

Abstract
Introduction
Our goal was to determine whether symptom progression in primary lateral sclerosis (PLS) was consistent with disease spread through axonal pathways or contiguous cortical regions.
Methods
The date of symptom onset in each limb and cranial region was obtained from 45 PLS patient charts. Each appearance of symptoms in a new body region was classified as Axonal, Contiguous, Possibly Contiguous, or Unrelated according to whether the somatotopic representations were adjacent in the cortex.
Results
Of 152 spread events, the first spread event was equally divided between Axonal (22) and Contiguous (23), but the majority of subsequent spread events were classified as Contiguous.
Discussion
Symptom progression in PLS patients is consistent with disease spread along axonal tracts and by local cortical spread. Both were equally likely for the first spread event, but local cortical spread was predominant thereafter, suggesting that late degeneration does not advance through long axonal tracts.
Keywords: Primary lateral sclerosis, motor neuron disorder, disease progression, chart review, symptom spread
Introduction
In amyotrophic lateral sclerosis (ALS) symptoms typically begin in one body region and progress to other regions, affecting upper and lower motor neurons of the same body segment.1,2 Several hypotheses have been proposed to explain how symptoms spread between upper and lower motor neurons and from one body region to another. Two competing hypotheses are that degeneration occurs between axonally connected regions and the more recent hypothesis of a prion-like propagation of misfolded proteins to proximate cells.2–6 In ALS, the progression of lower motor neuron impairment can sometimes mask involvement of upper motor neurons. Primary lateral sclerosis (PLS), a form of motor neuron disorder that selectively affects upper motor neurons,7,8 may present a clearer case to allow spread patterns within the motor cortex to be examined. Local spreading of misfolded proteins should produce symptoms in contiguous somatotopic representations of the motor cortex, whereas spread through axonal connections would produce symptoms in non-contiguous representations. Our objective was to determine whether patterns of symptom spread in patients with PLS were consistent with either of these proposed mechanisms, using natural history data collected on PLS patients in our clinic.
Materials and Methods
We reviewed the charts of 45 patients (27 men, 18 women) who fulfilled the diagnosis of clinically pure PLS7,8 followed between 2000 and 2013. All patients gave written, informed consent for an Institutional Review Board approved natural history study (NCT00015444). The initial site of symptom onset and early progression was determined by patient report and outside medical records. The timing and location of extremity and cranial symptoms (dysarthria, dysphagia, brisk gag and jaw jerks) at each visit were documented. In the event that a patient did not confirm symptoms in a particular segment at the time of the visit, but upper motor neuron signs were present, the date of the examination was noted as the time of symptom onset. Each onset of symptoms in a new body segment was termed a “spread event”. Spread events were classified as Axonal, Contiguous, Possibly Contiguous, or Unrelated (Supplementary Table 1). Spread of symptoms to the contralateral homologous limb was classified as Axonal. Spread to somatotopically adjacent regions was classified as Contiguous. Because cranial exams were not lateralized, spread of symptoms between one arm and cranial region were classified as Possibly Contiguous, and Contiguous when both arms were involved. Symptoms beginning simultaneously in two regions, or separated by more than 10 years, or spreading between cranial regions and legs, skipping the arms were classified as Unrelated. From each involved region, subsequent spreads were classified as separate events. When a spread event occurred with more than one region previously affected, the classification was based on the most recent preceding symptom.
Results
The mean age at symptom onset was 48 years (range 29 – 63 years). Symptoms began in segments as follows: leg in 35 patients; arm in 2 patients; cranial in 6 patients; leg and arm in 1 patient; leg and cranial in 1 patient. The total number of spread events was 152. Of the 152 spread events 39 were classified as Axonal, 77 as Contiguous, 6 as Possibly Contiguous, and 30 as Unrelated. The first spread events were nearly equally divided between Axonal (23) and Contiguous (22), with one Possibly Contiguous, spread event (Figure 1). Twenty-two of the Axonal spread events were from one leg to another. Following the initial spread events, more subsequent events were classified as Contiguous than Axonal. Among second spread events (S2), there were significantly fewer Axonal than Contiguous spread events (10 vs. 38; p < 0.0001, binomial test), with 2 events classified as Possibly Contiguous and 7 Unrelated. Among third spreads, there were 6 Axonal, 14 Contiguous, and 3 Possibly Contiguous spreads. Among fourth spreads, there were 3 Contiguous and no Axonal spread events. Second or third Axonal spread events were more common among patients with first Contiguous first spread events than first Axonal spread events (11 vs. 5). Spread between legs and cranial regions accounted for 18 Unrelated spread events, with 11 occurring in the rostro-caudal direction6. There was no difference in the time interval for spread in Axonal and Contiguous events (2.2 vs. 2.4 years; ANOVA, p=0.81).
Figure 1.
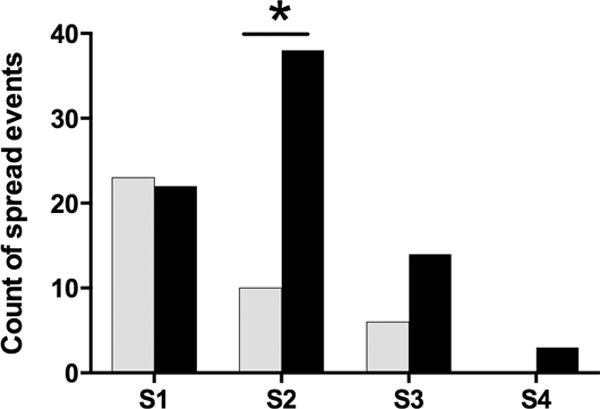
Frequency of axonal and contiguous spread events. The first spread events (S1) were classified as axonal (grey bars) and contiguous (black bars) in equal proportions. Significantly more second spread events (S2) were classified as contiguous (asterisks, binomial test, p <0.0001). A small number of third (S3) but no fourth (S4) events were classified as axonal.
Discussion
Progressive disability in PLS occurs by the spread of symptoms to new body segments and by increasing severity of affected regions.9 In this study, we found that the pattern of symptom spread to new body segments in PLS patients was consistent with more than one mechanism of spread between cortical regions, similar to findings in ALS.2,10 Although spread between somatotopically adjacent representations was the most common pattern overall, the initial spread of symptoms, from first to second affected body region, occurred with equal frequency to a non-contiguous cortical region. The majority of these first spread events, from one leg to another, were compatible with transcallosal spread. Spread along identifiable axonal pathways was much less common later in the disease, suggesting that later degeneration does not primarily advance through long axonal connections. These findings are consistent with single motor unit studies that found that cortical and spinal impairment were correlated at the earliest stages of the disease in ALS, but subsequently progressed independently in brain and spinal cord11. Reports from several large ALS clinics also found that an initial focal onset is most frequently followed by local spread, although distinct phenotypes with different spread patterns can be identified.2,10,12
There are several limitations of this study. First, the sequence or timing of symptom progression prior to the first clinic visit is subject to patient recall bias. Secondly, the patterns of spread depend on complex classification rules. The Axonal classification was essentially limited to the contralateral limbs, presumed to be connected through the corpus callosum. Patients with initial symptoms in the legs had fewer possibilities for later axonal spread. Additionally, we termed the non-contiguous spread between cranial and leg regions as unrelated, although it could represent spread through local axons. Similarly, contiguous spread may not require a prion-like direct cell-to-cell propagation, since the mechanism for local spread could be through short intracortical axonal connections. Nevertheless, our findings highlight that, within the cortex, degeneration appears to spread over time in PLS by both routes, as it is thought to do for ALS. Prospective observations will be useful to confirm the predominance of contiguous spread in later stages of disease progression in PLS.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, NIH (Z01 NS002976). The authors thank Dr. Tianxia Wu for statistical assistance.
Abbreviations
- ALS
Amyotrophic lateral sclerosis
- ANOVA
Analysis of variance
- PLS
Primary lateral sclerosis
References
- 1.Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet. 2011;377(9769):942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 2.Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology. 2009;73(10):805–811. doi: 10.1212/WNL.0b013e3181b6bbbd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swash M. How does ALS spread between neurones in the CNS? J Neurol Neurosurg Psychiatry. 2013;84(1):116–117. doi: 10.1136/jnnp-2012-303992. [DOI] [PubMed] [Google Scholar]
- 4.Verstraete E, van den Heuvel MP, Veldink JH, Blanken N, Mandl RC, Hulshoff Pol HE, van den Berg LH. Motor network degeneration in amyotrophic lateral sclerosis: a structural and functional connectivity study. PLoS One. 2010;5(10):e13664. doi: 10.1371/journal.pone.0013664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravits J, Appel S, Baloh RH, Barohn R, Rix Brooks B, Elman L, Floeter MK, Henderson C, Lomen-Hoerth C, Macklis JD, McCluskey L, Mitsumoto H, Przedborski S, Rothstein J, Trojanowski JQ, van den Berg LH, Ringel S. Deciphering amyotrophic lateral sclerosis: What phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(Suppl 1):5–18. doi: 10.3109/21678421.2013.778548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanouchi T, Ohkubo T, Yokota T. Can regional spreading of amyotrophic lateral sclerosis motor symptoms be explained by prion-like propagation? J Neurol Neurosurg Psychiatry. 2012;83(7):739–745. doi: 10.1136/jnnp-2011-301826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singer MA, Statland JM, Wolfe GI, Barohn RJ. Primary lateral sclerosis. Muscle Nerve. 2007;35(3):291–302. doi: 10.1002/mus.20728. [DOI] [PubMed] [Google Scholar]
- 8.Gordon PH, Cheng B, Katz IB, Pinto M, Hays AP, Mitsumoto H, Rowland LP. The natural history of primary lateral sclerosis. Neurology. 2006;66(5):647–653. doi: 10.1212/01.wnl.0000200962.94777.71. [DOI] [PubMed] [Google Scholar]
- 9.Floeter MK, Mills R. Progression in primary lateral sclerosis: a prospective analysis. Amyotroph Lateral Scler. 2009;10(5–6):339–346. doi: 10.3109/17482960903171136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korner S, Kollewe K, Fahlbusch M, Zapf A, Dengler R, Krampfl K, Petri S. Onset and spreading patterns of upper and lower motor neuron symptoms in amyotrophic lateral sclerosis. Muscle Nerve. 2011;43(5):636–642. doi: 10.1002/mus.21936. [DOI] [PubMed] [Google Scholar]
- 11.Attarian S, Vedel JP, Pouget J, Schmied A. Progression of cortical and spinal dysfunctions over time in amyotrophic lateral sclerosis. Muscle Nerve. 2008;37(3):364–375. doi: 10.1002/mus.20942. [DOI] [PubMed] [Google Scholar]
- 12.Gargiulo-Monachelli GM, Janota F, Bettini M, Shoesmith CL, Strong MJ, Sica RE. Regional spread pattern predicts survival in patients with sporadic amyotrophic lateral sclerosis. Eur J Neurol. 2012;19(6):834–841. doi: 10.1111/j.1468-1331.2011.03616.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.