

Summary
Matrix metalloproteinases (MMPs) play incompletely understood roles in health and disease. Knowing the MMP cleavage preferences is essential for a better understanding of the MMP functions and design of selective inhibitors. To elucidate the cleavage preferences of MMPs, we employed a high throughput multiplexed peptide-centric profiling technology involving the cleavage of 18,583 peptides by 18 proteinases from the main sub-groups of the MMP family. Our results enabled comparison of the MMP substrates on a global scale leading to the most efficient and selective substrates. The data validated the accuracy of our cleavage prediction software. This software allows us and others to locate, with a nearly 100% accuracy, the MMP cleavage sites in the peptide sequences In addition to increasing our understanding of both the selectivity and the redundancy of the MMP family, our study generated a roadmap for the subsequent MMP structural-functional studies and efficient substrate and inhibitor design.
Graphical Abstract
Introduction
Matrix metalloproteinases (MMPs) belong to a zinc endopeptidase, metzincin superfamily (Nagase and Fushimi, 2008; Nagase, et al., 2006). This superfamily is distinguished from other proteinases by the presence of a conserved HEXXHXXGXX(H/D) histidine sequence motif. This motif displays three histidine residues that chelate the active site zinc. The metzincin family is divided into four sub-families: seralysins, astacins, adamalysins [ADAMs (proteins with a disintegrin and a metalloproteinase domain) and ADAM-TS (ADAM with thrombospondin-like motif)] and MMPs (Gomis-Ruth, 2009; Gomis-Ruth, 2003).
There are 23 individual MMPs in humans from which seventeen proteinases are soluble and six are membrane-tethered [membrane type (MT)-MMPs] (Puente, et al., 2003). Normally, soluble MMPs are sub-divided into four major sub-families such as collagenases (MMP-1/interstitial collagenase, MMP-8/collagenase-2 and MMP-13/collagenase-3), stromelysins (MMP-3/stromelysin-1, MMP-10/stromelysin-2 and MMP-11/stromelysin-3), gelatinases (MMP-2/gelatinase A and MMP-9/gelatinase B) and matrilysins (MMP-7/matrilysin and MMP-26/matrilysin-2). In addition, the MMP family includes several MMPs that are not associated with the above four sub-families such as MMP-12/metalloelastase, MMP-19, MMP-20/enamelysin, MMP-21, MMP-23, MMP-27 and MMP-28/epilysin. Among the six MT-MMP sub-family members, four exhibit a transmembrane domain and a short cytoplasmic tail (MMP-14/MT1-MMP, MMP-15/MT2-MMP, MMP-16/MT3-MMP and MMP-24/MT5-MMP) and two attach to the cell membrane via a glycosylphosphatidyl inositol (GPI) anchor (MMP-17/MT4-MMP and MMP-25/MT6-MMP) (Egeblad and Werb, 2002; Nagase and Woessner, 1999). With the exception of MMP-7 and MMP-26 that consist of a catalytic domain alone, other MMPs have a C-terminal hemopexin-like domain linked to the catalytic domain by a flexible hinge region.
MMPs are synthesized as latent zymogens. To become active proteinases, the zymogens require proteolytic activation in which the N-terminal inhibitory prodomain is removed and the catalytic site of the emerging enzyme is exposed. Because of their high degrading activity and potentially disastrous effect on the microenvironment, cellular localization and activity of MMPs are tightly controlled, either positively or negatively, at both the transcriptional and post-transcriptional levels. In a feedback loop, some regulatory factors are either activated or inactivated by MMP proteolysis (Kajita, et al., 2001; McQuibban, et al., 2000; Mori, et al., 2002; Rodriguez, et al., 2010).
Evidence suggests that MMPs play an important role in the extracellular matrix proteolysis and tissue remodeling. In addition to the matrix, MMPs cleave growth factors and cytokines, and multiple adhesion and signaling cell receptors (Belkin, et al., 2001; Dean, et al., 2008; Deryugina, et al., 2002; Mori, et al., 2002). Enhanced expression of MMPs often directly correlates with malignant cell invasion and metastasis, and tumor neovascularization. Although our knowledge is expanding, we do not yet understand the precise functional role of the individual MMPs in normal versus pathological conditions. Knockouts of the individual MMP genes in mice, with the exception of MMP-14, do not elicit an easily recognized phenotype and are non-lethal, suggesting a functional redundancy among MMPs. MMP-14 knockout, in turn, has a profound effect: MMP-14 null mice develop dwarfism, extensive bone malformations and die before adulthood {Holmbeck, 2004 #738}. Mice lacking both MMP-2 and MMP-14 die immediately after birth (Oh, et al., 2004).
Despite multiple earlier studies, our abilities to quantitatively estimate functional redundancy among MMPs and to rank protein substrates according to their sensitivity to MMPs were limited. These deficiencies become especially important as several MMPs, including MMP-8 and, potentially, MMP-19 and MMP-25, demonstrate anti-tumor properties and thus should not be pharmacologically targeted in certain malignancies while the function of multiple additional MMPs remains unknown in disease context (Chernov, et al., 2010; Chernov and Strongin, 2011; Coussens, et al., 2002; Gutierrez-Fernandez, et al., 2008; Overall and Kleifeld, 2006; Shay, et al., 2015). Precise knowledge of the MMP cleavage preferences is needed to rationally relate MMPs to the cleaved substrates. Conversely, identification of the multiple cleavage sequences is required to recognize the MMPs’ cleavage signature (Jabaiah and Daugherty, 2011). This cleavage signature, if known, may directly relate the individual MMPs to their respective protein substrates (auf dem Keller, et al., 2010; Butler, et al., 2010; Butler, et al., 2009; Butler, et al., 2008; Butler and Overall, 2007; Dean and Overall, 2007; Doucet, et al., 2008; Doucet, et al., 2011; Dufour and Overall, 2013; Overall and Kleifeld, 2006; Ratnikov, et al., 2014; Schlage and Auf dem Keller, 2015).
To determine cleavage preferences of MMPs, we employed a high throughput multiplexed peptide-centric profiling technology that we developed (Kozlov, et al., 2012; Shiryaev, et al., 2014; Shiryaev, et al., 2013; Shiryaev, et al., 2013). In this study, we treated a pool containing 18,583 peptide-cDNA fusions with 18 MMPs (one proteinase per reaction). As a result, we generated a volume of the peptide cleavage data. These data enabled us to accomplish an in-depth comparative analysis of substrate recognition by MMPs. Therefore we were able, for the first time, to deliver comprehensive information on both substrate cleavage redundancy and distinction in the MMP family.
Results
Cleavage profiling technology
To characterize the cleavage preferences of the individual representative proteinases of the human MMP family, we used a high-throughput peptide-centric cleavage profiling technology. This technology includes cleavage of a large number of peptide-cDNA fusions designed in silico and then synthesized in vitro. The technology has been tested and validated by determining cleavage preferences of several viral, bacterial and human proteinases (Kozlov, et al., 2012; Shiryaev, et al., 2014; Shiryaev, et al., 2013; Shiryaev, et al., 2013; Shiryaev, et al., 2012).
Here, we used the high throughput multiplexed peptide-centric profiling of the 18 individual MMPs. These MMPs represented the main sub-groups in the MMP family (collagenases, gelatinases, stromelysins and MT-MMPs). Each MMP was allowed to cleave a library of 18,583 peptide-cDNA fusions. The rules for designing 15,000 10-mer peptide sequences were derived from our bioinformatics analysis of 1,369 peptide sequences that were identified as the most efficient substrates of MMPs in substrate phage display (Ratnikov, et al., 2014). This 15,000 peptide sub-set was generated by random number generator resulting in a random combination of the accepted residues at each of the individual P5–P5′ positions, followed by filtering out redundant sequences. Our 18,583 peptide set also included 598 sequences of the potential MMP cleavage sites derived from the crystal structures of human secretory proteins and, in addition, 2,985 control peptides, which included the substrate peptides of non-MMP enzymes and random sequences that were not cleaved by MMPs in our previous experiments.
In our tests, aliquots of the peptide library were subjected to cleavage by MMPs. The cDNA portions, which have been released by MMP proteolysis of the peptide-cDNA fusions, were collected. The nucleotide sequence of the released cDNA templates was then determined using a high-throughput DNA sequencer, thus providing the sequence of the encoded peptides. The raw cleavage data of 18,583 individual peptides we synthesized, cleaved by the individual MMPs and then analyzed the cleavage efficiency are presented in Supplemental Table S1.
Cleavage specificity of the MMP catalytic domain versus the full-length MMP enzyme
Because of its limited length, the 10-residue peptides can occupy the MMP catalytic cleft but they are not expected to protrude outside the catalytic domain margins. To exclude any significant, albeit unexpected, interactions of the peptide-DNA fusions with the structural domains, which are distinct from and additional to the MMP catalytic domain, the peptide-cDNA fusion library was co-incubated with the individual catalytic domains of MMP-2 and MMP-9, and with the full-length MMP-2 and MMP-9 enzymes. Following MMP proteolysis of the peptides and DNA sequencing of the samples, the individual peptide-cDNA fusions with at least 10 raw sequencing counts (1,902 and 2,167 substrates for MMP-2 and MMP-9, respectively) were analyzed further.
An average correlation coefficient among triplicate samples was 0.92 suggesting a reliable execution of our cleavage assays. An average correlation coefficient between the full-length MMP-2 and MMP-9 enzymes and the MMP-2 and MMP-9 catalytic domains Z-scores representing strength of the cleavage was 0.72 and 0.77, respectively (Fig. 1). These data suggested that there was a high level correlation between the cleavage preferences of the catalytic domains and the full-length enzymes of MMPs in our peptide cleavage assays. We concluded that the use of the MMP catalytic domains provides reliable information about the peptide cleavage preferences of MMPs and that the effect of the MMP auxiliary domains is insignificant in our peptide-centric assays.
Fig. 1. Cleavage parameters of the catalytic domains versus the full-length enzymes of MMP-2 and MMP-9.
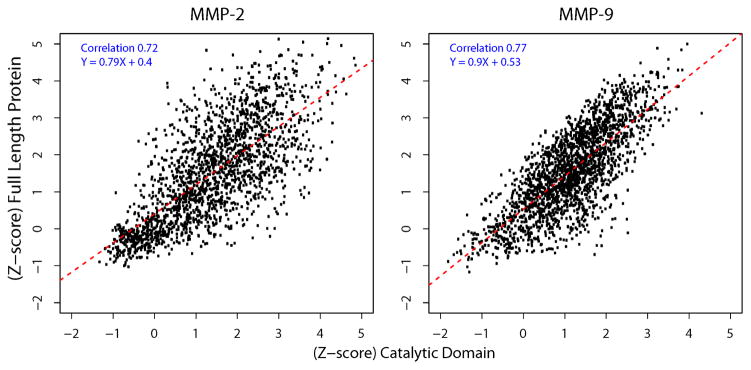
The X-axis and Y-axis denote the Z-score of substrate cleavage by the catalytic domain and by the full-length proteinase, respectively. Black dots represent selected substrates (1,902 for MMP-2 and 2,167 for MMP-9) with raw sequencing counts >10. Red dotted line represents a linear regression fitted line. Pearson’s correlation coefficient and regression equation are shown within the panels. Assays were performed in triplicate. Average correlation among the replicates was 0.92 (not shown). Average correlation between the full-length enzyme and the catalytic domain was 0.72 (MMP-2) and 0.77 (MMP-9).
MMP profiling
Our cleavage analysis of the peptide-cDNA fusion library is presented in Fig. 2. The scatter density plots show the relations of the log transformed raw sequencing counts versus the Z-scores of the individual peptide-cDNA fusions for the individual MMPs. For each MMP, except for MMP-7 and MMP-19, we recorded >500 substrates with Z-score >2.5, an indicative of the reliable cleavage.
Fig. 2. Scatter density plot for the 18 MMPs.

Density plots of Z-score versus raw sequencing counts of 18,583 peptide substrates. X-axis and Y-axis denote the Z-score of substrate cleavage and log2 raw DNA counts in an MMP digest sample, respectively. Darker blue represents a higher density of substrates, with outlying substrates shown as dots (the Z-score >2.5; p-value <0.0062 by one tail test). All of the MMPs, except MMP-7 and MMP-19, exhibited >500 substrates with the Z-score >2.5. Because of a limited number of the efficiently cleaved peptide substrates, MMP-7 and MMP-19 were not analyzed further.
We then directly compared our kinetics data (in which the cleavage read-out was expressed as kcat/KM) (Ratnikov, et al., 2014) with our current data in a Z-score format. This comparison allowed us to conclude that there was a direct correlation between both data sets. Thus, the kcat/KM value of 9,270 peptides from the substrate phage display assays corresponded to the Z-score equal or above of 2.35 for the same peptides in our current study. In general, these data indicated that from the kinetics perspectives the selected range of the Z-score values corresponded to the efficient cleavage peptides. However, for MMP-7 and MMP-19, in our peptide-centric assays we did not observe significant substrates passing the Z-score >2.5 threshold. Hence, MMP-7 and MMP-19 were not analyzed further.
The Pearson correlation coefficient matrix of the Z-scores (over 0.7) of 18,583 peptide substrates showed high correlation among the members of the individual MMP subfamilies (Fig. 3). Thus, MMP-14’s specificity highly correlated (an average correlation coefficient over 0.8) with that of MMP-15, MMP-16 and MMP-24, whereas no correlation was observed with those of all of soluble MMPs, and MMP-17 and MMP-25. Because the latter two are linked to the cell surface via a GPI-linker and, consequently, are in a distinct microenvironment, it is not surprising that MMP-17 and MMP-25 exhibit distinct cleavage preferences relative to the four other MT-MMPs.
Fig. 3. Pearson’s correlation coefficient matrix of the Z-scores of 18,583 peptide substrates cleaved by MMPs.

The bottom triangular matrix shows raw correlation coefficient. The top triangular matrix represents a degree of correlation in a form of an ellipse (a low minor axis denotes a high correlation). The color of the coefficient and ellipse also indicates the correlation strength to aid the eye. Cyan indicates low correlation (0 – 0.3), blue denotes medium correlation (0.3 – 0.8), while red denotes high correlation (0.8 – 1) among the MMP cleavage preferences.
Similarly, MMP-1’s cleavage preferences correlated with a correlation coefficient of 0.79 and 0.74 with those of MMP-8 and MMP-13, all three from the collagenase family, respectively. Conversely, the peptide cleavage pattern for the collagenase sub-family members did not well correlate with other MMPs, except MMP-11. According to our data, there was a significant, 0.86, correlation between the cleavage preferences of MMP-11 and the collagenase sub-family members such as MMP-1 and MMP-8. These correlations indicate that MMP-11 (stromelysin-3) shares substrate specificity with both the collagenase (MMP-1 and MMP-8) and the stromelysin (MMP-3 and MMP-10) sub-family members.
On the same note, from the cleavage perspectives MMP-12 resembled the members of the stromelysin sub-family, including MMP-3 and MMP-10. There was also a level of correlation between MMP-12 and MMP-20, both of which correlated with MMP-11, but not with other MMPs. In turn, MMP-12 shared sequence preferences with none the collagenases sub-family members.
As expected, gelatinases, MMP-2 and MMP-9, exhibited a significant similarity in their cleavage preferences (Bauvois, 2012). In addition, MMP-2’s cleavage preferences were similar with those of MMP-1, MMP-8 and MMP-13. This is not entirely surprising because gelatinases efficiently degrade gelatin (denatured collagen) and collagen is the main substrate of the collagenase sub-family members.
In general, there was roughly a 50% overlap among the cleavage preferences of all MMPs suggesting that approximately 50% of the peptide sequences that are cleaved by an individual MMP would be also cleaved by all other MMPs. This high level of cleavage preference redundancy explains the functional redundancy observed among the MMP family members. In addition, this cleavage pattern similarity among MMPs provides a fundamental rationale by which MMPs efficiently substitute for one another in cells/tissues, as a result, masking the knockout phenotype in the MMP knockout mice.
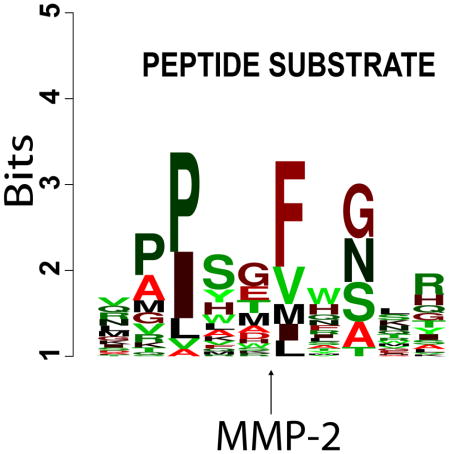
Based on the Z-score parameter, we next selected the top 100 substrates for each MMP. These substrates were considered to be the most efficiently cleaved substrate sequences. We used this peptide subset to identify the preferred cleavage sequences for the individual MMPs. A direct relationship between the peptide sequences and their susceptibility to MMP proteolysis was visualized in a form of sequence logos. Since the design of the peptide library was biased towards sequences containing the motif PXX-L, the logos were normalized for the bias. For each MMP, the most frequent 5-residue long P3-P2′ sequence motifs were selected (Fig. 4). Clearly, there is an overlap in the cleavage preferences of MMPs. For example, a cleavage preference overlap was evident for related MMP-1 and MMP-8 (collagenases-1 and -2, respectively). As a whole, all of MMPs were active against the peptides that exhibited the PXX-L cleavage motif at the P3-P1′ positions.
Fig. 4. Frequency plot of the efficient cleavage sequences of MMPs in an IceLogo format.

The height of a character is proportional to the frequency of the amino acid residue at the individual position of the cleaved peptide. 18,583 peptide substrates were cleaved by the individual MMPs. The scissile bond is between the P1 and P1′ residues. The Z-scores for the substrates were calculated and the substrates were ranked according to their Z-scores. The most efficient 100 substrates (Z-score >2; p-value <0.02) were selected for each MMP. Because the design of the peptide substrates was biased to the PXXL-containing sequences, the resulting position-specific matrix of the top 100 substrates was normalized at each position by using the amino acid residue frequency at this particular position in the entire 18,583 peptide library. The five most frequently occurring sequence motifs for each MMP are also shown.
There was also a level of distinction even between the most closely related MMPs. To highlight this level of distinction and to reveal the most selective, albeit not necessarily cleavage-efficient, peptide sequences, each substrate for each MMP was ranked based on the difference between the Z-score for the MMP of interest and the average Z-score for all other MMPs for that particular substrate. An additional constraint was applied to the Z-score of the selected peptides for the MMP of interest that it should exceed 1.65 (p-value <0.05). As a result, we selected the top 50 peptide substrates which were preferentially specific and significantly proteolyzed by each MMP. For each MMP, the most frequent 5-residue long P3-P2′ sequence motifs were also selected (Fig. 5).
Fig. 5. Frequency plot of the selective cleavage sequences of MMPs in an IceLogo format.

The height of a character is proportional to the frequency of the amino acid residue at the individual position of the cleaved peptide. Substrates were ranked according to the difference between their Z-score for the particular MMP and Z-score for all of other MMPs. Top 50 peptide substrates were selected for each MMP (Z-score >1.65; p-value <0.05). Because the design of the peptide substrates was biased to the PXXL-containing sequences, the resulting position-specific matrix of the top 50 substrates was normalized at each position by using the amino acid residue frequency at this particular position in the entire 18,583 peptide library. The three most frequent sequence motifs for each MMP are also shown.
To visualize both the similarity and difference in the cleavage preferences among MMPs, we further used the top 10 most specific peptides (160 substrates for 16 MMPs) for the hierarchical clustering analysis based on Euclidian distance and average linkage algorithm and presented as heat map (Fig 6; red, black and blue designate the high, medium and low Z-scores, respectively). The distinct diagonal pattern of the efficiently cleaved peptides (red) highlights these substrates that were most specifically cleaved by a particular MMP but that were inefficiently cleaved by other MMPs.
Fig. 6. Supervised clustering of the ten most selective peptide substrates for each of MMPs in a heatmap format.

Z-scores indicating cleavage activity of all 18,583 peptide substrates were calculated for each MMP, all substrates were ranked according to their Z-score difference relative to all other MMPs. Top 10 most selective substrates were chosen for each MMP (Z-score >1.65). X-axis shows MMPs. Y-axis shows 160 peptides (top 10 specific peptides for each MMP). Red, black and blue designate high, medium and low Z-scores, respectively.
Correlation with the PWM predictions
Recently, based on the substrate phage display data (Ratnikov, et al., 2014), we developed the PWM prediction methodology (http://cleavpredict.burnham.org). PWM was designed to predict the MMP cleavage sites in the peptide/protein sequences (Kumar, et al., 2015). To test the accuracy of our PWM software relative the current cleavage data, we analyzed all substrates with the Z-score >2.5 for eleven MMPs (MMP-2, MMP-3, MMP-8, MMP-9, MMP-10, MMP-14, MMP-15, MMP-16, MMP-17, MMP-24 and MMP-25). The level of correlation between the PWM predictions and the experimental Z-scores was generally very high. Nearly all of the substrates that were efficiently cleaved were predicted to be efficiently cleaved by PWM (92%–99.7%) (Table 1; Supplemental Fig. S1). A significant level of correlation of the experimental results published by others (Turk, et al., 2001) with the predicted PMW scores serves as additional validation of our CleavPredict software (Supplemental Table S2). Furthermore, multiple protein targets of MMP-2 and MMP-9 identified by others (Prudova, et al., 2010; Schilling and Overall, 2008; Schilling and Overall, 2007) were readily identified by CleavPredict, both with high accuracy and precision [discussed in (Kumar, et al., 2015)].
Table 1. Peptide substrates with the Z-score >2.5 for each MMP.
Substrates with the Z-score >2.5 that are not predicted by the PWM software are shown as red dots in Supplemental Fig. S1.
| MMP | Number of substrates with Z-score >2.5 | Number (%) of substrates predicted by PWM | Number (%) of substrates not predicted by PWM |
|---|---|---|---|
| MMP-2 | 499 | 457 (92%) | 42 (8%) |
| MMP-3 | 198 | 184 (93%) | 14 (7%) |
| MMP-8 | 418 | 415 (99.3%) | 3 (0.7%) |
| MMP-9 | 178 | 174 (97.8%) | 4 (2.2%) |
| MMP-10 | 334 | 310 (93%) | 24 (7%) |
| MMP-14 | 568 | 566 (99.7%) | 2 (0.3%) |
| MMP-15 | 164 | 163 (99.4%) | 1 (0.6%) |
| MMP-16 | 311 | 310 (99.7%) | 1 (0.3%) |
| MMP-17 | 177 | 175 (98.9%) | 2 (1.1%) |
| MMP-24 | 367 | 354 (96.5%) | 13 (3.5%) |
| MMP-25 | 402 | 400 (99.5%) | 2 (0.5%) |
Discussion
The capability of proteinases to catalytically cleave substrate proteins post-synthesis is essential for sustaining life in all of its forms, from viruses to humans. It is widely accepted that limited, site-specific proteolysis is one of the most important post-translational modifications, especially because this post-translational modification, in contrast to others, is irreversible under physiological conditions.
Understanding of the physiological role of a protease requires identification of both its cleavage substrates and its cleavage efficacy against the substrate(s) relative to others. Knowing these cleavage specificity and efficiency parameters can greatly improve our ability to rationally use proteinases in health research, medicine and biotechnology. In addition, this knowledge, once acquired experimentally and then transformed into user-friendly software, would be highly valuable for laboratory researchers.
As a step toward these goals, we used an unbiased approach to identify the relative primary cleavage specificity and efficiency parameters for an important group of human proteinases, the MMP family. MMPs play multiple diverse roles in both normal development and pathologies, especially in malignancy and metastasis. MMPs exhibit similar catalytic domain fold and highly homologous active site structures. These parameters contribute to the functional redundancy in the MMP family and cause difficulty in designing selective MMP inhibitors (Maskos, 2005; Zucker, et al., 2000). Comprehensive assessment of these cleavage parameters requires the availability of volumes of the cleavage data but so far these data were quite limited (Kazanov, et al., 2011; Ratnikov, et al., 2014).
Here, we employed the high throughput multiplexed peptide-centric profiling for multiple MMPs. The MMP species we analyzed included the members of the representative MMP sub-families (collagenases, gelatinases, stromelysins and MT-MMPs). As a result, we can now confidently conclude that that there is a roughly 50% overlap in the cleavage preferences on MMPs. This means that if a peptide substrate is cleaved by a single MMP there is a 50% probability that this particular peptide sequence will be cleaved by many others, if not all, MMPs.
In addition, our data highlighted the subtle differences in the cleavage preferences among MMPs and allowed us to determine the most efficient substrates and, conversely, the most selective peptide cleavage sequences for the individual, even closely related, MMPs. Thus, for example, the presence of Asp at the P1′ position would allow the discrimination of MMP-8 (collagenase-2) not only from the closely related MMP-1 (interstitial collagenase) but also from all other MMPs. Furthermore, in addition to the well-established structural homology parameters, we now can exploit the cleavage preferences perspective to additionally define the relations existing among the MMP sub-families and the individual proteinases. Thus, as judged from their sequence preferences, MMP-11 (stromelysin-3) is likely closely related to both the collagenase and stromelysin sub-families. In agreement, evolutionary pathways suggest that MMP-11 co-evolved closely to the MT-MMP subfamily and that the other two stromelysins (MMP-3 and MMP-10) are strongly diverged from MMP-11. In turn, as based on both the MMP cleavage preferences we recorded and the position of MMP-12 in the evolutionary tree (Fu, et al., 2009; Massova, et al., 1998), MMP-12 also appears to be close to the stromelysins. Our data summarized in Fig. 6 allow us and others to select the peptide sequences that are most biased to the individual MMPs and, at the same time, to forecast the interference of other individual MMP type in the cleavage of this individual peptide. Because of our unbiased approach, our results will facilitate exploring of many novel biological roles for multiple individual MMPs, a significant challenge for current modalities.
Furthermore, our cleavage data could be further exploited by bioinformatics researchers in an attempt to identify those variations in the MMP sequence and structure, which result in the distinct cleavage preferences among the MMP family members. Consequently, knowing these specificity-determining residues may ultimately lead to the design of mutant MMPs biased to the efficient proteolysis of the particular, clinically- or biotechnologically-relevant, protein targets.
Volumes of the cleavage data we generated also allowed us to critically assess the cleavage prediction PMW algorithm we designed based on the less voluminous data (Kumar, et al., 2015; Ratnikov, et al., 2014). According to our analysis, at the exhaustive proteolysis conditions, PMW correctly predicts, on average, over 93% of the cleavage sites in peptides, reaching a nearly 100% accuracy level for certain MMPs (Table 1). This high level of correlation implies that the open access PWM software (http://cleavpredict.burnham.org) exhibited a significant predictive power and that this software can be recommended for the use by others. We, however, admit that descriptors deduced directly from the amino acid sequence exhibit incomplete predictive capabilities. Because of the effect of the MMP auxiliary domains, structural constraints and special-temporal differences between the substrate and the protease, peptide cleavage in in vitro assays may not always predict that the same sequence is targeted in a natural protein. These limitations are discussed in a detail in our additional manuscripts (Belushkin, et al., 2014; Kazanov, et al., 2011; Kumar, et al., 2015).
Overall, in addition to increasing our fundamental understanding of MMP proteolysis, our results provide a roadmap for the design of both the most efficient and, alternatively, most selective cleavage substrates for the individual MMPs. These substrates can be used in various applications including design of quenched peptides, biosensors, drug delivery reagents and many other molecular tools in which MMP proteolysis is required for the emerging biological effect (Lu, et al., 2013; Ouyang, et al., 2010) We believe that our results will stimulate and focus functional studies by others in both biology and medicine. Thus, evidence suggests that 1) enhanced activities of MMPs are important to multiple pathologies, 2) measuring just one individual protease is unlikely to be representative of the proteolytic environment, 3) no diagnostic/theranostic tests are currently available to detect elevation in MMP activity levels, and 4) the development of diagnostic tests based on the MMP activities could dramatically change the provision of care, especially in outpatient settings. Ultimately, our work will bring us a step closer to the proteolysis-targeting personalized medicine.
Significance
Proteolysis is of paramount importance to biological regulation and life in all of its forms, in both normal development and disease. The significance of proteolysis is magnified because, in contrast to other post-translational modifications, proteolysis is irreversible under physiological conditions. Our study is a step forward towards determining the cleavage preferences of proteinases and then computationally connecting the proteinases with their previously unknown substrates in the proteome. Here, our focus is on the MMP family. Thus, we used an unbiased approach to identify the relative cleavage specificity and efficiency parameters for the members of the representative MMP sub-families (collagenases, gelatinases, stromelysins and MT-MMPs). For these purposes, we employed a high throughput multiplexed peptide-centric profiling technology that we have recently developed and employed successfully to determine the cleavage preferences of several bacterial, viral and human proteinases. In our current study, we treated a pool containing 18,583 peptide-cDNA fusions with 18 MMPs. As a result, we generated a volume of the peptide cleavage data. These data enabled us to accomplish a comparative analysis of substrate recognition by MMPs and to deliver comprehensive information on both substrate cleavage redundancy and distinction for the individual members of the MMP family. Based on our data, the more selective and efficient substrates and inhibitors of MMP can be designed. Volumes of the cleavage data we generated also allowed us to critically assess the cleavage prediction PMW algorithm we developed. Following our comprehensive tests, we are now confident that the open access PWM software (http://cleavpredict.burnham.org) exhibits a significant predictive power and that this software can be recommended for the use by others who are trying to connect the MMP family members with their novel respective substrates.
Experimental procedures
General reagents
The reagents were purchased from Sigma-Aldrich (St. Louis, USA) unless indicated otherwise. The broad-spectrum Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2 substrate was acquired from R&D Systems (Minneapolis, USA). The catalytic domains of MMP-1, -2, -3, -7, -9, -10, -11, -12, -13, -19 and -20 were from Enzo Life Sciences (Farmingdale, NY). The catalytic domains of MMP-8, -14, -15, -16, -24 and -25 were isolated in our laboratories (Shiryaev, et al., 2009; Shiryaev, et al., 2009). The full-length MMP-2 and MMP-9 were isolated and then activated by p-aminophenylmercuric acetate (Chen, et al., 2002).
MMP activity assay with the quenched peptide substrate
To standardize the activity for each MMP, cleavage activity assay was performed in triplicate in wells of a 96 well plate (0.2 ml reaction volume) in 50 mM HEPES buffer, pH 6.8, containing 10 mM CaCl2 and 50 μM ZnCl2. The Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2 substrate and enzyme concentrations were 10 μM and 10 nM, respectively. Initial reaction velocities were monitored continuously at λex=320 nm and λem=400 nm on a fluorescence spectrophotometer. The determined activity and amounts of each MMP was then normalized relative to the activity and amounts of the standard full-length MMP-2 enzyme solution of a known concentration. As a result of both these activity measurements and our preliminary tests and assays, a 2 nM equivalent of MMP-2 was used for each individual MMP in the multiplex peptide cleavage assays described below.
The substrate library
To elucidate the MMP cleavage preferences, we prepared a set of 18,583 peptide-cDNA fusions. Each fusion construct contained a 10-residue peptide substrate flanked by N- and C-terminal common sequences. Statements below regarding peptides refer only to the variable 10-mer portions of the peptides and disregard the flanking constant portions. The 18,583 set included several sub-sets. The main sub-set we designed in silico included approximately 15,000 peptide sequences that we expected to be cleaved efficiently by MMPs. The design rules were derived from our bioinformatics analysis of 1,369 peptide sequences that were identified as the most efficient substrates of MMPs using phage display methods (Ratnikov, et al., 2009; Ratnikov, et al., 2014). We used these validated substrate sequences to identify amino acid residue types that are accepted by the following MMPs: MMP-2, MMP-9, MMP-14, MMP-15, MMP-16, MMP-24 and MMP-25 at each of the individual P5–P5′ substrate positions. We then used a random number generator to create multiple peptide sequences. For this purpose, we used a random combination of those residues, which have accepted by MMPs and which have been identified in our earlier studies, at each of the individual P5–P5′ positions (Ratnikov, et al., 2014). Duplicate sequences, if they were present, were then filtered out. We also excluded peptides that were predicted to be inefficient MMP substrates based on the statistical substrate specificity profiling software predictions (CleavPredict.SanfordBurnham.org) (Kumar, et al., 2015).
Based on the specificity profiling software predictions and the crystal structures of secretory proteins available from the Protein Data Bank (PDB) database, we included 598 additional peptide sequences that were likely MMP cleavage substrates. The 18,583 peptide set also included 1,643 control peptides containing the furin cleavage motif. Additional positive and negative controls included known peptide substrates of thrombin, enterokinase, caspases, and West Nile and Dengue virus proteinases, and randomly generated sequences.
Multiplex peptide cleavage assay and data analysis
Biotinylated peptide-cDNA pool was prepared as reported earlier (Kozlov, et al., 2012). The template DNAs encoded 18,583 10-mer peptide substrates flanked by the N-terminal (Biotin-linker-Gly-Ala) and the C-terminal (Gly-Asn-Ala-Ser-Ala-Ser-Ala-Ala-Gly-Ala-linker-DNA) common sequences. Six biotinylated oligonucleotides of a known concentration were added to the peptide-cDNA pool as internal standards for normalization. Streptavidin-coated magnetic beads with the immobilized peptide-cDNA pool (1 pmol total, ~0.05 fmol/peptide) were co-incubated with the individual MMPs in triplicates (a 2 nM MMP-2 equivalent each) at 37°C for 30 min (3 μl reaction volume) in 50 mM HEPES, pH 6.8, containing 10 mM CaCl2 and 50 μM ZnCl2. Reactions without proteinases were used as controls. Following cleavage of the peptide-cDNA fusions by MMPs, the cleaved peptide-cDNA fragments were released from the beads and solubilized in the reaction solution. The reaction solutions were separated from the beads and collected. To identify cleaved peptides, DNA adapters required for sequencing were installed in the released cDNA by PCR. The obtained DNA constructs were sequenced using a MiSeq sequencing instrument (Illumina, San Diego, USA).
Total sequencing counts for each sample were normalized using the added internal standards. Abundance of each cleaved, solubilized peptide was quantified by counting the number of DNA reads corresponding to the peptide sequence resulting in the discrete count data of 18,583 peptide substrates for each of the 18 MMPs studied. The cleavage level (Z-score) for each substrate in a sample was then calculated as a function of the average counts of the three replicates in the proteinase-treated samples and the average counts in the control samples (no MMP, buffer only) as described below.
The cleavage level was estimated by comparing the log-transformed counts in the MMP-treated versus the control samples. To adjust for sequence-specific variance in the abundance levels of each peptide present in the pool, we used a locally weighted scatter plot smoothing fit as implemented in the lowess (locally weighted scatterplot smoothing) function from the statistical analysis package R. Our past data indicated that variance in our data was not constant but that it was rather a function of abundance; this is the primary reason for choosing lowess function rather than traditional linear regression. The residual of the lowess fit for each peptide, that is, the deviation from the lowess line, was obtained with positive residuals (peptide abundances, which are above the line) indicating a higher relative abundance in protease compared to buffer and negative residuals indicating a lower relative abundance. Each residual was then standardized and transformed into a Z-score by subtracting the mean of all residues and dividing by the standard deviation of all residuals {Z-score = [residuals(i) – mean(all residuals)]/st.dev(all residuals)}. After this transformation, Z-scores of the intact peptides were shown to have a standard normal distribution (mean = 0 and standard deviation = 1). Statistical significance was inferred by converting Z-scores into p-values by 1 – pnorm (Z-value) in R. A higher Z-score indicates a higher cleavage activity and a lower p-value of a peptide substrate. We chose a significance cut-off for each peptide of p-value <0.05 after adjusting for multiple testing corrections according to the method of Benjamini and Hochberg (Benjamini and Hochberg, 1995). Substrates with cleavage efficiency below the threshold (Z-score <1.65, which corresponded to p-value of 0.05) were considered resistant to MMP proteolysis.
The sequence logos for the selected MMP substrate peptides were created using the web-based IceLogo program (Colaert, et al., 2009). The listing of the most sensitive 100 peptides was compiled by taking 100 peptides for each MMP with the highest Z-scores (all values passed the significance cut-off of p-value <0.02 and Z-score >2). The listing of the most selective 50 substrates for each MMP was compiled by ranking substrates whose Z-score was higher than the average of all other MMPs for this particular substrate peptide (these top 50 peptides had Z-score >1.65 and p-value <0.05) indicating that this peptide was preferred by a particular MMP and was not preferred by any other MMP.
PWM matrix and substrate cleavage predictions for individual MMPs
We used the positional weight matrices (PWM) approach for calculating the cleavage score of the peptides (Kumar, et al., 2015). The previously cleavage data from the substrate phage display (Ratnikov, et al., 2014) were used to design peptide substrates for the eleven individual MMPs (MMP-2, MMP-3, MMP-8, MMP-9, MMP-10, MMP-14, MMP-15, MMP-16, MMP-24 and MMP-25). The PMW values were calculated for each MMP by a) aligning the peptide sequence with that of the potential cleavage site, b) calculating the frequencies (P(iAA,j)) of occurrence of each amino acid type (iAA) in each of the j-th position, ranging from P3 to P2′, and c) normalizing each amino acid residue position relative to the distribution of amino acid residue types in the set of the background sequences. The background sequences were obtained from 766 peptides for which cleavage frequency was below 10% in our substrate phage display studies (Ratnikov, et al., 2014). Thus, the final PWM values for each amino acid iAA at the j-th position were calculated as:
| (1) |
We used the log2 values of the appropriate PWM elements as the primary scoring function for substrate prediction. The actual cleavage recognition score was defined in eqs. 2 and 3 as a sum of log2 of PWM matrix elements for iAA amino acid type at the j-th position. Summation runs over the P3-P2′ positions in the substrate:
| (2) |
where
| (3) |
If any element of the PWM was equal to zero, then the offset value was used instead. This was done in order to avoid calculation of log2(0) and yet to add the sufficient penalty to the scoring function if an amino acid type iAA at j-th position was not observed in the training set. If the PWM score was above the cut-off value, the peptide bond was considered as a potential cleavage site. Both offset and cut-off values were specific for each MMP and optimized by using a two-dimensional 10-fold cross-validation in which the F1-score was maximized. This primary scoring function was used to screen every peptide bond in the peptide sequences and to predict whether the peptide may be cleaved by the individual MMP.
Supplementary Material
Highlights.
Knowing the cleavage preferences is essential for understanding the functions of MMPs
Profiling of >18,500 peptide sequences determined the cleavage preferences of 18 MMPs
Our results enabled comparison of the cleavage preferences of MMPs on a global scale
This study generated a roadmap for the structural-functional studies of MMPs
Acknowledgments
Our work was supported by R01CA83017, R01CA157328 and R01DE022757 (AYS), R44GM085884 (IAK), and R01GM098835 (PC) grants from the NIH. We would like to thank employees of Prognosys Biosciences for their help, especially, Petr Capek for preparation of oligonucleotides and Wayne Delport for sequencing data analysis.
Footnotes
Author Contribution. MK, performed the bioinformatics analyses of the cleavage data, corrected the manuscript, and discussed the data; SAS, purified and analyzed the individual proteinases, corrected the manuscript, and discussed the data; PC and SK, prepared the in silico peptide sequence sets, corrected the manuscript, and discussed the data; NM, performed the cleavage assays; DAR, corrected the manuscript and discussed the data; AVC and AGR, maintained the expression cell lines, and discussed the data; JWS, drafted and corrected the manuscript, and discussed the data; IAK, synthesized the peptide constructs, corrected the manuscript, and discussed the data; AYS, set up and coordinated the project, drafted the manuscript, and discussed the data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- auf dem Keller U, Prudova A, Gioia M, Butler GS, Overall CM. A statistics-based platform for quantitative N-terminome analysis and identification of protease cleavage products. Mol Cell Proteomics. 2010;9:912–927. doi: 10.1074/mcp.M000032-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauvois B. New facets of matrix metalloproteinases MMP-2 and MMP-9 as cell surface transducers: outside-in signaling and relationship to tumor progression. Biochim Biophys Acta. 2012;1825:29–36. doi: 10.1016/j.bbcan.2011.10.001. [DOI] [PubMed] [Google Scholar]
- Belkin AM, Akimov SS, Zaritskaya LS, Ratnikov BI, Deryugina EI, Strongin AY. Matrix-dependent proteolysis of surface transglutaminase by membrane- type metalloproteinase regulates cancer cell adhesion and locomotion. J Biol Chem. 2001;276:18415–18422. doi: 10.1074/jbc.M010135200. [DOI] [PubMed] [Google Scholar]
- Belushkin AA, Vinogradov DV, Gelfand MS, Osterman AL, Cieplak P, Kazanov MD. Sequence-derived structural features driving proteolytic processing. Proteomics. 2014;14:42–50. doi: 10.1002/pmic.201300416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- Butler GS, Dean RA, Morrison CJ, Overall CM. Identification of cellular MMP substrates using quantitative proteomics: isotope-coded affinity tags (ICAT) and isobaric tags for relative and absolute quantification (iTRAQ) Methods Mol Biol. 2010;622:451–470. doi: 10.1007/978-1-60327-299-5_26. [DOI] [PubMed] [Google Scholar]
- Butler GS, Dean RA, Smith D, Overall CM. Membrane protease degradomics: proteomic identification and quantification of cell surface protease substrates. Methods Mol Biol. 2009;528:159–176. doi: 10.1007/978-1-60327-310-7_12. [DOI] [PubMed] [Google Scholar]
- Butler GS, Dean RA, Tam EM, Overall CM. Pharmacoproteomics of a metalloproteinase hydroxamate inhibitor in breast cancer cells: dynamics of membrane type 1 matrix metalloproteinase-mediated membrane protein shedding. Mol Cell Biol. 2008;28:4896–4914. doi: 10.1128/MCB.01775-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler GS, Overall CM. Proteomic validation of protease drug targets: pharmacoproteomics of matrix metalloproteinase inhibitor drugs using isotope-coded affinity tag labelling and tandem mass spectrometry. Curr Pharm Des. 2007;13:263–270. doi: 10.2174/138161207779313524. [DOI] [PubMed] [Google Scholar]
- Chen EI, Kridel SJ, Howard EW, Li W, Godzik A, Smith JW. A unique substrate recognition profile for matrix metalloproteinase-2. J Biol Chem. 2002;277:4485–4491. doi: 10.1074/jbc.M109469200. [DOI] [PubMed] [Google Scholar]
- Chernov AV, Baranovskaya S, Golubkov VS, Wakeman DR, Snyder EY, Williams R, Strongin AY. Microarray-based transcriptional and epigenetic profiling of matrix metalloproteinases, collagens, and related genes in cancer. J Biol Chem. 2010;285:19647–19659. doi: 10.1074/jbc.M109.088153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernov AV, Strongin AY. Epigenetic regulation of matrix metalloproteinases and their collagen substrates in cancer. Biomolecular concepts. 2011;2:135–147. doi: 10.1515/BMC.2011.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaert N, Helsens K, Martens L, Vandekerckhove J, Gevaert K. Improved visualization of protein consensus sequences by iceLogo. Nat Methods. 2009;6:786–787. doi: 10.1038/nmeth1109-786. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- Dean RA, Cox JH, Bellac CL, Doucet A, Starr AE, Overall CM. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood. 2008;112:3455–3464. doi: 10.1182/blood-2007-12-129080. [DOI] [PubMed] [Google Scholar]
- Dean RA, Overall CM. Proteomics discovery of metalloproteinase substrates in the cellular context by iTRAQ labeling reveals a diverse MMP-2 substrate degradome. Mol Cell Proteomics. 2007;6:611–623. doi: 10.1074/mcp.M600341-MCP200. [DOI] [PubMed] [Google Scholar]
- Deryugina EI, Ratnikov BI, Postnova TI, Rozanov DV, Strongin AY. Processing of integrin alpha(v) subunit by membrane type 1 matrix metalloproteinase stimulates migration of breast carcinoma cells on vitronectin and enhances tyrosine phosphorylation of focal adhesion kinase. J Biol Chem. 2002;277:9749–9756. doi: 10.1074/jbc.M110269200. [DOI] [PubMed] [Google Scholar]
- Doucet A, Butler GS, Rodriguez D, Prudova A, Overall CM. Metadegradomics: toward in vivo quantitative degradomics of proteolytic post-translational modifications of the cancer proteome. Mol Cell Proteomics. 2008;7:1925–1951. doi: 10.1074/mcp.R800012-MCP200. [DOI] [PubMed] [Google Scholar]
- Doucet A, Kleifeld O, Kizhakkedathu JN, Overall CM. Identification of proteolytic products and natural protein N-termini by Terminal Amine Isotopic Labeling of Substrates (TAILS) Methods Mol Biol. 2011;753:273–287. doi: 10.1007/978-1-61779-148-2_18. [DOI] [PubMed] [Google Scholar]
- Dufour A, Overall CM. Missing the target: matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol Sci. 2013;34:233–242. doi: 10.1016/j.tips.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Fu L, Das B, Mathew S, Shi YB. Genome-wide identification of Xenopus matrix metalloproteinases: conservation and unique duplications in amphibians. BMC Genomics. 2009;10:81. doi: 10.1186/1471-2164-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis-Ruth FX. Catalytic domain architecture of metzincin metalloproteases. J Biol Chem. 2009;284:15353–15357. doi: 10.1074/jbc.R800069200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis-Ruth FX. Structural aspects of the metzincin clan of metalloendopeptidases. Mol Biotechnol. 2003;24:157–202. doi: 10.1385/MB:24:2:157. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Fernandez A, Fueyo A, Folgueras AR, Garabaya C, Pennington CJ, Pilgrim S, Edwards DR, Holliday DL, Jones JL, Span PN, et al. Matrix metalloproteinase-8 functions as a metastasis suppressor through modulation of tumor cell adhesion and invasion. Cancer Res. 2008;68:2755–2763. doi: 10.1158/0008-5472.CAN-07-5154. [DOI] [PubMed] [Google Scholar]
- Jabaiah A, Daugherty PS. Directed evolution of protease beacons that enable sensitive detection of endogenous MT1-MMP activity in tumor cell lines. Chem Biol. 2011;18:392–401. doi: 10.1016/j.chembiol.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajita M, Itoh Y, Chiba T, Mori H, Okada A, Kinoh H, Seiki M. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J Cell Biol. 2001;153:893–904. doi: 10.1083/jcb.153.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanov MD, Igarashi Y, Eroshkin AM, Cieplak P, Ratnikov B, Zhang Y, Li Z, Godzik A, Osterman AL, Smith JW. Structural determinants of limited proteolysis. J Proteome Res. 2011;10:3642–3651. doi: 10.1021/pr200271w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov IA, Thomsen ER, Munchel SE, Villegas P, Capek P, Gower AJ, Pond SJ, Chudin E, Chee MS. A highly scalable peptide-based assay system for proteomics. PLoS One. 2012;7:e37441. doi: 10.1371/journal.pone.0037441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Ratnikov BI, Kazanov MD, Smith JW, Cieplak P. CleavPredict: a platform for reasoning about matrix metalloproteinases proteolytic events. PloS One. 2015;10:e0127877. doi: 10.1371/journal.pone.0127877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Wang Y, Huang H, Pan Y, Chaney EJ, Boppart SA, Ozer H, Strongin AY, Wang Y. Quantitative FRET imaging to visualize the invasiveness of live breast cancer cells. PLoS One. 2013;8:e58569. doi: 10.1371/journal.pone.0058569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskos K. Crystal structures of MMPs in complex with physiological and pharmacological inhibitors. Biochimie. 2005;87:249–263. doi: 10.1016/j.biochi.2004.11.019. [DOI] [PubMed] [Google Scholar]
- Massova I, Kotra LP, Fridman R, Mobashery S. Matrix metalloproteinases: structures, evolution, and diversification. Faseb J. 1998;12:1075–1095. [PubMed] [Google Scholar]
- McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289:1202–1206. doi: 10.1126/science.289.5482.1202. [DOI] [PubMed] [Google Scholar]
- Mori H, Tomari T, Koshikawa N, Kajita M, Itoh Y, Sato H, Tojo H, Yana I, Seiki M. CD44 directs membrane-type 1 matrix metalloproteinase to lamellipodia by associating with its hemopexin-like domain. Embo J. 2002;21:3949–3959. doi: 10.1093/emboj/cdf411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H, Fushimi K. Elucidating the function of non catalytic domains of collagenases and aggrecanases. Connective tissue research. 2008;49:169–174. doi: 10.1080/03008200802151698. [DOI] [PubMed] [Google Scholar]
- Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Nagase H, Woessner JF., Jr Matrix metalloproteinases. J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- Oh J, Takahashi R, Adachi E, Kondo S, Kuratomi S, Noma A, Alexander DB, Motoda H, Okada A, Seiki M, et al. Mutations in two matrix metalloproteinase genes, MMP-2 and MT1-MMP, are synthetic lethal in mice. Oncogene. 2004;23:5041–5048. doi: 10.1038/sj.onc.1207688. [DOI] [PubMed] [Google Scholar]
- Ouyang M, Huang H, Shaner NC, Remacle AG, Shiryaev SA, Strongin AY, Tsien RY, Wang Y. Simultaneous visualization of protumorigenic Src and MT1-MMP activities with fluorescence resonance energy transfer. Cancer Res. 2010;70:2204–2212. doi: 10.1158/0008-5472.CAN-09-3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall CM, Kleifeld O. Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6:227–239. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- Prudova A, auf dem Keller U, Butler GS, Overall CM. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol Cell Proteomics. 2010;9:894–911. doi: 10.1074/mcp.M000050-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente XS, Sanchez LM, Overall CM, Lopez-Otin C. Human and mouse proteases: a comparative genomic approach. Nat Rev Genet. 2003;4:544–558. doi: 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- Ratnikov B, Cieplak P, Smith JW. High throughput substrate phage display for protease profiling. Methods Mol Biol. 2009;539:93–114. doi: 10.1007/978-1-60327-003-8_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnikov BI, Cieplak P, Gramatikoff K, Pierce J, Eroshkin A, Igarashi Y, Kazanov M, Sun Q, Godzik A, Osterman A, et al. Basis for substrate recognition and distinction by matrix metalloproteinases. Proc Natl Acad Sci U S A. 2014;111:E4148–4155. doi: 10.1073/pnas.1406134111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: What they do not do. New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803:39–54. doi: 10.1016/j.bbamcr.2009.09.015. [DOI] [PubMed] [Google Scholar]
- Schilling O, Overall CM. Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat Biotechnol. 2008;26:685–694. doi: 10.1038/nbt1408. [DOI] [PubMed] [Google Scholar]
- Schilling O, Overall CM. Proteomic discovery of protease substrates. Curr Opin Chem Biol. 2007;11:36–45. doi: 10.1016/j.cbpa.2006.11.037. [DOI] [PubMed] [Google Scholar]
- Schlage P, Auf dem Keller U. Proteomic approaches to uncover MMP function. Matrix Biol. 2015 doi: 10.1016/j.matbio.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Shay G, Lynch CC, Fingleton B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015 doi: 10.1016/j.matbio.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiryaev SA, Aleshin AE, Muranaka N, Kukreja M, Routenberg DA, Remacle AG, Liddington RC, Cieplak P, Kozlov IA, Strongin AY. Structural and functional diversity of metalloproteinases encoded by the Bacteroides fragilis pathogenicity island. FEBS J. 2014 doi: 10.1111/febs.12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiryaev SA, Chernov AV, Golubkov VS, Thomsen ER, Chudin E, Chee MS, Kozlov IA, Strongin AY, Cieplak P. High-resolution analysis and functional mapping of cleavage sites and substrate proteins of furin in the human proteome. PLoS One. 2013;8:e54290. doi: 10.1371/journal.pone.0054290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiryaev SA, Remacle AG, Chernov AV, Golubkov VS, Motamedchaboki K, Muranaka N, Dambacher CM, Capek P, Kukreja M, Kozlov IA, et al. Substrate Cleavage Profiling Suggests a Distinct Function of Bacteroides fragilis Metalloproteinases (Fragilysin and Metalloproteinase II) at the Microbiome-Inflammation-Cancer Interface. J Biol Chem. 2013;288:34956–34967. doi: 10.1074/jbc.M113.516153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiryaev SA, Remacle AG, Savinov AY, Chernov AV, Cieplak P, Radichev IA, Williams R, Shiryaeva TN, Gawlik K, Postnova TI, et al. Inflammatory proprotein convertase-matrix metalloproteinase proteolytic pathway in antigen-presenting cells as a step to autoimmune multiple sclerosis. J Biol Chem. 2009;284:30615–30626. doi: 10.1074/jbc.M109.041244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiryaev SA, Savinov AY, Cieplak P, Ratnikov BI, Motamedchaboki K, Smith JW, Strongin AY. Matrix metalloproteinase proteolysis of the myelin basic protein isoforms is a source of immunogenic peptides in autoimmune multiple sclerosis. PLoS One. 2009;4:e4952. doi: 10.1371/journal.pone.0004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiryaev SA, Thomsen ER, Cieplak P, Chudin E, Cheltsov AV, Chee MS, Kozlov IA, Strongin AY. New details of HCV NS3/4A proteinase functionality revealed by a high-throughput cleavage assay. PLoS One. 2012;7:e35759. doi: 10.1371/journal.pone.0035759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk BE, Huang LL, Piro ET, Cantley LC. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat Biotechnol. 2001;19:661–667. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- Zucker S, Cao J, Chen WT. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene. 2000;19:6642–6650. doi: 10.1038/sj.onc.1204097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.