | Egypt | 2X | 24 |
| C tinctorius L. | 306596 | Egypt | 2X | 24 |
| C tinctorius L. | 239041 | Morocco | 2X | 24 |
| C tinctorius L. | 348915 | Canada | 2X | 24 |
| C tinctorius L. | 537111 | Mexico | 2X | 24 |
| C tinctorius L. | 560169 | USA | 2X | 24 |
| C tinctorius L. | 560172 | USA | 2X | 24 |
| C tinctorius L. | 560175 | USA | 2X | 24 |
| C tinctorius L. var.inermis Schweinf | CART 87 | Romania | 2X | 24 |
| Cross-species amplification | ||||
| C. oxyacantha | 426184 | Afghanistan | 2X | 24 |
| C. palaestinus | 235663 | Israel | 2X | 24 |
| C. boissieri Halacsy | Cart 85 | Greece | 2X | 20 |
| C. tenuis subsp. Foliosus | Cart 91 | Cyprus | 2X | 20 |
| C. glaucus subsp. anatolicus | Cart 43 | Israel | 2X | 20 |
| C. lanatus | 235666 | Portugal | 4X | 44 |
| C. lanatus subsp. Creticus | CART 10 | - | 4X | 44 |
| C. lanatus subsp. Lanatus | CART7 | - | 4X | 44 |
| C. lanatus subsp. turkestanicus | 426181 | Afghanistan | 4X | 44 |
# Genotypes with CART ID are obtained from IPK while the rest have been procured from USDA.
Leaf material was harvested from 10-week-old plants of each accession and total genomic DNA was isolated using CTAB method [39]. The qualitative and quantitative analysis of extracted DNA was done by electrophoresis on a 0.8% agarose gel and using a NanoDrop spectrophotometer (NanoDrop, Wilmington, DE).
Next generation sequencing using Illumina HiSeqTM 2000 and de novo assembly
A 100bp-paired end sequencing run was performed on HiSeq 2000 platform (Illumina, USA) by Macrogen Inc. (Korea). The FastQ files containing the raw data were submitted to the sequence read archive (SRA) at National Centre for Biotechnology Information (NCBI) under the accession number SRP050023. High quality reads were identified from genomic sequencing data using NGS QC Toolkit at default parameters [40]. De novo assembly was performed using SOAPdenovo version 2.04 (http://soap.genomics.org.cn//soapdenovo.html) [41] at various K-mer values (21, 27, 33, 39, 45, 51, 57 and 63). The assembled contigs from each run were pooled and clustered using CD-HIT (http://weizhong-lab.ucsd.edu/cd-hit/) [42]. Contigs with >90% sequence similarities were considered redundant and removed.
Identification of microsatellites, functional annotation and development of primer pairs
The assembled sequences were mined for perfect microsatellites using an in-house developed Perl script (S1 Script). The script contains separate modules for different SSR types (di- to hexa-nucleotides) and provides the details in terms of SSR type, repeat number, start and end position of repeat in the query sequence, total length of repeat and the complete sequence. Clustering was performed using CD-HIT on the identified sequences harboring microsatellites (clustering criteria; similarity > = 90% and 80% length coverage) to remove redundancy. Imperfect and compound SSR types were not included in the analysis. Functional annotation of the retrieved microsatellite sequences was performed using web-based automated annotation pipeline, FastAnnotator using default parameters (http://fastannotator.cgu.edu.tw/) [43].
Sequences containing microsatellites with repeat length of ≥ 20 bases (10 units for di-; 7 units for tri-, 5 units for tetra- nucleotides) and optimal flanking regions (≥ 30 bases on both flanks of microsatellite) were used to design primers. The web-based program, BatchPrimer3 version 1.0 (http://probes.pw.usda.gov/batchprimer3/) [44] was used for designing primer pairs with following parameters: primer length 18–28 bases; product size ranging from 100bp-500bp; optimum annealing temperature between 50°C to 65°C and GC content of 40% to 80% with an optimum value of 60%. Other parameters were used at default setting. Blast+ version 2.2.26 (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/2.2.26/) was used to query the previously reported SSR markers [24–26, 34] against the marker set for which primers were designed in the current study.BLASTN hits with an E value less than 1x10-13 were considered significant.
Validation of microsatellites
Primers were synthesized at Integrated DNA Technology, USA. Genomic DNA of two safflower accessions (PI: 560172 and 560175) was used as template for standardization of PCR conditions. The PCR was conducted in a total reaction volume of 15μl containing 50 ng of template DNA, 1X PCR buffer, 2mM of MgCl2, 0.2mM of each dNTP, 0.3mM each of forward and reverse primers and 1.25 U of Taq DNA polymerase (Biotools, Spain). Amplifications were performed in a Veriti thermal cycler (Applied Biosystems, USA) with the following cycling conditions: Initial denaturation at 96°C for 5 mins followed by 28–30 cycles of 96°C for 45s, primer annealing temperature (Tm; optimized for each primer pair; ranging between 55°C to 65°C) for 30s, DNA extension at 72°C for 1 min and a final extension at 72°C for 7 mins. The generated amplicons were analyzed on 2% agarose gel for product size and amplicon quality.
Genotyping and cross species transferability
Primer pairs producing a clear unambiguous band were used for genotyping a panel of 23 safflower accessions (Table 1). The polymorphic markers were further assessed for their cross species transferability in nine wild relatives of C. tinctorius L. (Table 1). For polymorphic markers, M13 tailing of the PCR product was adopted as described earlier [45, 46]. The labeled PCR products were analyzed on 6.5% PAGE using 4300 DNA analyzer system (LICOR, USA).
Marker diversity analysis
Statistical analyses of genetic data [Average number of alleles per locus (Na), gene diversity per locus (He), observed heterozygosity (Ho) and polymorphic information content (PIC)] of microsatellite markers were evaluated using POWERMARKER version 3.25 (http://www.powermarker.net) [47]. Cluster analysis for polymorphic microsatellite loci across the tested panel was performed using DARwin version 5.0.158 (http://darwin.cirad.fr/darwin) [48] based on simple matching coefficient.
Results and Discussion
Genome sequencing of C. tinctorius L.
Illumina paired-end technology was used for sequencing the safflower genome. We obtained 48,502,680 raw reads with an average read length of 101 bases which provided ~3.5X coverage of the genome. The average quality score (Q) of raw reads was >25 in all the sequencing cycles (Fig 1). The raw sequences were checked for sequence artifacts such as low quality reads and adaptor contamination using NGS QC Toolkit [40]. A total of 44,164,564 (91.06%) high quality filtered reads were obtained with 98.1% bases showing a Q value of >20.
Fig 1. Quality score during sequencing cycles of HiSeqTM 2000.

Assembly of the quality filtered and trimmed sequences were performed using SOAPdenovo version 2.04 [41]. Various k-mer values (21, 27, 33, 39, 45, 51, 57 and 63) were used for assembly and assembled sequences at each k-mer were pooled resulting in 4,078,739 contigs. Redundancy in contigs was removed using CD-HIT program [42], which resulted in 2,043,956 contigs with an average contig length of 264bp. Around 90% of the contigs were found in the size range of 100bp to 499bp. Length distribution of the obtained contigs is given in Table 2. Contigs <100 bases in length were excluded from further analyses. The generated safflower genome showed an average GC content of 38%, which is in consonance with several plant species such as Arabidopsis (36%), grape (34.6%), tomato (36.2%), potato (35.6%), rubber (36.2%) and mungbean (34.69%) [49–53].
Table 2. Length distribution of clustered sequences.
| Read length (base pair) | Number |
|---|---|
| 100–499 | 1843988 |
| 500–999 | 183848 |
| 1000–1999 | 15330 |
| 2000–2999 | 586 |
| 3000–3999 | 126 |
| 4000–4999 | 46 |
| 5000–5999 | 19 |
| 6000–6999 | 9 |
| 7000–7999 | 1 |
| 8000–8999 | 1 |
| 9000–9999 | 1 |
| 10000–10999 | 1 |
| Total | 2,043,956 |
| Average length (base pair) | 264 |
| Total nucleotides clustered | 540749137 |
Discovery of microsatellites
An in-house developed Perl script (S1 Script) was used for mining perfect microsatellites from the clustered genomic data. Perfect repeats were selected as these are known to have higher mutation rates than imperfect loci and are expected to therefore, yield more polymorphism [54]. Additionally, more allelic variation is observed with increasing number of repeats [55]. Thus, sequences with repeat length < 20 bases were not analysed as these may not be significantly polymorphic. Following these criteria, we identified 31,390 microsatellite sequences which were further filtered to remove redundancy using CD-HIT and a non-redundant set of 23,067 putative microsatellite loci was obtained.
Significant heterogeneity was observed in frequency, motif type and repeat length of SSRs in safflower. Di-nucleotides were the most frequent type of repeats representing 71% of total SSRs, followed by tri- (10%), tetra- (5.8%), penta- (5.13%) and hexa-nucleotide repeats (4.26%; Fig 2). Di-nucleotides are known to be the most represented SSRs in genomes of several plant species viz., pigeonpea, mungbean, sweet potato and sesame [30,54,56,57]. The safflower genome was highly enriched with AT/TA repeats accounting for 57.65% of all the dinucleotide motifs followed by AG/TC (27.5%) and AC/GT (14.8%) repeats. This is in consonance with the observations of Lee et al., [34] who isolated microsatellites based on pyro-sequencing of the safflower genome. In general, plant genomes have been reported to be rich in AT repeats [58–60]. We did not obtain any CG/GC repeat in the analyzed data (Fig 3 and Fig 4A). Among tri-nucleotide repeats, AAT was the most common motif (35.6%) followed by AAG (25%). However, Lee et al., [34] reported ACC to be the most frequent tri-nucleotide repeat (27%) in safflower. This variation could be due to differences in the quantum of data generated in the two studies. While Lee et al. [34] reported 1100 contigs with SSRs, we obtained a significantly higher number of SSR-containing contigs (23,067) which may represent a more accurate distribution of SSR frequencies. Another factor could be the inherent bias observed in individual sequencing runs [61,62] that could have led to differences between the two studies. The least frequent tri-nucleotide motif was CGC for which only one locus was detected and it represented the only GC-rich trimer repeat obtained in the current study (Figs 3 and 4B). Low frequency of GC-rich repeats was also reported in genomic sequences of other crops [52, 53]. Among tetra-nucleotide repeats, the ACAT motif was the most predominant (Fig 3).
Fig 2. Distribution analysis of major classes of microsatellites in safflower genome.

Fig 3. Frequency distribution of SSRs with most and least represented repeat motif in each class.

Fig 4. Characterization of di- and tri-nucleotide microsatellites discovered in safflower genome.

The microsatellite motifs were also assessed for their repetitive unit length. The reiteration number of a SSR motif ranged from 4 to 24 and di-nucleotides were found to have greater number of reiteration units, which gradually decreased in higher motif types (Fig 5).
Fig 5. Reiteration units observed for various classes of SSRs in safflower genome.

Functional annotation of microsatellite sequences
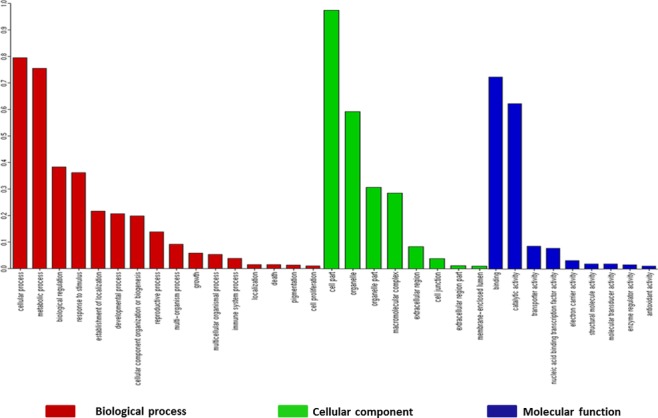
In order to study the potential functional significance of 23,067 microsatellite sequences, annotation was performed using FastAnnotator [43], which reported the average length and GC content of these sequences to be 300 bp and 30%, respectively. More than 50% of sequences (~ 13,000) were greater than 200 bp in length and the N50 of these sequences was 383 nucleotides. Out of the total set analyzed, 2,611 sequences were found to have similarity with sequences in the NCBI non-redundant protein database and 1,003 sequences (4.3%) were found to have at least one functional annotation. Around 738 sequences were assigned gene ontology (GO) while 99 sequences contained at least one domain. Ten sequences were found to be common among all annotation categories while 155 sequences were found to be common among GO and domain categories. Only one sequence was found to share the GO and enzyme annotation (Fig 6). S1 Table provides detailed information regarding annotated SSR sequences. Nine hundred and four contigs were mapped to gene ontology terms with 767, 695 and 757 assignments distributed under biological process, cellular component and molecular function ontology, respectively. Fig 7 shows GO classification (Level 2) of annotated microsatellite sequences. In the biological process ontology class, cellular and metabolic processes were predominant. Under molecular function class, binding and catalytic activity were the most abundant while cell part and organelle have the highest number of assignments under cellular component class. Similar results for distribution of GO terms were obtained in earlier studies on safflower floral transcriptome [63]. The present study thus reports a novel set of microsatellites, which might be correlated with the expressed components of safflower genome.
Fig 6. Functional annotation of microsatellite sequences.

Fig 7. Distribution of annotated genomic microsatellites of C. tinctorius L. among the Gene Ontology functional classes: Biological process, Cellular component and Molecular function (Level 2).
Validation of SSR markers
Primer pairs were designed for microsatellite loci using BatchPrimer3 version 1.0 [44] which allows detection of SSR motifs and designs primers from flanking regions. Out of 23,067 microsatellite loci identified above, 5,737 loci were recognized with sufficient flanking region and fulfilled the criteria for primer design (see Methods). Homology search of 5,737 microsatellite loci against the previously reported SSRs in safflower [24–26,34] was performed. The BLASTN results revealed that 14 and 7 sequences had significant similarity (E value < 1x10-13) with SSRs previously reported by Chapman et al. [24] and Lee et al. [34], respectively. These sequences were removed from the analysis leading to the identification of 5,716 novel microsatellites in safflower.
A subset of 325 microsatellite loci, designated as NGSaf_1 to NGSaf_325, was chosen for experimental validation and included di- (58), tri- (257) and tetra-nucleotide (10) repeats. These sequences have been submitted in the NCBI GenBank database under the accession numbers KM670560-KM670883 (S2 Table). High representation of trimeric repeats (79%) was selected to increase the probability of their presence in the coding regions [64]. It is believed that selective forces do not allow expansion of any repeat type other than trinucleotides in coding regions to avoid frame shift mutations that could alter protein functionality [60]. These repeats therefore, have a greater probability for stronger marker-gene/trait association and a high rate of transferability across species. Details of untested primer pairs are provided in S3 Table.
Out of 325 tested SSR primers, 294 (90.4%) generated high quality reproducible amplicons of expected size. Thirteen primer pairs failed to provide any PCR product and 18 primer pairs produced multiple amplicons, which were difficult to evaluate and were excluded from further analysis (S2 Table). The process of SSR development is subjected to attrition at each step. A mean 50% attrition between primer design and successful amplification of SSR loci has been reported in earlier studies [52, 65]. Lee et al. [34] tested 509 primer pairs in safflower, of which only 302 (59.3%) produced successful amplification. The higher rate of successful amplification in the current study (90.4%) could be due to improved generation and analysis of sequencing data.
Polymorphism analysis and cross species transferability of SSR markers
Based on their geographical origin, 23 safflower genotypes from 17 countries of the world (Table 1) were selected for testing the discriminatory potential of validated microsatellite markers. Out of the 294 microsatellite loci which produced robust amplification, 93 (31.6%) were polymorphic among the studied genotypes (Table 3). The average number of alleles (Na) per locus varied from 2 to 4 while the mean observed heterozygosity (Ho) and mean gene diversity (He) were 0.0494 and 0.3746, respectively. The low level of observed heterozygosity might be attributed to the highly self-pollinating nature of the crop. Sixty-eight polymorphic markers revealed 2 alleles, 23 markers yielded 3 alleles while 2 markers detected 4 alleles among the 23 accessions. In total, 213 alleles could be identified in the assessed genotypes using 93 polymorphic SSR loci. Polymorphism information content (PIC) was calculated for each marker and ranged from 0.0416 (NGSaf_43, 91, 115) to 0.5602 (NGSaf_69) with a mean PIC of 0.3075 (Table 3).
Table 3. Characteristics of 93 polymorphic SSR markers developed for Carthamus tinctorius L.
| Locus | Repeat motif | Forward primer sequence | Reverse primer sequence | Product size (bp) | Ta (°C) | Allele number (Na) | Gene diversity (He) | Heterozygosity (Ho) | PIC | GenBank accession number |
|---|---|---|---|---|---|---|---|---|---|---|
| NGSaf_2 | (TC)8 | ATCCTACGTGTCAACTTCCCAT | AATCAACAGCAACTAACGCTGA | 251 | 62 | 2 | 0.2580 | 0.0434 | 0.2247 | KM670561 |
| NGSaf_9 | (ACA)7 | AGTCCAACATAGCTCGGATTTC | GACCACTCTCTTCTCCGTCATC | 248 | 62 | 2 | 0.4848 | 0.0434 | 0.3673 | KM670568 |
| NGSaf_12 | (CGG)7 | TTCATCAACAAGTTACAAGGGC | ATTCCCTCTCCTACCCACCTAC | 288 | 62 | 2 | 0.3402 | 0 | 0.2823 | KM670571 |
| NGSaf_13 | (AGC)7 | GGCACAAAGGGATGTTTCTT | TTCTATGATGGGAGGAACTGGT | 108 | 58 | 2 | 0.0831 | 0 | 0.0797 | KM670572 |
| NGSaf_14 | (CAT)7 | ACCCATGTCAATGTACTCTTCGT | AATCTCCTCCATCTCCTCATCA | 196 | 58 | 2 | 0.1219 | 0.0434 | 0.1144 | KM670573 |
| NGSaf_15 | (TGT)7 | CACTCCCTCGTGCAGCTT | ATTCCCAATGCTCCAACAATC | 141 | 60 | 2 | 0.3147 | 0.0434 | 0.2652 | KM670574 |
| NGSaf_20 | (CATT)5 | AGCAAAGTGCTTGCTGACAA | AAGTTCAATTGAGCCCGATG | 243 | 60 | 2 | 0.4990 | 0 | 0.3745 | KM670579 |
| NGSaf_22 | (AG)9 | CCCTCGAGTAAAACTCAAGTCA | AAATGGATTGGTGGGTTTCA | 151 | 60 | 3 | 0.2344 | 0 | 0.2200 | KM670581 |
| NGSaf_23 | (AC)9 | ATATCCTCCTCCGCCGATAA | ATCAACAGGCGTCTCAACCT | 169 | 60 | 2 | 0.3856 | 0 | 0.3112 | KM670582 |
| NGSaf_28 | (ATC)7 | TATGATCTTGTCCGGCCTCT | ATGGCGGATGTTCATAAAGG | 177 | 62 | 2 | 0.0831 | 0 | 0.0797 | KM670587 |
| NGSaf_34 | (CAG)7 | CATCAAAATCATTGGTTGCTTG | TCTTTCACACACTTCTAAGGCAAA | 165 | 60 | 3 | 0.5950 | 0 | 0.5261 | KM670593 |
| NGSaf_39 | (GATG)5 | AATGCTCCAGCTTTCGACAT | TTCGGCCTTCGCTATGTAGT | 247 | 60 | 3 | 0.3553 | 0 | 0.3159 | KM670598 |
| NGSaf_43 | (CT)9 | AAAGCGGTGTTAGTGCTGTGTA | TTTATATGGAAGTGGAAAGGGG | 205 | 60 | 2 | 0.0425 | 0.0434 | 0.0416 | KM670602 |
| NGSaf_44 | (AGG)7 | CACTGTTGTGACCCTTGG | CACACACTTCTAAGGCAAACT | 110 | 62 | 2 | 0.4653 | 0 | 0.3570 | KM670603 |
| NGSaf_45 | (GCT)6 | ACGCCTCTTCTTCTTTCCTTCT | CATTCATGGGTTTAGGTGGC | 317 | 62 | 2 | 0.2268 | 0 | 0.2011 | KM670604 |
| NGSaf_48 | (CTT)6 | ACCCTGGATGACTGAAAACCTA | ATCAACACTCAGGCCCATATTC | 259 | 58 | 3 | 0.4990 | 0 | 0.4337 | KM670607 |
| NGSaf_49 | (CAT)6 | CTCCTTTTGCTTTCTTGATTGG | TCACTCTTGCTGATTTGCTTTG | 231 | 60 | 3 | 0.5368 | 0 | 0.4322 | KM670608 |
| NGSaf_56 | (GAT)6 | GCTAAAACAAGTGTGGCATCAA | AAGATGAAACCTCCCTCAAATG | 172 | 60 | 2 | 0.1587 | 0 | 0.1461 | KM670615 |
| NGSaf_63 | (GAA)6 | ATCCGTTCTTCTTCTAGCACCA | CAGGTGGCATGTATTTTGTTTG | 356 | 60 | 2 | 0.3147 | 0.0434 | 0.2652 | KM670622 |
| NGSaf_65 | (CTC)6 | GAGTGAAAAGAGGGAAGGGTTT | GGGGTTTGGAAAGGGTATTTA | 144 | 60 | 2 | 0.2580 | 0.0434 | 0.2247 | KM670624 |
| NGSaf_67 | (AGG)6 | CTGTTCCACAAGACAAAAGCAA | TCAAGTCCCAATCTCAACCTTC | 277 | 58 | 2 | 0.3147 | 0.0434 | 0.2652 | KM670626 |
| NGSaf_69 | (TAC)6 | TCTCATCAACGATAAAGCAGAATC | TCAACTTCATCTTTTCACGATTTC | 299 | 62 | 3 | 0.6353 | 0.0454 | 0.5602 | KM670628 |
| NGSaf_73 | (GTT)6 | CCAAGGTGACGTGTTGTTCTT | CATCCTTCGATCCACGATAACT | 225 | 60 | 2 | 0.1587 | 0.0869 | 0.1461 | KM670632 |
| NGSaf_83 | (AGA)6 | GCCAAACCCTAACACAGAATCA | CGGTTGTGCCCTAGCTTTTA | 400 | 62 | 2 | 0.4914 | 0 | 0.3707 | KM670642 |
| NGSaf_84 | (TCA)6 | CGCCATCTCTCTCCTCTTCTTA | GGTGTGGAATGGAATGATGATA | 133 | 60 | 2 | 0.3147 | 0.1304 | 0.2652 | KM670643 |
| NGSaf_89 | (TCT)6 | CTCGTAGCTGAGTTTATCGGTG | TGATTGCAGAGAGACTTGTTGA | 169 | 60 | 2 | 0.4536 | 0 | 0.3507 | KM670648 |
| NGSaf_91 | (TAT)6 | CCACTTCTAGTTCGGGTTTCTG | CTGCTGTCATTTCATAGGGTTG | 346 | 58 | 2 | 0.0425 | 0.0434 | 0.0416 | KM670650 |
| NGSaf_92 | (TAC)6 | AGAGGAGTCGATCTTGTGAAGG | GAGAGGTGATACGAGAAGCCAT | 107 | 58 | 2 | 0.2268 | 0 | 0.2011 | KM670651 |
| NGSaf_94 | (TCC)6 | CCATCGAAACTCTACAAAACCC | AGGACAAAAGAGGGAATGATGA | 400 | 60 | 2 | 0.4054 | 0.0434 | 0.3232 | KM670653 |
| NGSaf_98 | (TAC)6 | ATTCTCTGCATGTGGCTTTTCT | CTTGGTTACGGAGGAAGATTTG | 276 | 60 | 2 | 0.4834 | 0 | 0.3665 | KM670657 |
| NGSaf_101 | (TAT)6 | GATTCCGTGCATTCTACACAAA | GAGGAACGAACTAGGAAGGGTT | 141 | 62 | 3 | 0.4158 | 0 | 0.3741 | KM670660 |
| NGSaf_105 | (TGG)6 | CTGTAATTCTGCAACTGAGACCC | GAAGCCATTTTCCACGATTTC | 157 | 60 | 2 | 0.3856 | 0 | 0.3112 | KM670664 |
| NGSaf_111 | (CAA)6 | AATACCCCGCCATCATAGATTA | GAGCTATTCGACAACCAAATCC | 201 | 62 | 2 | 0.4914 | 0 | 0.3707 | KM670670 |
| NGSaf_114 | (AAT)6 | AGACAAATGCAACCACATTCAC | TGTTTATGATCCTTTCAGCCG | 377 | 60 | 2 | 0.1587 | 0 | 0.1461 | KM670673 |
| NGSaf_115 | (CTG)6 | CCATCCAATACTGCAAATCTCC | AGTACCAGCAAGCTCCACCTC | 240 | 62 | 2 | 0.0425 | 0.0434 | 0.0416 | KM670674 |
| NGSaf_117 | (GAA)6 | TTCCATTGAGTCCCATGAAGAT | TCTCTGTTCCACGTAGGGCT | 190 | 62 | 2 | 0.0831 | 0 | 0.0797 | KM670676 |
| NGSaf_130 | (TGG)6 | TGCGACTTGTGTTTCTTCTTCCC | AAAAGCCGTCCGGTGAAATTG | 371 | 62 | 2 | 0.4234 | 0 | 0.3337 | KM670689 |
| NGSaf_138 | (TGT)6 | AACCTGTGTACCATCTGCTAATTG | ATGAGATCCGAAGTCCATTGTT | 160 | 60 | 2 | 0.0831 | 0 | 0.0797 | KM670697 |
| NGSaf_142 | (TGT)6 | GATGTTAACCTGTGTACCATCTGC | CGTCTAATGAACACTCAATCCAAA | 442 | 60 | 3 | 0.4473 | 0.0454 | 0.3655 | KM670697 |
| NGSaf_145 | (GGA)6 | GAGCATGAAACGGAGAATTAGG | TCAACAGTAGCAGATCCTTCCA | 358 | 60 | 2 | 0.4990 | 0 | 0.3745 | KM670703 |
| NGSaf_148 | (CAT)6 | CCATGATTTTATGCTCATCGTG | AGCAATAGCAGGTGCAGTGATAG | 366 | 62 | 2 | 0.4536 | 0 | 0.3507 | KM670706 |
| NGSaf_151 | (GAT)6 | AGTCTTGGCCTGTAACCACTTATC | CATGAAGAGACGTTTGAAGCAG | 377 | 60 | 2 | 0.4914 | 0 | 0.3707 | KM670709 |
| NGSaf_152 | (CAA)6 | GTTGTTCGTTTGCCCCAATCTG | CAGAGCCACAAGATGCCGAATTA | 100 | 62 | 3 | 0.4682 | 0.0952 | 0.4149 | KM670710 |
| NGSaf_154 | (GAT)6 | TGATATCAATGGTATGATTTTCCTTT | GGATGGCGAACAAGATTACAA | 500 | 62 | 4 | 0.5826 | 0 | 0.4938 | KM670712 |
| NGSaf_155 | (AAG)6 | AAGTATTTGAACAAGTGTACCGGC | AGATGATGAAGGAAGGAGGTAATG | 335 | 58 | 2 | 0.3367 | 0 | 0.2800 | KM670713 |
| NGSaf_156 | (CAC)6 | AACTGCTTCTAGGGTTTTCCTCTT | CCCCAAATTACCAACTTCCATAC | 294 | 62 | 3 | 0.2637 | 0.0434 | 0.2385 | KM670714 |
| NGSaf_158 | (TAA)6 | ATGTTTGTCCCACTCGGTCTTC | TAAGTAGCTGAAGTGCAAGGTCGT | 274 | 60 | 2 | 0.1219 | 0.0434 | 0.1144 | KM670716 |
| NGSaf_164 | (TGT)6 | CATCTCAGCCTTTCTGTCTCCT | AAAATCGGTTGTTTGGTTGC | 313 | 60 | 2 | 0.2268 | 0 | 0.2011 | KM670722 |
| NGSaf_173 | (AGC)6 | GTGGTTCGAGTCTGTTTATTTCCA | CTGCACTTTTGAGTTGTGTGTGAT | 351 | 60 | 2 | 0.4536 | 0 | 0.3507 | KM670731 |
| NGSaf_178 | (ATC)6 | TAAGAAAGGGCACCATTGAAGTAG | GATATGAGCAGAGGAGTTTGTGC | 128 | 62 | 2 | 0.4763 | 0 | 0.3629 | KM670736 |
| NGSaf_181 | (GTA)6 | TATGGTGATCGAAGAAGAAGACAG | ACTGAGCAATGAAGAGTTCCAGA | 299 | 58 | 3 | 0.3279 | 0.0434 | 0.2954 | KM670739 |
| NGSaf_201 | (TAT)6 | GTTATTGTTGTCCGTGCAAGTAGT | GCTTGGTTCCTAGTCGTAGTTCAT | 309 | 58 | 2 | 0.4914 | 0.0869 | 0.3707 | KM670739 |
| NGSaf_204 | (AAC)6 | GCCATGCCCATATACAAACAGATA | AAGAAATGGTTCCACCGAGTCA | 350 | 60 | 2 | 0.3856 | 0 | 0.3112 | KM670762 |
| NGSaf_210 | (TCT)6 | TGATAGTAGCTTATTCCCTCAGCC | AACGGTGGTAGGATAGTTGACG | 199 | 60 | 2 | 0.4977 | 0 | 0.3738 | KM670768 |
| NGSaf_211 | (GAT)6 | GGTCTGCAAGAGTAAGTGGGAG | CCAAATCCCTGCTACAAAACAT | 132 | 60 | 2 | 0.4958 | 0.0909 | 0.3729 | KM670769 |
| NGSaf_236 | (ATC)6 | ACCTTGAGGGGTATTTGGGTAA | GTTGGTAGGGTGATCTTCGGT | 293 | 62 | 2 | 0.4921 | 0 | 0.3710 | KM670794 |
| NGSaf_237 | (ATT)6 | GACATTCCACTTATGCCGGTAG | TGGGACAATGACTCTGTTTGAG | 250 | 62 | 2 | 0.4962 | 0.9130 | 0.3731 | KM670795 |
| NGSaf_238 | (CAT)6 | AACAGTGGGCCTGATATGTTTT | CGGCTAATCCAAACCCTAGAAT | 343 | 62 | 2 | 0.4395 | 0.0434 | 0.3429 | KM670796 |
| NGSaf_239 | (ATT)6 | CAAAACTTCTCAACCGTGAAC | GTAACATACCGATTTTCTTGGC | 133 | 62 | 2 | 0.2777 | 0.2380 | 0.2391 | KM670797 |
| NGSaf_242 | (AAG)6 | GGAGGAGAGATTGAAGGTGAAG | CCAACCATCGTCTGAACTTTT | 129 | 62 | 2 | 0.1937 | 0.0434 | 0.1749 | KM670800 |
| NGSaf_245 | (ATG)6 | TGAACATGGTAAAAACCCATACA | TCTCAAAGGAATGGGAATGG | 109 | 62 | 2 | 0.4938 | 0 | 0.3718 | KM670803 |
| NGSaf_248 | (TCT)6 | CTGCATTTCTTCCCCATTATTATC | GTGTTTAAGGTTTCATTGCCTTCT | 173 | 62 | 2 | 0.0831 | 0 | 0.0797 | KM670806 |
| NGSaf_255 | (TCA)6 | AGGTGTTGGGAGGACACTAAAA | CCGCTCTTAGCTAAACTCTTGC | 224 | 62 | 2 | 0.4848 | 0.0434 | 0.3673 | KM670813 |
| NGSaf_257 | (ACA)6 | ACAGCACCTGCCATCTTCATTA | AGGCATGAGCTTCGATTTTAGC | 239 | 62 | 2 | 0.3856 | 0 | 0.3112 | KM670815 |
| NGSaf_259 | (ATT)6 | ATCGGGCATCCACTACTAGCTT | TGTGAAGTGAAAGATGAAATTCAAGA | 236 | 62 | 2 | 0.4763 | 0 | 0.3629 | KM670817 |
| NGSaf_261 | (TTC)6 | CGAATCTACCAAGATCTACCAAATCC | GATATGGCGGAGAAACCGTAAA | 102 | 60 | 2 | 0.4990 | 0.0869 | 0.3745 | KM670819 |
| NGSaf_262 | (TAT)6 | AAACCTGTACGGACACGTATCAA | CGAATTCCATCTCGATTTCTATATTTG | 132 | 60 | 3 | 0.5207 | 0.0434 | 0.4059 | KM670820 |
| NGSaf_264 | (TTA)6 | GATGATGAATTTTGGTCGAACG | CGTTTAATTGACGATAATGCATGTG | 152 | 62 | 3 | 0.5051 | 0.0454 | 0.4511 | KM670822 |
| NGSaf_265 | (AAT)6 | GGTAAAGACGAAGTTCACGATGG | TCTATCGCCGCCTTCTTCAC | 155 | 62 | 2 | 0.2508 | 0.0588 | 0.2193 | KM670823 |
| NGSaf_266 | (TGA)6 | TGGGAGTAAATTGGTGTTGAAGC | AAACGAAAACGGGAGAGATGAA | 211 | 62 | 3 | 0.1975 | 0.0434 | 0.1847 | KM670824 |
| NGSaf_273 | (GAA)6 | ATAGTAATTTGTAGCGAAAAGCGAAA | TGGATTCCAGGATTAACATATTCTT | 150 | 62 | 2 | 0.4914 | 0 | 0.3707 | KM670831 |
| NGSaf_276 | (AGA)6 | CCCATTTCAACTAAATTTCAAGC | TCTTCTTTCTGGGTGGCTTC | 110 | 62 | 2 | 0.0867 | 0 | 0.0830 | KM670834 |
| NGSaf_279 | (AAG)6 | AGCCTCGAGTTAAGCCATGA | TTCACGCGTTCTCTGTAGGA | 154 | 60 | 2 | 0.3254 | 0.0454 | 0.2724 | KM670837 |
| NGSaf_281 | (AG)22 | CAGGGACATCAGTTTGAGGAG | TGTTGGTGCTAACAAAGAACC | 108 | 62 | 3 | 0.6258 | 0 | 0.5509 | KM670839 |
| NGSaf_282 | (AG)18 | TACCCTCAATGGGAGTACCG | GGTCATGAGCTCGAATGAGG | 185 | 58 | 3 | 0.405 | 0 | 0.3679 | KM670840 |
| NGSaf_286 | (GA)16 | GCAAAATGGGAACACATTCA | TCTCCAGTGTAGCGTGTGAAA | 243 | 58 | 3 | 0.6124 | 0 | 0.5432 | KM670844 |
| NGSaf_289 | (AT)16 | TGCCATTTCATTAGTCACGA | ACTTGTTTTTACATCGAATCCTTT | 139 | 60 | 3 | 0.6200 | 0.9565 | 0.5435 | KM670847 |
| NGSaf_292 | (CT)16 | CGTAACCACATAATACTTGGAGAA | TCCATATCCTTGAAAACCCATT | 175 | 62 | 2 | 0.3856 | 0 | 0.3112 | KM670850 |
| NGSaf_294 | (CA)15 | AAGTAACAGATAAGATTTCAACAGCTC | CGATGGTATGAATAGTCGTCGT | 155 | 62 | 3 | 0.3494 | 0.0714 | 0.3084 | KM670852 |
| NGSaf_295 | (CT)15 | GCCTCTGCCTCTCTCTCTATTTT | CCTGTCGGTAGATCGAAGAAG | 131 | 62 | 2 | 0.4716 | 0 | 0.3604 | KM670853 |
| NGSaf_296 | (TC)15 | TCTCTGACGCTATGGGACAA | TGTGAGGATATGTAGAATCCCATT | 158 | 62 | 2 | 0.4536 | 0 | 0.3507 | KM670854 |
| NGSaf_300 | (CA)13 | TGCCTGATTCTTCACCTTCTC | TTAGCTGAAGCACTTTTGCTCTT | 100 | 62 | 3 | 0.5141 | 0 | 0.4608 | KM670858 |
| NGSaf_301 | (CA)12 | TCTTTTCAAGTGTTGCATGAGC | TGATGGTCTTCTCGCTACCA | 249 | 60 | 2 | 0.4567 | 0 | 0.3524 | KM670859 |
| NGSaf_306 | (CT)14 | GTCCAAGACCCGAGATGAAG | AGATTGGATTGCCGATAAATG | 152 | 60 | 2 | 0.1244 | 0 | 0.1167 | KM670864 |
| NGSaf_307 | (CT)14 | ACCCAAATCATGCTTCAACA | GGTGTGGATAATCAACCACTTTC | 151 | 58 | 2 | 0.3046 | 0 | 0.2582 | KM670865 |
| NGSaf_308 | (CT)13 | TAACCCGCATTCTTGGTTTT | CCCTATCTGTGTGAGCCTGA | 157 | 60 | 2 | 0.4990 | 0 | 0.3745 | KM670866 |
| NGSaf_309 | (GA)18 | TTGCAACCCTTCAACAAAAA | TCGAAGCCTTACCTTCCTCA | 172 | 62 | 3 | 0.4390 | 0.0454 | 0.3746 | KM670867 |
| NGSaf_310 | (GA)16 | GTCGATGGGAGTTTGAGAGG | CGATTGTGAGAGGGGTAGGA | 204 | 62 | 2 | 0.4958 | 0 | 0.3729 | KM670868 |
| NGSaf_313 | (GT)13 | GGGGTTGTTTTCCAAGAGTTT | CAGACAACTTTATCCAACATAGGAC | 155 | 60 | 3 | 0.4297 | 0 | 0.3854 | KM670871 |
| NGSaf_314 | (GT)12 | TTTTCCGACTAAAACCCCTTT | AGGGTGTTACACGCAAACTTC | 100 | 62 | 3 | 0.4917 | 0 | 0.4076 | KM670872 |
| NGSaf_322 | (TC)15 | AGCCGAAGAGCAAGCAAAT | GAGATGTTATGGCGGTGGTG | 105 | 62 | 2 | 0.5 | 0 | 0.375 | KM670880 |
| NGSaf_323 | (TC)13 | AAAGACTGTATTTGGGCTACGG | TTGAGATGCAACATGAATTGAA | 168 | 62 | 2 | 0.4501 | 0.6842 | 0.3488 | KM670881 |
| NGSaf_324 | (TG)15 | TGTTTTCCGGTTATTTGAGGTT | GAAGATGCTCTAACGGACCAA | 156 | 60 | 4 | 0.6228 | 0.0434 | 0.5457 | KM670882 |
| Mean | 2.29 | 0.3746 | 0.0494 | 0.3075 |
In our data, some repeat motifs were found to be more polymorphic among the studied accessions than other repeat types. The repeat motif ‘AG’ exhibited highest polymorphism (57.1%) among all analyzed repeats followed by ‘AAT’ (46.4%). Detailed information regarding motif type and percent polymorphism is given in Table 4. It has been reported that different taxa exhibit different preferences for SSR types [59]. This information would help in selection of motif types and increase the probability of finding polymorphic markers in safflower.
Table 4. Repeat motif type and their rate of polymorphism observed in the present study.
| Repeat Motif type | Total primer per motif | Polymorphic primers | Percentage polymorphism |
|---|---|---|---|
| Dinucleotides | |||
| AT | 9 | 1 | 11 |
| AG | 28 | 16 | 57 |
| AC | 21 | 7 | 33 |
| Trinucleotides | |||
| AAT | 26 | 11 | 42 |
| AAC | 32 | 10 | 31 |
| AAG | 62 | 12 | 19 |
| ACC | 16 | 3 | 19 |
| ACG | 10 | 2 | 20 |
| ACT | 39 | 10 | 25.6 |
| AGC | 15 | 3 | 20 |
| AGG | 21 | 5 | 24 |
| ATC | 26 | 8 | 30.7 |
| CCG | 8 | 1 | 12 |
The cross species transferability of polymorphic SSR loci was also assessed in nine wild species of safflower (Table 1). Each primer pair, except NGSaf_307, was found to be amplifiable in two or more of the tested wild relatives (S4 Table). Twenty five SSR markers showed 100% transferability to all the wild relatives. The highest rate of cross transferability of markers was observed in C. oxyacantha (97%) followed by C. palaestinus (87%) while markers were found to be least transferable in C. tenuis (42%) (Fig 8). Based on cytogenetic studies, C oxyacantha and C. palaestinus, had been proposed as the possible progenitors of cultivated safflower [66]. The high rate of cross species amplification of SSR markers obtained in the present study supports the earlier observations on homology between these species and their possible contribution to the safflower genome [67].
Fig 8. Cross-species transferability of 93 polymorphic safflower microsatellite markers in various species of genus Carthamus.

Cluster analysis for assessment of phylogenetic relationships in Carthamus sp.
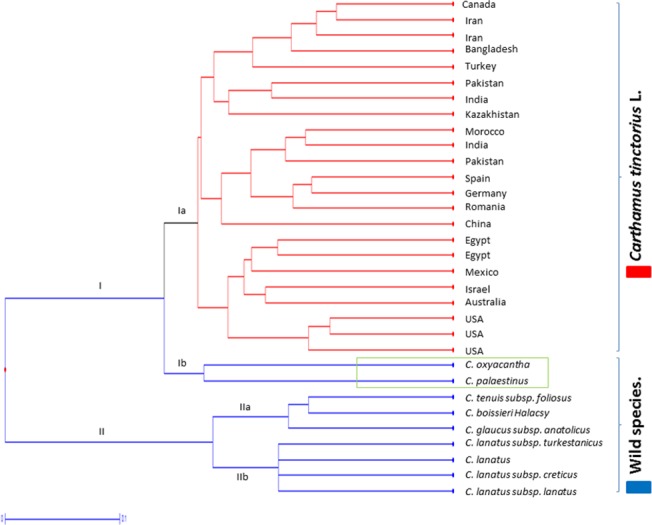
Cluster analysis based on simple matching coefficient was used to assess the genetic relationships between C. tinctorius (safflower) genotypes and related wild species (Fig 9). The analysis grouped the studied accessions into two major clusters (I and II). All safflower genotypes, irrespective of their geographical origin, clustered in a single group (subgroup Ia) although some indicative groupings were observed for genotypes from USA and the European gene pool. Inclusion of more accessions from these geographical zones might be useful in identifying regional gene pools in the crop. The two wild species, C. oxyacantha and C. palaestinus, grouped together in subgroup Ib of cluster I. The clustering of C. oxyacantha and C. palaestinus along with C. tinctorius genotypes in cluster I supports the hypothesis that these wild species are more closely related to cultivated safflower than the other wild relatives. All the other wild species grouped together in Cluster II. Distinct clustering of accessions with differences in chromosome number was also observed. All Carthamus accessions with chromosome number n = x = 12 grouped together in cluster I. Cluster II segregated into two subgroups. Cluster IIa contained the diploid wild species with basic chromosome number = 10 (C. glaucus subspecies anatolicus, C. boissieri Halacsy and C. tenuis) while the tetraploid relatives (C. lanatus subspecies creticus, C. lanatus, C. lanatus subspecies turkestanicus and C. lanatus subspecies lanatus; basic chromosome number = 11) grouped in Cluster IIb.
Fig 9. Phylogenetic dendrogram based on 93 polymorphic microsatellite markers, elucidating the genetic diversity and relationships among and between safflower accessions and its wild relatives.
Cross species amplification of microsatellite markers improves with decreasing phylogenetic distances [68]. The family Asteraceae is reported to have a low level of genetic conservation resulting in limited transferability of microsatellite markers across different genera [69]. Cross-genera transferability of microsatellite markers from sunflower has been shown to be inadequate and of limited use in safflower [14, 70]. Molecular markers developed in the current study demonstrated a high rate of interspecific amplification (ranging from 42% to 97%) within the genus Carthamus. We have also established the efficiency of these markers in elucidating the genetic relationships between members of the genus Carthamus. These markers could also be used for synteny studies between cultivated and wild species of safflower.
Conclusion
In conclusion, our study provided an insight into the microsatellite components of safflower genome. Using next generation sequencing data, a large set of 5,716 novel microsatellite primers were designed of which, 325 markers were experimentally validated. Ninety-three markers were found to be polymorphic among the studied accessions. These markers were successfully used for genetic analysis in C. tinctorius L. and also showed significant cross species transferability in related wild species. Our data supports C. oxyacantha and C. palaestinus as the possible progenitors of cultivated safflower. We were also able to distinguish between various wild species with differing basic chromosome numbers. Markers generated in this study will enhance the current repository for safflower and would be useful in crop improvement programs. The current study also supports the efficiency of next generation sequencing data in providing faster and reliable resources for marker development in non-model crops.
Supporting Information
(ZIP)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
This work was funded by the DST-PURSE grant of the Department of Science and Technology, Government of India to the University of Delhi. HA was supported by a research fellowship from University Grants Commission, India. We are thankful for the support of our technical and field staff in completion of the work.
Data Availability
All relavant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by Delhi University- Department of Science and Technology grant, Government of India to SG, AJ, AK and MA, and grant no. Dean(R)/2009/868. HA was supported by a research fellowship from University Grants Commission, India, grant no. Sch.No./JRF/AA/208/2010-11. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Garnatje T, Garcia S, Vilatersana R, VallÈS J. Genome size variation in the genus Carthamus (Asteraceae, Cardueae): systematic implications and additive changes during allopolyploidization. Ann Bot 2006, 97(3):461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weiss EA. Safflower Weiss, EA Oilseed crops. London, Longman; 1983, 216–281. [Google Scholar]
- 3.FAOSTAT 2013. Available: http://faostat.fao.org/site/567/default.aspx#ancor.
- 4. Dajue L, Mündel HH. Safflower Carthamus tinctorius L. Promoting the conservation and use of underutilized and neglected crops. 7 Institute of Plant Genetics and Crop Plant Research, Gatersleben: International Plant Genetic Resources Institute, Rome 1996. [Google Scholar]
- 5. Velasco L, Pérez‐Vich B, Fernández‐Martínez JM. Identification and genetic characterization of a safflower mutant with a modified tocopherol profile. Plant Breed 2005, 124(5):459–463. [Google Scholar]
- 6. Ilkılıç C, Aydın S, Behcet R, Aydin H. Biodiesel from safflower oil and its application in a diesel engine. Fuel Process Technol 2011, 92(3):356–362. [Google Scholar]
- 7. McPherson MA, Yang RC, Good AG, Nielson RL, Hall LM. Potential for seed-mediated gene flow in agroecosystems from transgenic safflower (Carthamus tinctorius L.) intended for plant molecular farming. Transgenic Res 2009, 18(2): 281–299. 10.1007/s11248-008-9217-0 [DOI] [PubMed] [Google Scholar]
- 8. Flider FJ. Development and commercialization of GLA safflower oil. Lipid Technol 2013, 25(10):27–229. [Google Scholar]
- 9. Carlsson AS, Zhu LH, Andersson M, Hofvander P. Platform crops amenable to genetic engineering–a requirement for successful production of bio-industrial oils through genetic engineering. Biocatal Agric Biotechnol 2014, 3(1):58–64. [Google Scholar]
- 10.Nimbkar N. Issues in safflower production in India. Safflower: Unexploited potential and world adaptability Proceedings of the Seventh International Safflower Conference, Wagga Wagga, New South Wales, Australia; 2008.
- 11. Collard BCY, Jahufer MZZ, Brouwer JB, Pang ECK. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 2005, 142(1–2):169–196. [Google Scholar]
- 12. Varshney RK, Mahendar T, Aggarwal RK, Börner A. Genic molecular markers in plants: development and applications In Genomics-assisted crop improvement (pp.13–29). Springer; Netherlands: 2007. [Google Scholar]
- 13. Golkar P. Breeding improvements in safflower (Carthamus tinctorius L.): a review. Australian Journal of Crop Science 2014, 8(7): 1079–1085 [Google Scholar]
- 14. García-Moreno María J, Velasco L, Begoña Pérez-Vich. Transferability of non-genic microsatellite and gene-based sunflower markers to safflower. Euphytica 2010, 175(2):145–150. [Google Scholar]
- 15. Hamdan YAS, García‐Moreno María J, Redondo‐Nevado J, Velasco L, Begoña Pérez‐Vich. Development and characterization of genomic microsatellite markers in safflower (Carthamus tinctorius L.). Plant Breed 2011, 130(2):237–241. [Google Scholar]
- 16. Sehgal D, Rajpal VR, Raina SN, Sasanuma T, Sasakuma T. Assaying polymorphism at DNA level for genetic diversity diagnostics of the safflower (Carthamus tinctorius L.) world germplasm resources. Genetica 2009,135(3):457–470. 10.1007/s10709-008-9292-4 [DOI] [PubMed] [Google Scholar]
- 17. Johnson RC, Kisha TJ, Evans MA. Characterizing safflower germplasm with AFLP molecular markers. Crop Sci 2007, 47(4):1728–1736. [Google Scholar]
- 18. Yang YX, Wu W, Zheng YL, Chen L, Liu RJ, Huang CY. Genetic diversity and relationships among safflower (Carthamus tinctorius L.) analyzed by inter-simple sequence repeats (ISSRs). Genet Resour Crop Evol 2007, 54(5):1043–1051. [Google Scholar]
- 19. Peng S, Feng N, Guo M, Chen Y, Guo Q. Genetic variation of Carthamus tinctorius L. and related species revealed by SRAP analysis. Biochem Systematics Ecol 2008, 36(7):531–538. [Google Scholar]
- 20. Kumar S, Ambreen H, Murali TV, Bali S, Agarwal M, Kumar A, et al. Assesment of genetic diversity and population structure in a global reference collection of 531 accessions of Carthamus tinctorius L. (Safflower) using AFLP markers. Plant Mol Biol Rep 2014, 10.1007/s11105-014-0828-8 [DOI] [Google Scholar]
- 21. Cardle L, Ramsay L, Milbourne D, Macaulay M, Marshall D, Waugh R. Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics 2000, 156(2):847–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Song QJ, Shi JR, Singh S, Fickus EW, Costa JM, Lewis J, et al. Development and mapping of microsatellite (SSR) markers in wheat. Theor Appl Genet 2005, 110(3):550–560. [DOI] [PubMed] [Google Scholar]
- 23. Kalia RK, Rai MK, Kalia S, Singh R, Dhawan AK. Microsatellites markers: an overview of the recent progress in plants. Euphytica 2011, 177:309–334. [Google Scholar]
- 24. Chapman MA, Hvala J, Strever J, Matvienko M, Kozik A, Michelmore RW, et al. Development, polymorphism, and cross-taxon utility of EST–SSR markers from safflower (Carthamus tinctorius L.). Theor Appl Genet 2009, 120(1):85–91. 10.1007/s00122-009-1161-8 [DOI] [PubMed] [Google Scholar]
- 25. Naresh V, Yamini KN, Rajendrakumar P, Kumar VD. EST-SSR marker-based assay for the genetic purity assessment of safflower hybrids. Euphytica 2009, 170(3):347–353. [Google Scholar]
- 26. Hamdan YAS, García‐Moreno M J, Redondo‐Nevado J, Velasco L, Pérez‐Vich B. Development and characterization of genomic microsatellite markers in safflower (Carthamus tinctorius L.). Plant Breed 2011, 130(2):237–241. [Google Scholar]
- 27. Zane L, Bargelloni L, Patarnello T. Strategies for microsatellite isolation: a review. Mol Ecol 2002, 11(1):1–16. [DOI] [PubMed] [Google Scholar]
- 28. Zalapa JE, Cuevas H, Zhu H, Steffan S, Senalik D, Zeldin E, et al. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am J Bot 2012, 99(2):193–208. 10.3732/ajb.1100394 [DOI] [PubMed] [Google Scholar]
- 29. Huang D, Zhang Y, Jin M, Li H, Song Z, Wang Y, et al. Characterization and high cross‐species transferability of microsatellite markers from the floral transcriptome of Aspidistra saxicola (Asparagaceae). Mol Ecol Resource 2014, 14(3):569–577. [DOI] [PubMed] [Google Scholar]
- 30. Dutta S, Kumawat G, Singh BP, Gupta DK, Singh S, Dogra V, et al. Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea [Cajanus cajan (L.) Millspaugh]. BMC Plant Biol 2011, 11(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang H, Jiang J, Chen S, Qi X, Peng H, Li P, et al. Next-generation sequencing of the Chrysanthemum nankingense (Asteraceae) transcriptome permits large-scale unigene assembly and SSR marker discovery. PloS one 2013, 8(4): e62293 10.1371/journal.pone.0062293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang H, Walla JA, Zhong S, Huang D, Dai W. Development and cross-species/genera transferability of microsatellite markers discovered using 454 genome sequencing in chokecherry (Prunus virginiana L.). Plant Cell Rep 2012, 31(11):2047–2055. 10.1007/s00299-012-1315-z [DOI] [PubMed] [Google Scholar]
- 33. Yang T, Jiang J, Burlyaeva M, Hu J, Coyne CJ, Kumar S, et al. Large-scale microsatellite development in grasspea (Lathyrus sativus L.), an orphan legume of the arid areas. BMC Plant Biol 2014, 14(1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee GA, Sung JS, Lee SY, Chung JW, Yi JY, Kim YG, et al. Genetic assessment of safflower (Carthamus tinctorius L.) collection with microsatellite markers acquired via pyrosequencing method. Mol Ecol Resource 2013, 14(1):69–78. [DOI] [PubMed] [Google Scholar]
- 35. Pearl SA, Bowers JE, Reyes-Chin-Wo S, Michelmore RW, Burke JM. Genetic analysis of safflower domestication. BMC Plant Biol 2014, 14(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ashri A. Evaluation of the world collection of safflower, Carthamus tinctorius L. II. Resistance to the safflower fly, Acanthophilus helianthi R. Euphytica 1971, 20(3):410–415. [Google Scholar]
- 37. Sabzalian MR, Saeidi G, Mirlohi A, Hatami B. Wild safflower species (Carthamus oxyacanthus): A possible source of resistance to the safflower fly (Acanthiophilus helianthi). Crop Prot 2010, 29(6): 550–555. [Google Scholar]
- 38. Majidi MM, Tavakoli V, Mirlohi A, Sabzalian MR. Wild safflower species (Carthamus oxyacanthus Bieb.): A possible source of drought tolerance for arid environments. Australian Journal of Crop Science 2011, 5(8):1055. [Google Scholar]
- 39. Doyle J. DNA protocols for plants—CTAB total DNA isolation In: Hewitt GM, Johnston A (eds) Molecular techniques in taxonomy. Springer, Berlin, 1991. pp 283–293. [Google Scholar]
- 40. Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PloS one 2012, 7(2):e30619 10.1371/journal.pone.0030619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1:18 10.1186/2047-217X-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li WZ, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22:1658–9. [DOI] [PubMed] [Google Scholar]
- 43. Chen TW, Gan RCR, Wu TH, Huang PJ, Lee CY, Chen YYM, et al. FastAnnotator-an efficient transcript annotation web tool. BMC Genomics 2012, 13(Suppl 7):S9 10.1186/1471-2164-13-S7-S9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. You FM, Huo N, Gu YQ, Luo MC, Ma Y, Hane D, et al. BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinformatics 2008, 9(1):253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Perry DJ. Identification of Canadian durum wheat varieties using a single PCR. Theor Appl Genet 2004, 109:55–61. [DOI] [PubMed] [Google Scholar]
- 46. Bali S, Raina SN, Bhat V, Aggarwal RK, Goel S. Development of a set of genomic microsatellite markers in tea (Camellia L.)(Camelliaceae). Mol Breeding 2013, 32(3):735–741. [Google Scholar]
- 47. Liu K, Muse SV. POWERMARKER: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21:2128–2129. [DOI] [PubMed] [Google Scholar]
- 48.Perrier X, Jacquemoud-Collet J (2006) DARwin software. Available: http://darwin.cirad.fr/darwin.
- 49. Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana . Nature 2000, 408(6814):796 [DOI] [PubMed] [Google Scholar]
- 50. Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449(7161): 463–467. [DOI] [PubMed] [Google Scholar]
- 51. Zhu W, Ouyang S, Lovene M, O'Brien K, Vuong H, Jiang J, et al. Analysis of 90 Mb of the potato genome reveals conservation of gene structures and order with tomato but divergence in repetitive sequence composition. BMC Genomics 2008, 9(1):286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pootakham W, Chanprasert J, Jomchai N, Sangsrakru D, Yoocha T, Tragoonrung S, et al. Development of genomic‐derived simple sequence repeat markers in Hevea brasiliensis from 454 genome shotgun sequences. Plant Breed 2012, 131(4):555–562. [Google Scholar]
- 53. Tangphatsornruang S, Somta P, Uthaipaisanwong P, Chanprasert J, Sangsrakru D, Seehalak W, et al. Characterization of microsatellites and gene contents from genome shotgun sequences of mungbean (Vigna radiata L. Wilczek). BMC Plant Biol 2009, 9(1):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jin L, Macaubas C, Hallmayer J, Kimura A, Mignot E. Mutation rate varies among alleles at a microsatellite locus: phylogenetic evidence. Proc Natl Acad Sci USA 1996, 93(26):15285–15288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Queller DC, Strassmann JE, Hughes CR. Microsatellites and kinship. Trends Ecol Evol 1993, 8(8):285–288. 10.1016/0169-5347(93)90256-O [DOI] [PubMed] [Google Scholar]
- 56. Wang Z, Fang B, Chen J, Zhang X, Luo Z, Huang L, et al. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genomics 2010, 11(1):726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wei W, Qi X, Wang L, Zhang Y, Hua W, Li D, et al. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics 2011, 12(1):451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Powell W, Machray GC, Provan J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci 1996, 1(7):215–222. [Google Scholar]
- 59. Tóth G, Gáspári Z, Jurka J. Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res 2000, 10(7):967–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ellegren H. Microsatellites: Simple sequences with complex evolution. Nat Rev Genet 2004, 5:435–445. [DOI] [PubMed] [Google Scholar]
- 61. Aird D, Ross MG, Chen WS, Danielsson M, Fennell T, Russ C, et al. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol 2011, 12(2): R18 10.1186/gb-2011-12-2-r18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, et al. Characterizing and measuring bias in sequence data. Genome Biol 2013, 14(5): R51 10.1186/gb-2013-14-5-r51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lulin H, Xiao Y, Pei S, Wen T, Shangqin H. The first Illumina-based de novo transcriptome sequencing and analysis of safflower flowers. PloS one 2012, 7(6):e38653 10.1371/journal.pone.0038653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Morgante M, Hanafey M, Powell W. Microsatellites are preferentially associated with non-repetitive DNA in plant genomes. Nat Genet 2002, 30(2):194–200. [DOI] [PubMed] [Google Scholar]
- 65. Squirrell J, Hollingsworth PM, Woodhead M, Russell J, Lowe AJ, Gibby M, et al. How much effort is required to isolate nuclear microsatellites from plants? Mol Ecol 2003, 12(6):1339–1348. [DOI] [PubMed] [Google Scholar]
- 66. Ashri A, Knowles PF. Cytogenetics of safflower (Carthamus tinctorius L.) species and their hybrids. Agron J 1960, 52:11–17. [Google Scholar]
- 67. Sehgal D, Rajpal VR, Raina SN. Chloroplast DNA diversity reveals the contribution of two wild species to the origin and evolution of diploid safflower (Carthamus tinctorius L.). Genome 2008, 51(8): 638–643. 10.1139/G08-049 [DOI] [PubMed] [Google Scholar]
- 68. Peakall R, Gilmore S, Keys W, Morgante M, Rafalski A. Cross-species amplification of soybean (Glycine max) simple sequence repeats (SSRs) within the genus and other legume genera: implications for the transferability of SSRs in plants. Mol Biol Evol 1998, 15(10):1275–1287. [DOI] [PubMed] [Google Scholar]
- 69. Whitton J, Rieseberg LH, Ungerer MC. Microsatellite loci are not conserved across the Asteraceae . Mol Biol Evol 1997, 14:204–209. [DOI] [PubMed] [Google Scholar]
- 70. Heesacker A, Kishore VK, Gao W, Tang S, Kolkman JM, Gingle A, et al. SSRs and INDELs mined from the sunflower EST database: abundance, polymorphisms, and cross-taxa utility. Theor Appl Genet 2008, 117(7):1021–1029. 10.1007/s00122-008-0841-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(ZIP)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All relavant data are within the paper and its Supporting Information files.