

Abstract
Objective
To detect rare coding variants underlying loci detected by genome‐wide association studies (GWAS) of late onset Alzheimer disease (LOAD).
Methods
We conducted targeted sequencing of ABCA7, BIN1, CD2AP, CLU, CR1, EPHA1, MS4A4A/MS4A6A, and PICALM in 3 independent LOAD cohorts: 176 patients from 124 Caribbean Hispanics families, 120 patients and 33 unaffected individuals from the 129 National Institute on Aging LOAD Family Study; and 263 unrelated Canadian individuals of European ancestry (210 sporadic patients and 53 controls). Rare coding variants found in at least 2 data sets were genotyped in independent groups of ancestry‐matched controls. Additionally, the Exome Aggregation Consortium was used as a reference data set for population‐based allele frequencies.
Results
Overall we detected a statistically significant 3.1‐fold enrichment of the nonsynonymous mutations in the Caucasian LOAD cases compared with controls (p = 0.002) and no difference in synonymous variants. A stop‐gain mutation in ABCA7 (E1679X) and missense mutation in CD2AP (K633R) were highly significant in Caucasian LOAD cases, and mutations in EPHA1 (P460L) and BIN1 (K358R) were significant in Caribbean Hispanic families with LOAD. The EPHA1 variant segregated completely in an extended Caribbean Hispanic family and was also nominally significant in the Caucasians. Additionally, BIN1 (K358R) segregated in 2 of the 6 Caribbean Hispanic families where the mutations were discovered.
Interpretation
Targeted sequencing of confirmed GWAS loci revealed an excess burden of deleterious coding mutations in LOAD, with the greatest burden observed in ABCA7 and BIN1. Identifying coding variants in LOAD will facilitate the creation of tractable models for investigation of disease‐related mechanisms and potential therapies. Ann Neurol 2015;78:487–498
The first large‐scale genome‐wide association studies (GWAS) using common single nucleotide polymorphisms (SNPs) identified CLU, PICALM, CR1, and BIN1 as late onset Alzheimer disease (LOAD) susceptibility loci,1, 2, 3 which were widely confirmed by others.4, 5 The effect sizes of these genetic associations were much smaller than for APOE,2, 6 with odds ratios ranging from 1.16 to 1.20. Follow‐up GWAS identified additional LOAD susceptibility variants.7, 8 Although the known function of the genes implicated in these GWAS encode proteins implicating disruptions of lipid metabolism, immune response, and endocytosis or intracellular trafficking as potential mechanisms in LOAD, only a handful of disease‐associated variants in these genes, such as SORL1,9, 10 have been identified.
Surprisingly, targeted exome sequencing of large multiplex pedigrees with LOAD identified mutations in APP, PSEN1, and PSEN2,11, 12 indicating that rare coding sequence variants even in genes associated with early onset Alzheimer disease (AD) may account for a portion of disease risk in LOAD. Also, rare coding sequence variants in ADAM10,13 TREM2,12, 14 and PLD3 15 have been found in patients with LOAD. Because the majority of loci detected by SNP‐based GWAS of LOAD have not been investigated for rare coding sequence variants, we conducted targeted sequencing of the top 8 genetic loci frequently associated with LOAD,7, 8, 16, 17, 18 with the exception of the CD33 locus, which was not well replicated in subsequent large meta‐GWAS.18
Subjects and Methods
Sample Selection
All participants (Table 1) were recruited after providing informed consent and with approval by the relevant institutional review boards. Persons deemed unaffected were required to have had documented cognitive testing and clinical examination to verify their clinical status and diagnosis. Families in which patients had known mutations in APP, PSEN1, PSEN2, GRN, or MAPT were excluded. All selected probands came from families with ≥4 affected individuals.
Table 1.
Demographics of the Samples in the Targeted Sequencing Experiment
| Status | No. | Mean Age at Onset or Last Examination, yr ± SD | Women, No. (%) | APOE ε4, % | |
|---|---|---|---|---|---|
| Targeted sequencing | |||||
| NIA‐LOAD | Affected | 120 | 75.1 ± 8.3 | 77 (64.2) | 13.6 |
| Unaffected | 33 | 82.2 ± 10.8 | 24 (72.7) | 34.1 | |
| Toronto | Affected | 210 | 73.9 ± 7.3 | 106 (50.4) | 35.7 |
| Unaffected | 53 | 80.3 ± 3.6 | 33 (62.3) | 22.4 | |
| Hispanics | Affected | 176 | 74.8 ± 8.3 | 111 (63.1) | 25.3 |
| Follow‐up genotyping | |||||
| Hispanics | Unaffected | 444 | 81.2 ± 7.1 | 302 (68.0) | 12.9 |
| WHICAP | Unaffected | 300 | 84.7 ± 5.5 | 174 (58.0) | 10.0 |
| Toronto | Unaffected | 238 | 73.1 ± 9.4 | 137 (57.5) | 14.4 |
LOAD = late onset Alzheimer disease; NIA = National Institute on Aging; SD = standard deviation; WHICAP = Washington Heights‐Inwood Community Aging Project.
National Institute on Aging–LOAD/National Cell Repository for Alzheimer's Disease Study
Affected and unaffected individuals (n = 153) from 129 families within the National Institute on Aging (NIA)‐LOAD Family Study5 were selected for targeted sequencing analysis, including 120 individuals with LOAD and 33 similarly aged unaffected subjects (see Table 1). Patients had a mean age of onset of 75.1 ± 8.3 years with 38% frequency of the APOE ε4 allele, and unaffected participants were older (mean age = 82.2 ± 10.8 years) with 13% frequency of the APOE ε4 allele.
Family Study of Genetic Influence in AD
Recruitment for the Family Study of Genetic Influence in AD (Estudio Familiar de Influencia Genetica en Alzheimer) began in 1998, and was restricted to Caribbean Hispanics,19 mostly from the Dominican Republic. A total of 176 affected patients from 124 families were selected for targeted sequencing (the mean age of onset was 74.8 ± 8.3 years; 63.1% were woman).
Toronto LOAD Study
Targeted sequencing of GWAS loci was conducted in 210 well‐characterized sporadic LOAD patients and 53 normal controls of European ancestry from the GenADA study based on sufficient quantity/quality DNA samples. The mean age of onset in cases was 73.9 ± 7.3 years, 50.4% were women, and APOE ε4 allele frequency was 35.7%. These patients were either clinically diagnosed with probable LOAD (n = 169) or autopsy‐confirmed LOAD cases from the brain bank at the Tanz Centre for Neurodegenerative Research in Toronto (n = 41). Mean age at the time of examination in controls was 80.3 ± 3.6 years, 62.3% of them were women, and the APOE ε4 allele frequency was 22.4%.
Exome Aggregation Consortium
The Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org) data set was used as a reference data set of population‐based allele frequencies. It contains data from 60,706 unrelated adult individuals sequenced as part of various disease‐specific and population genetic studies from 6 different ethnic groups. LOAD was not among the diseases investigated. We used the non‐Finnish European and Latino/American cohorts to compare the frequencies of variants found in Caucasian and Caribbean Hispanic cohorts, respectively.
Sample Preparation
High molecular weight DNA was isolated from either fresh or frozen samples that had been stored at −80oC. Blood genomic DNA was isolated using the Gentra Puregene and FlexiGene kits (Qiagen, Valencia, CA), and saliva genomic DNA was isolated using prepIT.L2P (DNA Genotek, Kanata, Ontario, Canada). When high‐quality DNA derived from blood was unavailable, lymphocyte cell lines were used (in a total of 13 probands). DNA concentration was determined by NanoDrop (Thermo Fisher Scientific, Waltham, MA) for most analyses.
Targeted Sequencing
Target enrichment of the samples was performed using the Agilent Technologies (Santa Clara, CA) SureSelect system (for Hispanics and the NIA‐LOAD data set), and Roche (Basel, Switzerland) NimbleGen SeqCap EZ Designs‐custom (for the Toronto data set). Custom oligonucleotide baits captured exonic regions and splice sites of the genes of interest and amplified them. For the SeqCap EZ approach, the sequencing library was hybridized to the SeqCap EZ Oligo pool that was made against the target regions of interest. The end product was subjected to high throughput sequencing. After the DNA samples were prepared, they were multiplexed with index “barcode” primers and pooled for sequencing in batches of up to 12 samples.
Sequencing of all samples occurred on Illumina's Genome Analyzer IIx, HiSeq 2000, and MiSeq platforms (http://www.illumina.com). Paired‐end reads were performed over 82 to 307 sequencing cycles. Data files were demultiplexed by barcode to separate pooled samples into individual probands. We were able to obtain high coverage at an average depth of >1,000× per sample and interval region captured.
Follow‐up Genotyping
Caribbean Hispanics with mutations also observed in 1 of the other 2 data sets underwent genotyping or Sanger sequencing to confirm nonsynonymous variants. To determine the population frequencies for variants discovered within our data sets, we genotyped unrelated controls of the same ethnic background (see Table 1). We also conducted validation genotyping in 13 Caribbean Hispanic families (n = 148) of the patients where the variants were discovered. Additionally, to compare the allele frequencies for novel variants identified in this study from unaffected persons in the Caribbean Hispanic population, we genotyped 444 unaffected and unrelated persons (68.0% women, mean age at examination = 81.2 ± 7.1, APOE ε4 allele frequency = 12.9%) of the same ancestry. We also genotyped 300 white, non‐Hispanic controls (58.0% women, mean age at time of examination = 84.7 ± 5.5 years, APOE ε4 allele frequency = 10.0%) in the NIA‐LOAD data set to estimate population frequencies for the mutations discovered. The controls were determined to be of the same ethnic background as the familial cases using methods described previously.19 Follow‐up genotyping was also done on 238 normal controls matched to the Toronto sporadic LOAD data set by ethnic origin, sex, and age (57.5% women, mean age at time of examination = 73.1 ± 9.4 years, APOE ε4 allele frequency = 14.4%). Genotypes were generated using MassArray iPLEX technology (Sequenom, San Diego, CA), following the manufacturers’ instructions. The system involves multiplex polymerase chain reaction and minisequencing assays, followed by matrix‐assisted laser desorption/ionization time of flight mass spectrometry analysis.
Analytical Methods
We aligned the reads obtained from the pooled sequencing to the human reference genome build 37 using the Burrows–Wheeler Aligner20 (http://bio‐bwa.sourceforge.net). Quality control of the sequencing data was done using established methods, including base alignment quality calibration and refinement of local alignment around putative indels using the Genome Analysis Toolkit (GATK).21 Variants were called and recalibrated using multisample calling with GATK's UnifiedGenotyper and VariantRecalibrator modules. Reliably called variants were annotated by ANNOVAR22 including in silico functional prediction using POLYPHEN23 and extent of cross‐species conservation using PHYLOP.24
Burden Tests
We estimated the burden of different classes of mutations (loss of function, all nonsynonymous, and all synonymous variants) using a binomial test as described in Neale et al.25 To determine whether a class of mutations was enriched in cases, we used a binomial test with probability of success equal to the frequency of mutation class in controls (background or expected frequency). Also, any bias introduced in the test due to an unbalanced case–control set was compared to observations from the synonymous mutation class that was used to set the background expectation.
Individual Single Nucleotide Variant Significance Tests
To test the association of individual single nucleotide variants (SNVs) with LOAD, we compared the allele frequencies of SNVs in patients with unaffected samples from follow‐up genotyping combined with the publicly available ExAC data using Fisher exact test. We used this data set to provide a much larger and more representative estimate of allele frequencies than could be ascertained from the smaller NIA‐LOAD and Toronto data sets alone. Because of the lack of an optimal ethnically matched control data set for Caribbean Hispanics, we used the Latino American cohort for an estimate of allele frequencies of rare variants. Additionally, for the Caribbean Hispanic cohort only, we tested segregation and LOAD association in this data set using generalized estimation equations to adjust for familial correlation.
Results
Sequencing
We identified 12 coding mutations in 7 genes in at least 2 of the 3 data sets, including 7 autopsy‐confirmed LOAD cases (Table 2). These 12 coding mutations included: 4 mutations in ABCA7, 2 each in CD2AP and PICALM, and 1 each in BIN1, CLU, EPHA1, and MS4A6A. Three rare coding mutations were observed in cases from all 3 data sets: rs138047593 in BIN1, rs202178565 in EPHA1, and rs138650483 in MS4A6A; the EPHA1 and BIN1 mutations were subsequently confirmed by follow‐up genotyping in the Hispanic and Caucasian cohorts. The EPHA1 variant was absent in the genotyped Caucasian controls and the BIN1 variant had the same frequency as the Caucasian LOAD cases. Thus, we assessed the association of these variants independently in Caucasian and Hispanic cohorts by comparing them against the population‐based allele frequencies available in the ExAC database and by testing family‐based association in Caribbean Hispanic families.
Table 2.
Annotation of Rare or Novel Nonsynonymous Single Nucleotide Polymorphisms Found in at Least 2 of the 3 Data Sets
| Chr | Position | ID | Ref | Alt | Function | Gene | AA Change | POLYPHEN | SIFT |
|---|---|---|---|---|---|---|---|---|---|
| 2 | 127808046 | rs138047593a | T | C | Nonsynonymous | BIN1 | K358R | D | D |
| 6 | 47563608 | rs138727736 | A | G | Nonsynonymous | CD2AP | T374A | B | B |
| 6 | 47591941 | rs116754410 | A | G | Nonsynonymous | CD2AP | K633R | D | D |
| 7 | 143095499 | rs202178565b | G | A | Nonsynonymous | EPHA1 | P460L | D | P |
| 8 | 27462662 | rs41276297 | G | A | Nonsynonymous | CLU | T203I | B | B |
| 11 | 59940500 | rs138650483a | C | T | Exonic/splicing | MS4A6A | V218M | D | D |
| 11 | 85687719 | rs147556602 | G | C | Nonsynonymous | PICALM | P495A | D | D |
| 11 | 85701307 | rs117411388 | T | C | Nonsynonymous | PICALM | H458R | D | D |
| 19 | 1041971 | rs201665195 | T | G | Nonsynonymous | ABCA7 | L101R | D | D |
| 19 | 1051006 | rs143718918 | G | A | Nonsynonymous | ABCA7 | R880Q | D | D |
| 19 | 1057343 | rs117187003 | G | A | Nonsynonymous | ABCA7 | V1599M | D | D |
| 19 | 1058154 | novel | G | T | Stop‐gain | ABCA7 | E1679X | — | — |
Found in all 3 data sets.
Found in all 3 data sets, not found in any unaffected subjects in targeted sequencing.
Caucasians
For the 12 variants detected in the NIA‐LOAD and Toronto data sets (Table 3), we compared the frequency of SNVs between 330 cases in these data sets and the 33,370 non‐Finnish Europeans from ExAC using a Fisher exact test. A stop‐gain mutation in ABCA7 (E1769X) and missense mutation in CD2AP (K633R) were highly significant after correction for multiple testing (p = 5.3E‐04 and 5.3E‐08, respectively). Of the remaining variants discovered in multiple data sets, 1 rare variant in both EPHA1 and PICALM was nominally significant (p = 0.03 and 0.007, respectively). The p.K358R variant in BIN1 was observed in 1.8% of the ExAC database Caucasians, which is similar to the frequency we observed in the cohort of patients here with LOAD. The remaining variants were extremely rare (minor allele frequency [MAF] < 0.5%) in the ExAC database Caucasians.
Table 3.
Allele Frequency and Fisher Tests of Single Nucleotide Polymorphisms in Caucasians
| Targeted Sequencing, Carriers | Targeted Sequencing Frequency | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | ID | NIA‐LOAD | Toronto (autopsy cases) | NIA‐LOAD | Toronto | ||||
| Unaffected | LOAD | Unaffected | LOAD | LOAD | LOAD | ExAC Frequency in Europeans | p, Fisher Test | ||
| BIN1 | rs138047593 | 0 | 1 | 0 | 7 (4) | 0.004386 | 0.016667 | 1.82E‐02 | 3.69E‐01 |
| CD2AP | rs138727736 | 0 | 0 | 0 | 4 (2) | 0 | 0.009524 | 4.73E‐03 | 1.37E‐01 |
| CD2AP a | rs116754410 | 0 | 0 | 0 | 1 (1) | 0 | 0.002381 | 3.06E‐05 | 5.33E‐08 |
| EPHA1 a | rs202178565 | 0 | 1 | 0 | 1 | 0.004386 | 0.002381 | 4.05E‐04 | 3.07E‐02 |
| CLU | rs41276297 | 0 | 0 | 0 | 2 | 0 | 0.004762 | 2.51E‐03 | 2.82E‐01 |
| MS4A6A | rs138650483 | 1 | 1 | 0 | 1 | 0.00431 | 0.002381 | 3.76E‐03 | 1.00E+00 |
| PICALM | rs147556602 | 0 | 0 | 1 | 0 | 0 | 0 | 3.61E‐04 | 1.00E+00 |
| PICALM a | rs117411388 | 0 | 2 | 0 | 2 | 0.00885 | 0.004762 | 1.11E‐03 | 6.84E‐03 |
| ABCA7 | rs201665195 | 0 | 1 | 0 | 1 | 0.004348 | 0.002381 | 1.14E‐03 | 1.82E‐01 |
| ABCA7 | rs143718918 | 0 | 1 | 0 | 1 | 0.004425 | 0.002381 | 2.11E‐03 | 3.89E‐01 |
| ABCA7 | rs117187003 | 0 | 4 | 1 | 1 | 0.017857 | 0.002381 | 4.16E‐03 | 2.01E‐01 |
| ABCA7 a | 19:1058154 | 0 | 1 | 0 | 1 | 0.004425 | 0.002381 | 3.02E‐05 | 5.34E‐04 |
Nominally significant single nucleotide variants.
ExAC = Exome Aggregation Consortium; LOAD = late onset Alzheimer disease; NIA = National Institute on Aging.
Caribbean Hispanics
For the 7 variants found in this data set (and at least 1 other Caucasian data set; Table 4), we tested segregation and LOAD association using validation‐genotyping data in 13 families and 444 independent case–controls. Furthermore, we compared the frequency of the variants in LOAD patients with the Latino allele frequencies (n = 5,789) in the ExAC database. The p.P460L in EPHA1 and p.K358R in BIN1 were significantly associated with LOAD when compared to both internally genotyped Caribbean Hispanic controls and population Latino controls in the ExaC database after correction for multiple testing (Table 4). Notably, the EPHA1 rs202178565 variant (P460L) was observed in only 1 of the 444 unaffected Caribbean Hispanic individuals and none of the Caucasian controls. This EPHA1 mutation also segregated completely in 4 affected members of a Caribbean Hispanic family (Fig 1). The variant was significant both in the Fisher exact test (p = 2.6E‐03) and regression model (p = 8.64E‐05) in Caribbean Hispanics and nominally significant in the Caucasian cohort (p = 0.03).
Table 4.
Allele Frequency and Association Tests in Hispanics
| Gene | ID | Targeted Sequencing, Affected Carriers | Targeted Sequencing, Affected Carriers, Frequency | Control Frequency | Familial Case Frequency | Familial Control Frequency | Beta | p | ExAC Frequency in Latinos | p, Fisher Test |
|---|---|---|---|---|---|---|---|---|---|---|
| BIN1 a | rs138047593 | 6 | 0.01705 | 0.0084 | 0.0859 | 0.0641 | 2.03 | 1.27E‐05 | 2.60E‐03 | 5.85E‐04 |
| CD2AP | rs138727736 | 2 | 0.00568 | 0.0108 | 0.0238 | 0.0128 | 1.26 | 3.04E‐02 | 3.78E‐03 | 3.91E‐01 |
| CD2AP | rs116754410 | 2 | 0.00568 | 0.0160 | 0.0323 | 0.0128 | 0.69 | 3.37E‐01 | 1.77E‐03 | 1.41E‐01 |
| EPHA1 a | rs202178565 | 2 | 0.00568 | 0.0011 | 0.0078 | 0 | 3.44 | 1.25E‐04 | 8.64E‐05 | 2.55E‐03 |
| CLU | rs41276297 | 1 | 0.00284 | 0.0012 | 0 | 0 | 1.04E‐03 | 3.23E‐01 | ||
| MS4A6A | rs138650483 | 1 | 0.00284 | 0.0049 | 0.0085 | 0 | 0.63 | 4.56E‐01 | 4.16E‐03 | 1.00E+00 |
| PICALM | rs147556602 | 1 | 0.00472 | 0.0037 | 0.0154 | 0 | 1.21 | 1.81E‐01 | 7.84E‐04 | 1.67E‐01 |
Nominally significant single nucleotide variants.
ExAC = Exome Aggregation Consortium.
Figure 1.
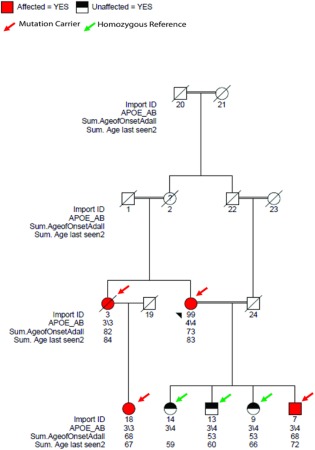
Missense damaging mutation rs202178565 in EPHA1 (ephrin type‐A receptor 1). This mutation was not found in any external controls. Import ID = internal subject ID; APOE_AB = APOE ε4 status; Sum.AgeofOnsetAdAll = age at onset of disease; Sum.Age last seen2 = age of the last examination of the subject.
Follow‐up genotyping of the BIN1 p.K358R mutation revealed that it was predominantly found in affected members with LOAD from 6 Caribbean Hispanic families. We observed BIN1 p.K358R in 11 LOAD patients and only 3 elderly controls (>65 years) in these families. We also observed the mutation in 9 unaffected members younger than 65 years (average age = 54 years). We observed a higher frequency of the mutation in the families (0.085 in familial cases and 0.069 in familial controls) compared to genotyped Caribbean Hispanic controls (0.0084) and Latino population controls from the ExAC database (0.0026). This variant was significantly associated with Caribbean Hispanic LOAD families in both a regression model (p = 1.27E‐05) and by Fisher exact test (p = 5.85E‐04). The BIN1 p.K358R allele frequencies in Caucasian and Caribbean Hispanic population controls were similar. However, we found much higher frequency of this variant in families, suggesting that the effect of this variant in multiplex families may be due to epistasis with other genetic or environmental risk factors. Further investigation of this mutation is required to evaluate the effect of this variant in LOAD pathogenesis.
Other Mutations
In addition to mutations observed in multiple data sets, a total of 88 rare damaging mutations were found to be present in individual data sets and were only detected in patients with LOAD: 21 in NIA‐LOAD, 37 in the Toronto data set, and 30 in the Caribbean Hispanics. When compared to the ExAC population frequencies, 38 of 88 variants were nominally significant at p < 0.05 (Supplementary Table), 21 of which were observed in ABCA7 and 5 in EPHA1. All the nominally significant variants were extremely rare in the general population (max MAF = 0.05%), and a majority of them were predicted to be deleterious to the coding protein.
Burden Tests
We calculated the overall burden of these novel or rare coding nonsynonymous mutations (including SNVs and short indels) compared with the burden of synonymous mutations in cases and controls for each gene in the 3 data sets (Table 5). Combining the observations from the NIA‐LOAD and Toronto Caucasian data sets, we detected a statistically significant 3.1‐fold enrichment of the nonsynonymous mutations in cases versus controls (p = 0.002). The LOAD cases also carried 2.76 times more loss of function mutations (stop‐loss, stop‐gain, frameshift, or splicing) and damaging missense mutations compared to controls (p = 0.02). In contrast, we did not observe a difference in synonymous mutations for LOAD cases in the 2 Caucasian data sets compared with controls (1.07‐fold, p = 0.59). The mutation rate per Caribbean Hispanic LOAD patient was comparable to that in the Caucasian data set across all genes (see Table 5).
Table 5.
Number of Mutations (Mutation Rate per Subject) in the 3 Different Mutation Classes for Each Gene in the NIA‐LOAD, Toronto, and Hispanic Data Sets
| Cases | Controls | |||||
|---|---|---|---|---|---|---|
| Data Set | C1 | C2 | C3 | C1 | C2 | C3 |
| NIA‐LOAD data set | ||||||
| ABCA7 | 12 (0.100) | 15 (0.125) | 2 (0.0170) | 0 | 0 | 0 |
| BIN1 | 1 (0.008) | 2 (0.017) | 3 (0.025) | 0 | 0 | 2 |
| CD2AP | 2 (0.017) | 2 (0.017) | 2 (0.017) | 1 | 1 | 1 |
| CLU | 1 (0.008) | 1 (0.008) | 1 (0.008) | 0 | 0 | 0 |
| CR1 | 0 | 1 (0.008) | 2 (0.017) | 0 | 0 | 0 |
| EPHA1 | 3 (0.025) | 3 (0.025) | 2 (0.017) | 0 | 0 | 0 |
| MS4A6A | 1 (0.008) | 1 (0.008) | 0 | 1 | 1 | 0 |
| PICALM | 0 | 3 (0.025) | 0 | 0 | 0 | 0 |
| Total | 20 | 28 | 12 | 2 | 2 | 3 |
| Toronto data set | ||||||
| ABCA7 | 16 (0.075) | 16 (0.075) | 5 (0.023) | 1 | 1 | 0 |
| BIN1 | 8 (0.038) | 8 (0.038) | 4 (0.019) | 0 | 0 | 4 |
| CD2AP | 4 (0.019) | 8 (0.038) | 3 (0.014) | 1 | 2 | 0 |
| CLU | 0 | 2 (0.009) | 5 (0.023) | 0 | 0 | 1 |
| CR1 | 1 (0.005) | 1 (0.005) | 9 (0.042) | 0 | 0 | 1 |
| EPHA1 | 1 (0.005) | 5 (0.023) | 6 (0.028) | 0 | 0 | 2 |
| MS4A6A | 1 (0.005) | 2 (0.009) | 0 | 0 | 0 | 0 |
| PICALM | 2 (0.009) | 2 (0.009) | 1 (0.005) | 1 | 1 | 0 |
| Total | 33 | 44 | 33 | 3 | 4 | 8 |
| Burden test | ||||||
| No. | 53 | 72 | 45 | 5 | 6 | 11 |
| Enrichment (p) | 2.76 (0.02) | 3.1 (0.002) | 1.07 (0.59) | |||
| Hispanic data set | ||||||
| ABCA7 | 15 (0.085) | 17 (0.097) | 12 (0.068) | |||
| BIN1 | 2 (0.011) | 9 (0.051) | 8 (0.045) | |||
| CD2AP | 0 | 4 (0.023) | 0 | |||
| CLU | 1 (0.006) | 4 (0.023) | 5 (0.028) | |||
| CR1 | 0 | 4 (0.023) | 2 (0.011) | |||
| EPHA1 | 8 (0.045) | 8 (0.045) | 6 (0.034) | |||
| MS4A6A | 0 | 1 (0.006) | 0 | |||
| PICALM | 0 | 1 (0.006) | 0 | |||
| Total | 26 | 48 | 33 | |||
C1 = class I: loss of function (stop‐gain, stop‐loss) and damaging missense mutations; C2 = class II: all nonsynonymous mutations; C3 = class III: all synonymous mutations; LOAD = late onset Alzheimer disease; NIA = National Institute on Aging.
In total, 11.1% of all patients with LOAD from 3 data sets were carriers of at least 1 coding ABCA7 mutation. Remarkably, 47% of all potentially damaging mutations were observed in the ABCA7 gene. Of the rare mutations, 8% were detected in EPHA1, affecting 3.1% of investigated LOAD cases and only a single Caribbean Hispanic control. These results are striking because based on a recent study26 of thousands of exomes, ABCA7 and EPHA1 are highly conserved genes and ranked in the top 2nd and 11th percentiles, respectively, for intolerance toward mutation in the general population. The high mutation rate in LOAD compared to controls in the highly conserved ABCA7 and EPHA1 implies a putative functional role in the pathogenesis of LOAD.
BIN1 was a strong contributor to the increased mutation rate in cases compared to controls, showing damaging variants in 19 cases (3.75%) but none in controls (see Table 2 and Supplementary Table). The most frequent mutation in the patients was in BIN1 (p.K358R), where we identified carriers in 8 Caucasian (including 4 autopsy cases) and 6 Hispanic patients.
There is prior evidence of increased expression of ABCA7, BIN1, and MS4A6A in LOAD brains,27 and increased ABCA7 expression is associated with clinical dementia rating,28 with higher expression being associated with more advanced cognitive decline. BIN1 expression levels were associated with disease progression, where higher expression was associated with a delayed age at onset. However, there was no evidence of differential expression of EPHA1 in LOAD compared with controls.28
Discussion
The results presented here imply that the loci from GWAS associated with LOAD likely contain multiple rare, damaging mutations that can be recurrent among unrelated patients and in some instances can segregate within families. The dense coverage we used for targeted sequencing allowed for the identification of variants that might not have been detectable with the more sparse coverage used in current whole exome or whole genome approaches. Variants in BIN1 (p.K358R), EPHA1 (p.P460L) were found in patients with LOAD from all 3 data sets and were statistically significant in follow‐up genotyping in the Caribbean Hispanic dataset. In the 2 Caucasian data sets, we found statistically significant variants in CD2AP and ABCA7. The nominally significant variants from individual data sets (see Supplementary Table) were observed in the ExAC data set at very low frequencies, providing further support that greater depth of targeted sequencing allows identification of very rare events.
In 3 data sets enriched by families multiply affected with LOAD, we sequenced 8 GWAS loci with consistent SNP‐based associations with LOAD across multiple investigations.18 Analysis of 2 Caucasian data sets revealed a significantly greater burden of rare and novel nonsynonymous (including SNVs and indels) alterations (p = 0.002) in cases compared to controls, whereas the mutation rate of synonymous variants was the similar in cases and controls. In LOAD we also observed a significant (p = 0.02) 3‐fold enrichment in the subset of alterations that were predicted to be damaging (by POLYPHEN or SIFT).
The greatest burden of damaging sequence variants was found in ABCA7. Among Caucasian LOAD cases, we detected 38 carriers of rare variants (20 in NIA‐LOAD and 18 in the Toronto data set), constituting 11.8% of 330 investigated cases, whereas only 1 carrier of such a variant was found among the 86 sequenced controls (1.2%; see Table 2 and Supplementary Table). In addition to nonsynonymous ABCA7 variants, we observed a splice site, a stop mutation, and frameshift deletions, suggesting a loss‐of‐function mechanism associated with LOAD. Our recent functional studies of ABCA7 strongly support such a possibility,29, 30 because suppression of ABCA7 in vitro and in vivo resulted in an elevation of amyloid production. The complex function of ABCA7 includes mediation of the biogenesis of high‐density lipoprotein with cellular lipid and helical apolipoproteins,31 as well as function in apolipoprotein‐mediated phospholipid and cholesterol efflux from cells.32 Finally, a direct role of ABCA7 in APP processing may be associated with its primary biological function to regulate endocytic pathways.30 Importantly, we previously identified ABCA7 as a major genetic risk LOAD locus in African Americans,33 and a whole‐genome sequencing study in a large Icelandic cohort identified excess burden of rare loss of function variants in ABCA7 in LOAD.34 We confirmed 2 ABCA7 loss of function variants reported in that study (c.4416+2T>G and p.Leu1403Argfs*7) and discovered 3 additional variants (p.708_710del, p.R1489X, and E1679X). Our analyses confirm that ABCA7 has the highest burden of deleterious variants in LOAD, but differences in the observed mutations could be due to ethnicity, capture, and coverage differences in the 2 studies.
BIN1 was also strongly associated in the burden analysis, with damaging variants in 17 cases (5.1%) but no controls. Several SNPs upstream of the BIN1 locus have been identified in different GWAS, with the largest effect sizes after APOE (eg, rs6733839, with a population attributable fraction of 8.1%35). BIN1 transcript levels were increased among LOAD brains compared to controls,36 but coding mutations have not been widely explored. So far, there are only 4 BIN1 coding variants with clinical significance listed in the ClinVar database (p.K575*, p.R154Q, p. D151N, and p.K35N) and all were reported under autosomal recessive centronuclear myopathy. Recently, Tan et al reported that a novel BIN1 missense mutation p.P318L among the Han Chinese could increase risk of developing AD,37 which was not detected in our data sets. The BIN1 mutations reported here included p.K358R, identified in 8 Caucasian and 6 Hispanic LOAD patients, as well as p.S267L and p.S202T, each identified in a single LOAD patient. None of these mutations were found in controls or unaffected family members. We observed a strong association between LOAD and BIN1 p.K358R only in the Caribbean Hispanics. The allele frequency of this variant in the Caucasian patients was similar to the general population. BIN1 p.K358R is a good candidate for functional studies based on its relatively high frequency in familial LOAD cases and segregation in Caribbean Hispanic families. Importantly, BIN1 p.K358R likely contributes to LOAD independently from the GWAS SNPs, because it is mapped to a different linkage disequilibrium block (Fig 2).
Figure 2.

Linkage disequilibrium (LD) plot of BIN1 in Hispanics. The LD plot is generated using 32 genotyped single nucleotide polymorphisms (SNPs) in 1,675 elderly subjects of Caribbean Hispanic ancestry. The reported genome‐wide significant hit in Lambert et al (rs6733839) is 27.1 kilobases (KB) upstream of BIN1.
We also identified 6 nonsynonymous variants in EPHA1, including p.H888Y, p.R791H, p.V514I, p.R471Q, p.P460L, and p.R337Q. The damaging EPHA1 variant p.P460L (rs202178565) was identified in cases in all 3 data sets and nearly absent among our controls as well as in 1000 Genomes and the ExAC server data set. This variant segregated with the LOAD in a Caribbean Hispanic family from the Dominican Republic (see Fig 1), supporting its causative role. The EPHA1 p.P460 amino acid is highly conserved in all mammals and predicted to have a damaging effect on the protein by POLYPHEN estimation. However, the biological impact of this mutation remains to be investigated, because there is only limited information on the function of the protein. Ephrin receptor A1 encoded by EPHA1 belongs to the ephrin receptor subfamily of the protein–tyrosine kinase family and plays roles in cell and axonal guidance and synaptic plasticity.
A rare variant was found in MS4A6A, which affects splicing of 1 transcript of the gene (NM_152852: exon8: c.651+1G>A) and is a missense mutation in another transcript (NM_022349: exon6: c.G652A: p.V218M). The MS4A6A p.V218M variant was detected in a single unaffected Caucasian. MS4A6A is located among several genes at Chr11q12 that all are associated with the inflammatory response. MS4A6E mRNA expression and an SNP nearby the gene (rs670139) are associated with more advanced Braak stages of tangle and plaques in AD brain tissue.28 However, until now a functional variant in this region has not been identified, and the current study might provide the first clue.38
We identified other rare damaging variants among LOAD associated genes, including CD2AP (p.I104N, p.R403G, p.L487V, p.M496I, p.S623N, and p.K633R) and CLU (p.V434M). CD2AP is an adaptor molecule involved in dynamic actin remodeling and membrane trafficking, and CLU encodes clusterin, which is a molecular chaperone,39 is present in senile plaques, and has been shown to modulate Aβ oligomer assembly.40 We previously reported rare SNPs and small structural variants within the CLU gene that were associated with LOAD.41
Taken together, the results here imply that multiple rare coding mutations are present in genes identified as LOAD‐associated GWAS loci. Common variants identified in GWAS frequently occur in noncoding sequences within or between genes, and as a result, their functional relationship to disease risk is often hard to define. The data reported here reveal that GWAS loci could harbor both rare damaging variants and common noncoding variants that are independently associated with LOAD (eg, in CLU).41 Thus, targeted sequencing within GWAS loci may enable the discovery of coding variants underlying or contributing to the association with LOAD. The use of noncoding variants to build cellular and animal models of disease is confounded by uncertainties surrounding the temporally and cell type–specific effects of these noncoding variants on the regulation of gene expression. By contrast, disease‐associated coding sequence variants can be used to build facile, tractable cellular and animal models by a variety of simple methods including both standard transgenesis and clustered, regularly interspaced, short palindromic repeat (CRISPR)‐CAS–based methods. Such models can be used to investigate the underlying molecular mechanisms of these genes in the pathogenesis of LOAD.
The individual effect of these rare variants is expected to be small, and different variants are likely to be causal in different patients and families. For example, the p.K538R variant in BIN1 has a strong effect in the Hispanic families but was not associated with LOAD in the Caucasian cohort. It is likely that such variants confer modified risk of disease or depend on other interacting genes or environmental factors. Identification of such rare coding variants could thus aid in understanding the biology of the disease.
The strengths of this study are the 3 independent cohorts and the careful phenotyping. That some of the same mutations were observed in 2 or 3 of the cohorts adds validity to our observations. Although there appears to be increased expression associated with some of the genes containing mutations, further studies are required to examine mutation‐specific expression and to understand the mechanisms by which these mutations lead to disease.
Authorship
B.N.V. and M.G. contributed equally to the article. Senior authors P.S.G.‐H. and R.M. contributed equally to the article. Conception and design of the study: R.M., P.S.G.‐H., and E.R. Data collection: R.M., P.S.G.‐H, E.R., R.L., D.R.‐D., M.M., and I.Z.J.‐V. Data analysis: B.N.V., M.G., C.S., A.K., S.S., and R.C. Writing of the final manuscript: B.N.V., R.M., M.G., P.S.G.‐H, E.R, S.B., J.H.L., and C.R.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Additional Supporting Information can be found in the online version of this article.
Supplementary Information
Acknowledgment
This work was supported by grants from the NIH National Institute on Aging (R37AG15473; P01AG07232; R01AG041797; U24AG026395, R.M.), Canadian Institutes of Health Research (E.R., P.S.G.‐H.), Wellcome Trust, Medical Research Council, Ontario Research Fund, and Alzheimer Society of Ontario (P.S.G.‐H.). Funding was received from the NIH National Institute on Aging (U24AG21886) for the NIA‐LOAD Cohort.
Correction added on 21 August 2015, after first online publication: the amino acid substitution “T374A” changed to “K633R” in abstract/text
References
- 1. Harold D, Abraham R, Hollingworth P, et al. Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 2009;41:1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lambert JC, Heath S, Even G, et al. Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 2009;41:1094–1099. [DOI] [PubMed] [Google Scholar]
- 3. Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome‐wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010;303:1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jun G, Naj AC, Beecham GW, et al. Meta‐analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol 2010;67:1473–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wijsman EM, Pankratz ND, Choi Y, et al. Genome‐wide association of familial late‐onset Alzheimer's disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet 2011;7:e1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997;278:1349–1356. [PubMed] [Google Scholar]
- 7. Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late‐onset Alzheimer's disease. Nat Genet 2011;43:436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 2011;43:429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pottier C, Hannequin D, Coutant S, et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early‐onset Alzheimer disease. Mol Psychiatry 2012;17:875–879. [DOI] [PubMed] [Google Scholar]
- 10. Vardarajan BN, Zhang Y, Lee JH, et al. Coding mutations in SORL1 and Alzheimer's disease. Ann Neurol 2015;77:215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee JH, Kahn A, Cheng R, et al. Disease‐related mutations among Caribbean Hispanics with familial dementia. Mol Genet Genomic Med 2014;2:430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med 2013;368:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim M, Suh J, Romano D, et al. Potential late‐onset Alzheimer's disease‐associated mutations in the ADAM10 gene attenuate {alpha}‐secretase activity. Hum Mol Genet 2009;18:3987–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 2013;368:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cruchaga C, Karch CM, Jin SC, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer's disease. Nature 2014;505:550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome‐wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010;303:1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harold D, Abraham R, Hollingworth P, et al. Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 2009;41:1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee JH, Cheng R, Barral S, et al. Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Arch Neurol 2011;68:320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc Hum Genet 2013;Chapter 7:Unit 7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010;20:110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Neale BM, Rivas MA, Voight BF, et al. Testing for an unusual distribution of rare variants. PLoS Genet 2011;7:e1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 2013;9:e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Myers AJ, Gibbs JR, Webster JA, et al. A survey of genetic human cortical gene expression. Nat Genet 2007;39:1494–1499. [DOI] [PubMed] [Google Scholar]
- 28. Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, Goate AM. Expression of novel Alzheimer's disease risk genes in control and Alzheimer's disease brains. PLoS One 2012;7:e50976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. WS Kim, H Li, K Ruberu, et al. Deletion of Abca7 increases cerebral amyloid‐beta accumulation in the J20 mouse model of Alzheimer's disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 2013;33:4387–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. K Satoh, S Abe‐Dohmae, S Yokoyama, P St. George‐Hyslop, P Fraser. ATP‐binding cassette transporter A7 (ABCA7) effects on amyloid processing and relevance to Alzheimer's disease. Alzheimer's & Dementia: The Journal of the Alzheimer's Association 8:P473. [Google Scholar]
- 31. Tanaka N, Abe‐Dohmae S, Iwamoto N, Yokoyama S. Roles of ATP‐binding cassette transporter A7 in cholesterol homeostasis and host defense system. J Atheroscler Thromb 2011;18:274–281. [DOI] [PubMed] [Google Scholar]
- 32. Chan SL, Kim WS, Kwok JB, et al. ATP‐binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J Neurochem 2008;106:793–804. [DOI] [PubMed] [Google Scholar]
- 33. Reitz C, Jun G, Naj A, et al. Variants in the ATP‐binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late‐onset Alzheimer disease in African Americans. JAMA 2013;309:1483–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Steinberg S, Stefansson H, Jonsson T, et al. Loss‐of‐function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet 2015;47:445–447. [DOI] [PubMed] [Google Scholar]
- 35. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chapuis J, Hansmannel F, Gistelinck M, et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry 2013;18:1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tan MS, Yu JT, Jiang T, Zhu XC, Guan HS, Tan L. Genetic variation in BIN1 gene and Alzheimer's disease risk in Han Chinese individuals. Neurobiol Aging 2014;35:1781.e1–1781.e8. [DOI] [PubMed] [Google Scholar]
- 38. Omenn GS, Guan Y, Menon R. A new class of protein cancer biomarker candidates: differentially expressed splice variants of ERBB2 (HER2/neu) and ERBB1 (EGFR) in breast cancer cell lines. J Proteomics 2014;107:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wyatt A, Yerbury J, Poon S, Dabbs R, Wilson M. Chapter 6: The chaperone action of Clusterin and its putative role in quality control of extracellular protein folding. Adv Cancer Res 2009;104:89–114. [DOI] [PubMed] [Google Scholar]
- 40. Narayan P, Orte A, Clarke RW, et al. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid‐beta(1‐40) peptide. Nat Struct Mol Biol 2012;19:79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bettens K, Brouwers N, Engelborghs S, et al. Both common variations and rare non‐synonymous substitutions and small insertion/deletions in CLU are associated with increased Alzheimer risk. Mol neurodegener 2012;7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information can be found in the online version of this article.
Supplementary Information