

Abstract
Fatty acid ethanolamides such as palmitoylethanolamide (PEA) and oleoylethanolamide (OEA) are lipid-derived mediators that potently inhibit pain and inflammation by ligating type-α peroxisome proliferator-activated receptors (PPAR-α). These bioactive substances are preferentially degraded by the cysteine hydrolase, N-acylethanolamine acid amidase (NAAA), which is highly expressed in macrophages. Here we describe a new class of β-lactam derivatives that are potent, selective and systemically active inhibitors of intracellular NAAA activity. The prototype of this class deactivates NAAA by covalently binding the enzyme's catalytic cysteine, and exerts profound anti-inflammatory effects in both mouse models and human macrophages. This agent may be used to probe the functions of NAAA in health and disease and as a starting point to discover better anti-inflammatory drugs.
Keywords: palmitoylethanolamide, oleoylethanolamide, peroxisome proliferator-activated receptor-α, N-acylethanolamine acid hydrolase, inflammation
Peripheral nociceptive neurons and macrophages, two cell types that are crucially involved in the control of pain and inflammation, generate the bioactive lipid amides palmitoylethanolamide (PEA) and oleoylethanolamide (OEA)1 (1 and 2, Figure 1), which exert profound analgesic and anti-inflammatory effects by engaging nuclear peroxisome proliferator-activated receptor-α (PPAR-α).2–4
Figure 1.

The endogenous N-acylethanolamine acid amidase (NAAA) substrates, palmitoylethanolamide (PEA) (1) and oleoylethanolamide (OEA) (2)
These effects are terminated through enzyme-mediated hydrolysis catalyzed either by fatty acid amide hydrolase (FAAH), a ubiquitous serine enzyme whose functional significance is widely recognized5, or by the less-well understood cysteine amidase, N-acylethanolamine acid amidase (NAAA).6 Evidence suggests that NAAA contributes to the regulation of pain7,8 and innate immunity,9 but progress in testing this hypothesis has been slow due to the lack of adequate pharmacological tools.10–13
Here, we disclose the prototype of a new class of potent, selective and systemically active NAAA inhibitors based on a β-lactam scaffold, and elucidate the mechanism through which this compound interacts with NAAA. We further show that the new inhibitor suppresses lung inflammation in mice via a mechanism that requires PEA- and OEA-mediated PPAR-α activation, and blocks endotoxin-induced inflammatory responses in human macrophages. This agent may be used to probe the functions of NAAA and inspire the design of novel anti-inflammatory drugs.
Results and Discussion
We have previously shown that the serine β-lactone N-[(3S)-2-oxo-3-oxetanyl]-3-phenylpropanamide (3, Figure 2) inhibits NAAA activity with submicromolar potency (IC50, for rat NAAA (r-NAAA) ∼420 nM).9 Since our initial structure-activity relationship (SAR) studies had demonstrated that the ability of 3 to block NAAA depends on the presence of the β-lactone moiety,9 we focused subsequent SAR investigations on molecules containing this electrophilic warhead. The work led us to identify the threonine β-lactone 4 (ARN077, Figure 2) as the first potent NAAA inhibitor (IC50 for r-NAAA ∼50 nM and for human NAAA (h-NAAA) ∼7 nM).14 We further demonstrated that 4 interacts covalently with NAAA's catalytic cysteine15 and displays marked analgesic properties in rodent models.7 We also found, however, that the intrinsic lack of stability of this compound (t1/2 in plasma <1 min) and all other components of the same chemical class, which is due to hydrolytic cleavage of the β-lactone ring, restricts their in vivo use to topical application.14
Figure 2.

Chemical structures of current N-acylethanolamine acid amidase (NAAA) inhibitors.
To obviate the instability of β-lactone-based NAAA inhibitors such as 3 and 4, we designed a series of molecules that retain key structural features of those compounds, but in which a more stable β-lactam moiety replaces the labile β-lactone. A first training set yielded inhibitors of moderate potency (e.g. 5; IC50 for h-NAAA ∼0.34 μM).16 Nevertheless, functionalization of the amino group on the 3-aminoazetidin-2-one ring as a carbamic acid ester, along with systematic modification of the O-alkyl substituent, progressively increased activity and eventually yielded the potent inhibitor 6 (ARN726; Figure 2 and 4A; IC50: r-NAAA, 63±15 nM; h-NAAA, 27±3 nM; n=3 each). Our SAR studies will be reported elsewhere.
Figure 4.
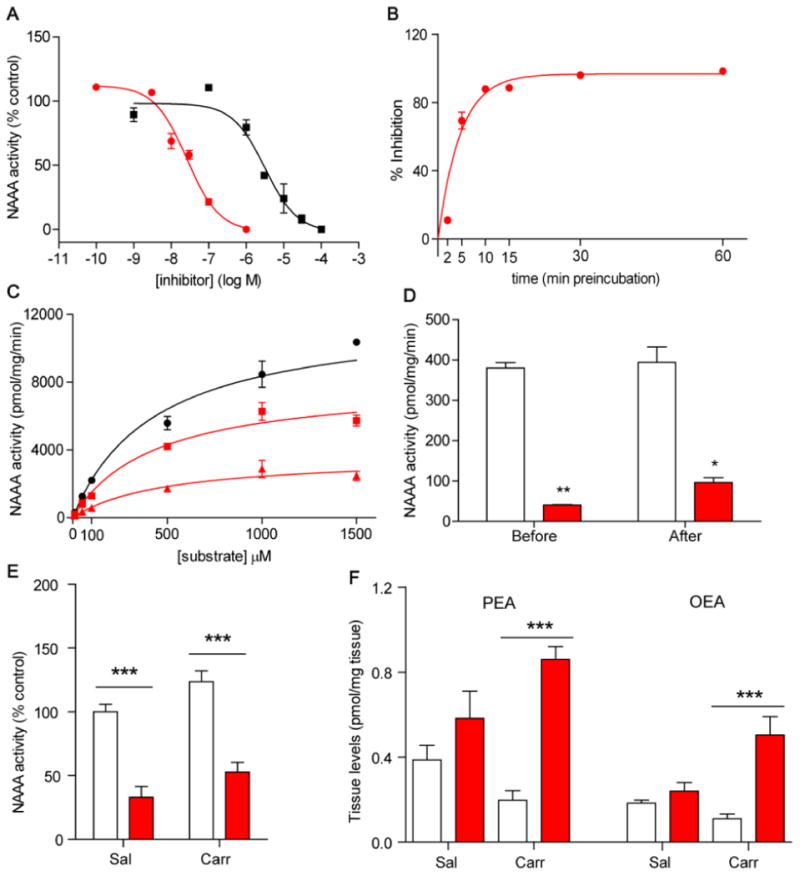
Compound 6 inhibits N-acylethanolamine acid amidase (NAAA) activity. A) Concentration-response curve for inhibition of h-NAAA by 6 (red circle) and its (R)-enantiomer 7 (black square). B) Time-course of h-NAAA inhibition by 6 (100 nM). C) Kinetic analysis of h-NAAA inhibition by 6 (vehicle, black circle; 10 nM, red square; 30 nM, red triangle). D) Effects of centrifugation dialysis on the inhibition of h-NAAA by 6. E) Effects of 6 (30 mg kg−1, oral) on ex vivo NAAA activity and F) palmitoylethanolamide (PEA) and oleoylethanolamide (OEA) levels in mouse lungs (vehicle, open bars; 6 red bars); saline (Sal) and carrageenan (Carr). Data are represented as mean ± SEM; * P<0.05; ** P<0.01; *** P<0.001.
As illustrated in Figure 3, the synthesis of (S)-6 and its less-active (R)-enantiomer 7 (IC50 for r-NAAA, ∼3.1±0.9 μM; for h-NAAA, ∼3.5±2 μM; n = 3) (Figure 4A) was accomplished in an enantioselective fashion by a strategy consisting in the coupling reaction of (S)- or (R)-2-oxoazetidin-3-yl ammonium acetate16 (11 and 12, respectively) with activated 4-cyclohexylbutan-1-ol (8).14
Figure 3.

Syntheses of compounds 6 (ARN726) and 7. Reaction conditions: a) 4-Dimethylaminopyridine (DMAP), di-2-pyridyl carbonate (2-DPC), dry CH2Cl2, room temperature, 16 h. b) (S)-11 or (R)-12, N,N-Diisopropylethylamine (DIPEA), dry CH2Cl2, room temperature, 16 h.
In vitro experiments with recombinant h-NAAA showed that the inhibitory effects of 6 (a) were maximal after 10 min of incubation with the enzyme (Figure 4B); (b) displayed non-competitive Michaelis-Menten kinetics (Figure 4C and Supplementary Table 1); and (c) were not affected by centrifugation dialysis (Figure 4D). These data suggest that 6 rapidly and irreversibly interacts with NAAA, possibly via covalent binding to the catalytic nucleophile (Cys131 in rodents, Cys126 in humans), as previously shown for the β-lactone 4.15
To test this idea, we incubated HEK-293 cells overexpressing r-NAAA with 6 or 7 for 1 h. After cell lysis, proteins were digested and the resulting peptides were analyzed by LC-MS/MS. Because 6 contains 3 sites susceptible to nucleophilic attack by cysteine, the compound could theoretically form three distinct adducts with NAAA. As illustrated in Figure 5A, nucleophilic attack of Cys131 on the endocyclic carbonyl would result in adduct 1, while attack on the carbamate moiety or the 2-position of the β-lactam ring would yield adducts 2 or 3, respectively. The results show that 6 reacts with the N-terminal cysteine of NAAA to form a covalent product (Figure 5B, red trace), which was unambiguously identified as adduct 1 (C131TSIVAQDSQGR, Figure 5C, Supplementary Figures 1A, B). Incubations with 7 produced only negligible amounts of the adduct (Figure 5B, black trace), while control incubations with DMSO yielded the unmodified native peptide (data not shown). Further MS/MS analyses ruled out formation of other potential adducts (Supplementary Figure 1).
Figure 5.
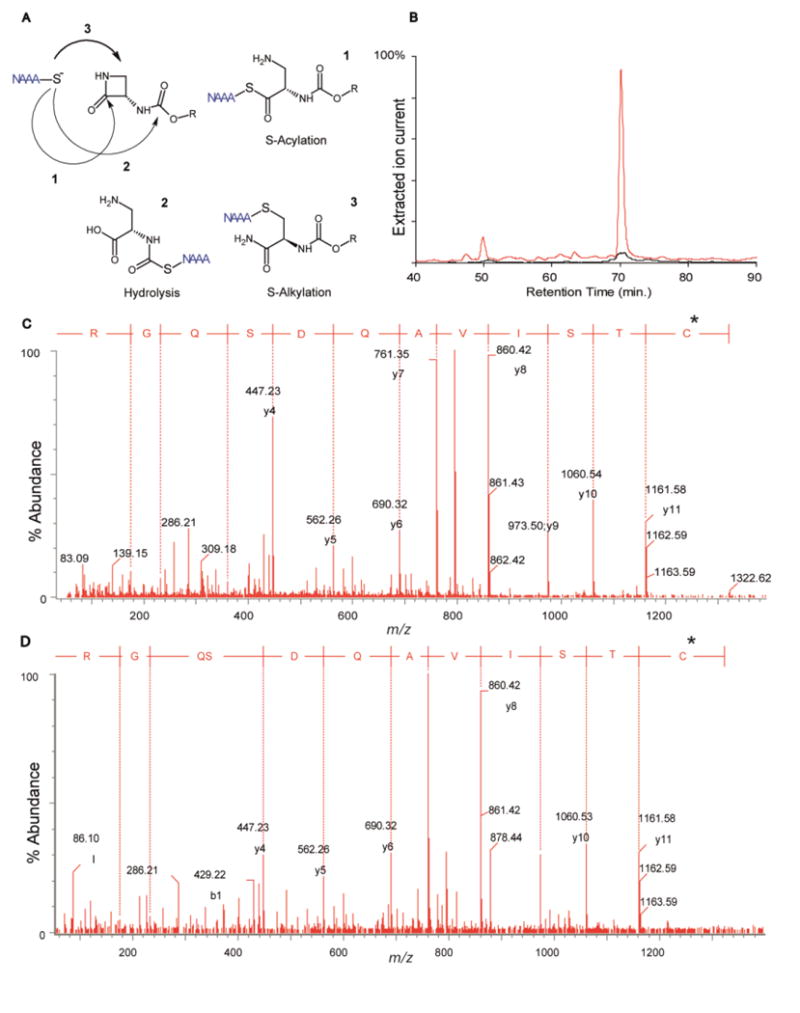
Compound 6 inhibits N-acylethanolamine acid amidase (NAAA) via S-acylation of the enzyme's catalytic cysteine. A) Nucleophilic attack of Cys131 on 6 can theoretically yield the three adducts 1–3. B) Extracted ion chromatograms of doubly-charged ion (m/z = 795.40) corresponding to the formation of adducts 1 of r-NAAA N-terminus with 6 (red trace) or 7 (black trace). C). MS/MS spectrum of adduct 1 of 6 with r-NAAA. The mass increase introduced by 6 (*) is carried by Cys131, confirming the proposed inhibition mechanism. D) MS/MS spectrum of the same adduct from rat lung, 3 h after intravenous administration of 6.
Under conditions that guarantee full NAAA inhibition (incubation time >10 min), compound 6 had no effect on rat FAAH (IC50>100 μM) and only marginally affected the activity of rat acid ceramidase (IC50 = 12.5±0.7 μM, n = 3), a cysteine amidase that is structurally related to NAAA.17 Furthermore, under similar conditions and a concentration of 10 μM, 6 did not interact with a panel of 28 biologically relevant targets comprising lipid-metabolizing and inflammation-related enzymes (Supplementary Table 2). Even further, 6 had no significant effect on 79 commonly targeted ion channels, membrane transporters and receptors (Supplementary Table 3). Collectively, the results outlined above indicate that 6 inhibits NAAA potently and selectively through a mechanism that requires S-acylation of the enzyme's catalytic cysteine.
Pharmacokinetic analyses revealed that 6 is quickly eliminated in vivo. After intravenous administration in mice (3 mg kg−1), the compound reached a maximal concentration in plasma (Cmax) of 1,608 ng mL−1 and disappeared from circulation with a t1/2 of ∼15 min. Its volume of distribution was 3,097 mL kg−1 and its clearance was 139 mL min−1 kg−1. Consistent with these data, 6 was efficiently metabolized in plasma (t1/2 =41±7 min in mouse; 12±6 min in rat; n = 3) and liver microsomes (t1/2<5 min in rat and mouse). Its primary metabolite in microsomes was identified as the product of β-lactam hydrolysis (data not shown).
Despite its rapid elimination, 6 effectively engaged NAAA in vivo. Intravenous injection of the compound in rats resulted in the rapid formation of covalent adduct NAAA in lungs (Figure 5D). Similarly, intraperitoneal (3–30 mg kg−1) (Supplementary Figure 2) or oral (30 mg kg−1) (Figure 4E) administration of 6 in mice was accompanied by a substantial inhibition of NAAA activity in various organs. As expected from prior work,9 the compound had no effect on PEA and OEA levels in the lungs of naïve mice, but markedly increased such levels in mice challenged with the inflammatory stimulus, carrageenan (Figure 4F). This state-dependent effect suggests that NAAA comes into contact with its endogenous substrates only during inflammation.9
Encouraged by the systemic activity of 6, we examined whether the compound affects inflammatory responses elicited in mice by carrageenan (Carr) or lipopolysaccharide (LPS). Intrapleural injection of carrageenan, but not its saline vehicle (Sal), triggered an inflammatory reaction characterized by elevated lung tissue myeloperoxidase (MPO) activity (Figure 6A), a marker of neutrophil infiltration, and tumor necrosis factor-α (TNF-α) levels in pleural exudate (Figure 6B). Compound 6 prevented both responses in a dose-dependent manner (1–30 mg kg−1, oral), whereas its (R)-enantiomer 7 (30 mg kg−1) had no such effect (Figure 6A, B). The maximal effect of 6 was comparable to that of the potent anti-inflammatory steroid dexamethasone (0.5 mg kg−1, intraperitoneal) (Figure 6A, B). Similarly to carrageenan, intranasal instillation of LPS increased leukocyte numbers in mouse lung bronchoalveolar lavage (Supplementary Figure 3A–C) as well as MPO activity (Supplementary Figure 3D) and expression of inducible nitric oxide synthase (iNOS) and MPO in lung tissue (Supplementary Figure 3E, F). Compound 6 (30 mg kg−1, oral) and dexamethasone (0.5 mg kg−1, i.p.) each suppressed these responses (Supplementary Figure 3A–F).
Figure 6.

Compound 6 blunts inflammatory responses in mouse lungs in vivo and human macrophages ex vivo. Effects of 6 (1–30 mg kg−1, oral), its (R)-enantiomer 7 (30 mg kg−1, oral), or dexamethasone (0.5 mg kg−1, i.p.) on carrageenan (Carr)-induced inflammation assessed as A) myeloperoxidase (MPO) activity in lung tissue and B) tumor necrosis factor (TNF)-α levels in pleural exudate. Peroxisome proliferator-activated receptor (PPAR)-α deletion eliminates the anti-inflammatory effects of 6: C) MPO activity in lungs and D) TNF-α levels in pleural exudate of PPAR-α-null mice or C57BL6/J wild-type (WT) littermates. E) N-acylethanolamine acid amidase (NAAA) expression in human granulocytes (CD15+), monocytes (CD14+), T-lymphocytes (CD3+), B-lymphocytes (CD19+), and NK cells (CD56+). F) NAAA expression in monocytes and monocyte-derived macrophages. G) Concentration-dependent effects of 6 on inducible nitric oxide synthase (iNOS) and H) TNF-α expression and I) TNF-α release by LPS-activated human macrophages. (A–D) Data are represented as mean ± SEM; # P<0.05 vs saline-treated mice (open bar; same genotype); * P<0.05, ** P<0.01, and *** P<0.0001 vs carrageenan-treated mice (shaded bar; same genotype); °°° P<0.0001 indicates differences in the responses between C57BL/6J WT and PPAR-α-null mice. (E–I) MFI=mean fluorescence intensity; # P<0.05 vs control macrophages (open bar) and * P<0.05 vs LPS-stimulated macrophages (shaded bar).
The effects of 6 were likely due to heightened PEA- and OEA-mediated signaling at PPAR-α. Supporting this conclusion, we found that 6: (a) increased PEA and OEA levels in carrageenan-treated mice (Figure 4F); and (b) did not prevent carrageenan-induced inflammatory responses in mutant PPAR-α-null mice (Figure 6C, D) which, as previously shown,18 were more sensitive to carrageenan than were wild-type controls (Figure 6C, D). Additionally, antagonism of CB1 or CB2 receptors did not prevent the anti-inflammatory effects of 6 (Supplementary Figure 4).
To test whether NAAA blockade by 6 modulates inflammatory reactions in human subjects, we first localized the enzyme in human blood-derived leukocytes using flow cytometry. As shown in Figure 6E, intracellular staining revealed high levels of mean fluorescence intensity (MFI) for NAAA in monocytes (571.7±29.04) and B lymphocytes (571.3±55.52), and moderate levels in T lymphocytes (232.7±32.24). Relatively low NAAA levels were found in granulocytes (94.72±19.54) and natural killer cells (53.91±7.67) (Supplementary Figure 5). The differentiation of human monocytes into macrophages was accompanied by an accrual in NAAA levels (Figure 6F). Stimulation of CD68+ macrophages with LPS increased both expression of iNOS and release of TNF-α, as assessed by flow cytometry and enzyme-linked immunoassay, respectively. Inclusion of 6 in the incubation medium resulted in a significant concentration-dependent inhibition of these responses (Figure 6G–I).
In sum, our results identify the β-lactam 6 as the first potent, selective and systemically active inhibitor of intracellular NAAA activity. We find that 6 blocks NAAA by covalently binding to the enzyme's catalytic cysteine, and displays pronounced anti-inflammatory properties in mouse models and human macrophages. Persistent inflammatory pathologies such as chronic obstructive pulmonary disease, rheumatoid arthritis and inflammatory bowel disease remain a medical challenge due to the limited efficacy and frequent side effects of existing therapies.19–22 Our work suggests that NAAA inhibitors such as 6 might inspire the discovery of new medicines that could fill this gap.
Methods
Chemicals
4-Cyclohexylbutyl-N-[(S)-2-oxoazetidin-3-yl]carbamate (compound 6)
4-Cyclohexylbutyl 2-pyridyl carbonate and 4-cyclohexylbutyl 2-oxopyridine-1-carboxylate
Under nitrogen atmosphere, to a stirred solution of commercially available 4-cyclohexyl-1-butanol (0.89 mL, 5.13 mmol) in dry CH2Cl2 (30 mL), DMAP (0.063 g, 0.51 mmol) and di-2-pyridyl carbonate (2-DPC) (1.33 g, 6.16 mmol) were added. The reaction mixture was stirred at r.t. for 16 h, then diluted with CH2Cl2 (50 mL) and sequentially washed with sat. NH4Cl solution (25 mL), sat. NaHCO3 solution (3 × 25 mL) and brine (25 mL). The organic layer was dried over Na2SO4, filtered and concentrated to dryness affording a pale yellow oil (1.45 g, quant.), as a mixture (1:1.7 ratio) of 4-cyclohexylbutyl 2-pyridyl carbonate and 4-cyclohexylbutyl 2-oxopyridine-1-carboxylate. The mixture of isomers was not separated and used in the next step without any further purification. MS (ESI) m/z: 278 [M-H]+, 300 [M-Na]+, 36 [M-K]+.
4-Cyclohexylbutyl-N-[(-S)-2-oxoazetidin-3-yl]carbamate
Under nitrogen atmosphere, to a suspension of [(S)-2-oxoazetidin-3-yl]-ammonium acetate (0.050 g, 0.34 mmol) in dry CH2Cl2 (4.0 mL), DIPEA (0.067 mL, 0.41 mmol) was added dropwise. Subsequently, the crude mixture (0.254 g) containing 4-cyclohexylbutyl 2-oxopyridine-1-carboxylate (0.094 g, 0.34 mmol) in dry CH2Cl2 (2.0 mL) was added. The reaction mixture was stirred at r.t. for 16 h, and then concentrated to dryness giving an oily residue (0.33 g). Purification by silica gel flash chromatography (cyclohexane/EtOAc 30:70) afforded pure 4-cyclohexylbutyl N-[(S)-2-oxoazetidin-3-yl]carbamate (0.053 g, 58%), as a white solid. -13.54 (c 0.09, MeOH). MS (ESI) m/z: 269 [M-H]+, 291 [M-Na]+, 307 [M-K]+. 1H NMR (DMSO-d6): δ 7.90 (s, 1H), 7.78 (d, 1H, J = 8.8 Hz), 4.67-4.50 (m, 1H), 3.94 (t, 2H, J = 6.7 Hz), 3.37 (t, 1H, J = 5.4 Hz), 3.06 (dd, 1H, J = 5.4, 2.8 Hz), 1.72-1.45 (m, 7H), 1.36-1.07 (m, 8H), 0.91-0.77 (m, 2H). 13C NMR (DMSO-d6): 168.2, 155.6, 64.1, 58.3, 42.6, 36.9, 36.5, 32.8, 28.9, 26.1, 25.8, 22.6.
4-Cyclohexylbutyl-N-[(R)-2-oxoazetidin-3-yl]carbamate (compound 7)
Under nitrogen atmosphere, to a suspension of [(R)-2-oxoazetidin-3-yl]-ammonium acetate (0.050 g, 0.34 mmol) in dry CH2Cl2 (4.0 mL), DIPEA (0.067 mL, 0.41 mmol) was added dropwise. Subsequently, the crude mixture (0.25 g) containing 4-cyclohexylbutyl 2-oxopyridine-1-carboxylate (0.094 g, 0.34 mmol) in dry CH2Cl2 (2.0 mL) was added. The reaction mixture was stirred at r.t. for 16 h, concentrated to dryness affording an oily residue (0.50 g). Purification by silica gel flash chromatography (cyclohexane/EtOAc 30:70) afforded pure 4-cyclohexylbutyl-N-[(R)-2-oxoazetidin-3-yl]carbamate (0.048 g, 53%), as a white solid. +12.87 (c 0.08 MeOH). MS (ESI) m/z: 269 [M-H]+, 291 [M-Na]+, 307 [M-K]+. 1H NMR (DMSO-d6): δ 7.90 (s, 1H), 7.78 (d, 1H, J = 8.8 Hz), 4.67-4.50 (m, 1H), 3.94 (t, 2H, J = 6.7 Hz), 3.37 (t, 1H, J = 5.4 Hz), 3.06 (dd, 1H, J = 5.4, 2.8 Hz), 1.72-1.45 (m, 7H), 1.36-1.07 (m, 8H), 0.91-0.77 (m, 2H). 13C NMR (DMSO-d6): 168.2, 155.6, 64.1, 58.3, 42.6, 36.9, 36.5, 32.8, 28.9, 26.1, 25.8, 22.6.
All of the commercially available reagents and solvents were used as purchased from vendors without further purification. Dry CH2Cl2 was purchased from Sigma-Aldrich. Optical rotations were measured on a Rudolf Research Analytical Autopol II Automatic polarimeter using a sodium lamp (589 nm) as the light source; concentrations are expressed in g/100 mL using CHCl3 as a solvent and a 1 dm cell. Column chromatography was performed on pre-packed silica cartridges (5 g) from Biotage. NMR spectra were recorded on a Bruker Avance III 400 system (400.13 MHz for 1H, and 100.62 MHz for 13C), equipped with a BBI inverse probe and Z-gradients, using deuterated solvents. Splitting parameters are designated as singlet (s), doublet (d), double doublet (dd), triplet (t), and multiplet (m). NMR coupling constants (J) are in Hertz.
UPLC-MS analyses were run on a Waters ACQUITY UPLC-MS system consisting of a SQD (Single Quadrupole Detector) Mass Spectrometer equipped with an Electrospray Ionization interface and a Photodiode Array Detector. UV spectra acquisition range was 210–400 nm. Analyses were performed on an ACQUITY UPLC HSS T3 C18 column (5X32.1 mm i.d., particle size 1.8 μm) with a VanGuard HSS T3 C18 pre-column (532.1 mm i.d., particle size 1.8 μm). Mobile phase was either 10mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN-H2O (95:5) at pH 5. Electrospray ionization in positive and negative mode was applied. All final compounds showed ≥ 95% purity by NMR and UPLC-MS analysis.
Human NAAA activity
Recombinant HEK-hNAAA cells were rinsed with PBS (pH 7.4), scraped from flasks, collected in 50 mL falcon tubes on ice and centrifuged at 150×g for 10 minutes at 4°C. The cell pellets were then suspended in 20 mM Tris-HCl buffer pH 7.4, 0.32 M sucrose, and sonicated. Samples were centrifuged at 800×g for 15 min at 4°C and the resulting supernatants were centrifuged at 12,000×g for 30 min at 4°C. The pellets were suspended in PBS on ice and subjected to 2 freeze/thaw cycles at −80°C. The suspensions were centrifuged at 105,000×g for 1 h at 4°C. Protein concentration was measured and samples stored at −80°C until use. Assay: as previously described for rat NAAA activity. Dialysis: Recombinant human NAAA was incubated in a buffer consisting of 100 mM NaH2PO4, 100 mM Sodium Citrate, 0.1% Triton-X 100, 3 mM DTT, pH 4.5 containing either vehicle (DMSO, 1%) or 6 (100 nM in DMSO 1%) at 37°C for 30 min. A sample was collected to determine NAAA activity (t=0) and the remaining was injected into dialysis cassettes (10 kDa molecular weight cut-off; Thermo Scientific) and dialyzed overnight in assay buffer under moderate stirring. DTT (3 mM) was added 1 h before the end of dialysis. After 16 h of dialysis, the samples were retrieved and assayed for NAAA activity.
Mouse NAAA activity
C57BL/6J male mice were treated with 6 or vehicle and 2 h later were killed for samples collection. Lung, spleen, and brain samples were dissected, minced over ice, and transferred into ice-cold Tris-HCl buffer (50 mM, pH 7.5) containing 0.32 M sucrose (final volume-to-weight ratio, 9:1). Samples were homogenized, centrifuged at 1,000×g for 15 minutes at 4°C, and the supernatants were ultracentrifuged at 12,000×g for 30 minutes at 4°C. The pellets were suspended in 10 mM phosphate-buffered saline (pH 7.4) on ice and subjected to two freeze/thaw cycle at −80°C. Suspensions were centrifuged at 105,000×g for 1 hour at 4°C. Protein concentration was measured in the supernatant, and samples were stored at −80°C until used. Protein preparations (50 μg for lung and spleen, 100 μg for brain) were suspended in NAAA assay buffer (0.1 M NaH2PO4, 0.1 M sodium citrate, 0.1% Triton-X 100, 3 mM dithiothreitol [DTT], pH 4.5) and mixed with the enzyme substrate (10-cis-heptadecenoylethanolamide, 50 μM). Reactions (in duplicate) were incubated for 30 minutes at 37°C and stopped by the addition of 0.2 mL ice-cold methanol containing 1 nmol heptadecanoic acid (NuChek Prep) as internal standard. Analyses of the newly formed heptadecenoic acid (17:1) were conducted by liquid chromatography/mass spectrometry.
Lipid extractions
Tissue PEA and OEA levels were quantified as previously described.23 Briefly, frozen lungs were weighed (approximately 70 mg) and homogenized in methanol (1 mL) containing [2H4]-PEA and [2H4]-OEA as internal standards. Lipids were extracted with chloroform (2 mL) and washed with water (1 mL). Following centrifugation (3000 rpm for 15 min at 4°C), organic phases were collected and dried under a stream of nitrogen. The organic extracts were fractionated by silica gel column chromatography. PEA and OEA were eluted with chloroform/methanol (9:1, v/v). Organic phases were evaporated under nitrogen and reconstituted in 100 μL of chloroform/methanol (1:3, v/v). Levels of PEA and OEA were measured using a Xevo TQ UPLC-MS/MS system (Waters), equipped with a reversed phase BEH C18 column (Waters), using a linear gradient of acetonitrile in water. Quantification was performed monitoring the following MRM transitions (parent m/z ->daughter m/z, collision energy eV): OEA 326->62,20; OEAd4 330->66,20; PEA 300->62,20; PEAd4 304->66,20. Analyte peak areas were compared with a standard calibration curve (1nM to 10 μM).
NAAA acylation in r-NAAA-overexpressing HEK-293 cells
HEK-293 cells stably transfected with r-NAAA were incubated for 1 hour at 37°C with 10 μM of 6 or 7 (0.1% final DMSO). After washing and cell lysis, the total protein extract was reduced with 50 mM Dithiothreitol (55°C for 30 minutes) and then alkylated with 100 mM iodacetamide (RT for 45 min). The proteins were then precipitated in cold acetone and the resulting pellet was resuspended in 50 mM ammonium bicarbonate pH 8. Trypsin (Porcine, proteomics grade; Sigma Aldrich) was then added to a final 1 to 50 ratio (w/w) with the protein mixture and incubated overnight at 37°C. The resulting peptides were then analyzed by high-resolution LC-MS/MS: an aliquot (5 μL) of the surnatant was loaded on a UPLC nano-LC chromatographic system equipped with a BEH C18 reversed phase column (0.75×100 mm). Peptides were eluted with a linear gradient of acetonitrile in water (both added with 0.1% formic acid) from 3 to 50% in 50 minutes. Flow rate was set to 300 nL per minute. Eluted peptides were analyzed in positive ion mode by high resolution tandem mass spectrometry on a Synapt G2 qTOF mass spectrometer (UPLC, column and qTOF; Waters). A linear ramp of the collision energy from 15 to 45 eV was used to induce backbone fragmentation of the eluted peptides.
In vivo NAAA acylation
Compound 6 was dissolved in PEG400/Tween 80/Saline solution at 10/10/80 % (v/v) respectively and administered intravenously (i.v.) to rats at 10 mg kg−1. After 1 h, rats were sacrificed and lungs were immediately dissected, frozen on dry ice, and stored at −80°C until analyses. Lungs were then homogenized in PBS pH 7.4 containing 0,32M sucrose using an IKA T-18 Ultraturrax homogenizer. Samples were then centrifuged 25 min at 800×g. The supernatant was further centrifuged for 30 min at 12,000×g at 4°C. The obtained pellets were resuspended in two volumes of PBS and subjected to two freeze/thaw cycles at −80°C to solubilize proteins. The suspensions were centrifuged at 105,000×g for 1 h at 4°C and the soluble fraction was taken. Protein concentration was measured by BCA protein assay (Euroclone) and samples were stored at −80°C until use. Protein samples (30 μg/lane) were first analyzed by western blot to detect NAAA expression (anti-hASAHL, R&D Systems) and then resolved by SDS-PAGE (3 lanes per sample, 50 μg each) and stained with coomassie blue method. Gel slices corresponding to NAAA molecular weight were excised, washed by cycles of dehydration with acetonitrile and rehydration with 100 mM NH4HCO3, reduced with 10 mM DTT, and alkylated with 55 mM IAA. Finally dried gel slices were rehydrated with a solution of 50 mM NH4HCO3, 5 mM CaCl2 and incubated with 12.5 ng μL−1 trypsin overnight at 37°C. Tryptic peptides were recovered and gel slices were further extracted with 5% of formic acid, the extracts were dried in speed vac. The resulting peptides were then analyzed by high-resolution LC-MS/MS: an aliquot (5 μL) of the supernatant was loaded on a UPLC nano-LC chromatographic system equipped with a BEH C18 reversed phase column (0.75X100 mm). The resulting peptides were then analyzed by high-resolution LC-MS/MS: an aliquot (5 μL) of the surnatant was loaded on a UPLC nano-LC chromatographic system equipped with a BEH C18 reversed phase column (0.75×100 mm). Peptides were eluted with a linear gradient of acetonitrile in water (both added with 0.1% formic acid) from 3 to 50% in 50 minutes. Flow rate was set to 300 nL per minute. Eluted peptides were analyzed in positive ion mode by high resolution tandem mass spectrometry on a Synapt G2 qTOF mass spectrometer (UPLC, column and qTOF; Waters). A linear ramp of the collision energy from 15 to 45 eV was used to induce backbone fragmentation of the eluted peptides.
Compound 6 in vitro selectivity profile
Compound 6 selectivity profile was conducted on 107 different enzymes, receptors, ion channels, and transporters (see Table S1 and S2) by a contract research organization (CEREP) using established protocols. IC50 were determined in case significant activity was observed at 10 μM (≥50% inhibition). For detailed information of the experimental conditions of the assays refer to the following URL: http://www.cerep.fr/cerep/users/pages/catalog/p_Catalogue.asp?classetest=33&TypCall=cat.
Pharmacokinetic analyses
Compound 6 was dissolved in PEG400/Tween 80/Saline solution at 10/10/80 % (v/v) and administered by the route (3 mg kg−1). Blood samples (200 μL) were collected after decapitation at 0, 15, 30, 60, and 120 min after treatment into tubes containing heparin sodium salt (20 μL, 5000 U mL−1; Sigma Aldrich) as anticoagulant. Plasma fractions were immediately separated by centrifugation (3000×g for 10 min, 4°C), frozen on dry ice, and stored at −20°C until analyses. Sample preparation: after a short centrifugation, mouse plasma samples (50 μL) were transferred into Eppendorf tubes and diluted with 150 μL of acetonitrile spiked with I.S. to a final 200 nM concentration. After vortexing (30 s) samples were centrifuged at 3,000×g for 10 min at 4°C; 80 μL of supernatant was diluted in a 1:1 proportion (v/v) with water. Calibration curve and quality controls: 6 was spiked in net solvent (PBS (pH 7.4) with 10% acetonitrile) to prepare a calibration curve ranging from 1 nM to10 μM. Three quality control samples were also prepared by spiking mouse plasma with 6 to final concentrations of 20 nM, 200 nM and 2000 nM. Calibrators and quality controls were crashed with acetonitrile spiked with I.S. as described for plasma samples. Calibrators, quality controls and samples were analyzed in triplicate. Plasma levels of 6 were measured with a Xevo TQ UPLC-MS/MS system (Waters), using a calibration curve and Warfarin as an internal standard (I.S.). Chromatography was carried out on an Acquity BEH C18 column (2.1 mm × 30 mm, 1.7 μm particle size; Waters). Flow rate was at 0.7 mL per min with a gradient from 10% solvent B to 100% solvent B in 1 min, and hold for 20 s. After the gradient, system was reconditioned at 10% solvent B for 30 sec (Solvent A = water plus formic acid 0.1%; solvent B = acetonitrile plus formic acid 0.1%). Mass spectrometry parameters: Positive ion mode. Capillary 3 KV, Cone 30 V, Source temperature 120°C, Cone gas 20 L/Hr, Desolvation gas 800 L/Hr, Desolvation temperature 400°C. The following multiple reaction monitoring transitions were followed: 6: 269 -> 241, 241 -> 103, 241 -> 59 at 25 eV of collision energy; Warfarin (I.S.): 309 -> 163 and 309 -> 251 at 18 eV and 16 eV of collision energy, respectively. The linear regression of the calibration curve yielded an R2 value of 0.995. The recovery of the quality controls (back-calculated from the regression curve) ranged from 85 to 95%. Pharmacokinetic data calculation: PK data were calculated on the basis of the time versus plasma concentration profiles of 6 using PK Solutions software version 2.0 (Summit Research Services).
Animals and treatments
Male CD1 mice (25–30 g) and male C57BL/6J mice homozygous for the PparαtniJGonz-targeted mutation and wild type (WT) littermates were used in all experiments of carrageenan-induced inflammation. C57BL/6J mice (22–25 g) were used in all experiments of LPS-induced inflammation. All mice were obtained from Charles River laboratories. Naïve Sprague-Dawley male rats, weighing 175–200 g (Charles River) were used for extraction of r-NAAA from lungs. Animals were housed in a temperature and humidity controlled room under a 12 h light/dark cycle with water and food ad libitum. All procedures were performed in accordance with the Italian regulations on the protection of animals used for experimental and other scientific purposes (D.M. 116192), and European Economic Community regulations (O.J. of E.C. L 358/1 12/18/1986). Animals were treated with 6 (0.1–30 mg kg−1), 7 (30 mg kg−1) or vehicle (PEG400/Tween 80/Saline solution at 10/10/80 %, v/v) by oral gavage 30 min before carrageenan or LPS. Dexamethasone (0.5 mg kg−1; Sigma Aldrich) or saline solutions were given i.p. 1 h before the inflammatory insult. CB1 (AM251, Sigma Aldrich) and CB2 (AM630, Tocris) receptor antagonists were administered at 1 mg kg−1 (i.p.) 30 min before the treatment with 6 or vehicle.
Carrageenan-induced inflammation
CD1 mice received saline or λ-carrageenan solution (2%, 0.1 mL; Sigma Aldrich) into the pleural cavity at the level of sixth intercostal space.24 Four h later, the animals were sacrificed by CO2 inhalation. The chest was opened and lungs were collected following perfusion with sterile PBS (10 mL), snap-frozen in liquid nitrogen and stored at −80°C until analysis.
Myeloperoxidase activity
Myeloperoxidase (MPO) activity in lung tissue was assayed as described elsewhere.25 Lung samples were homogenized in potassium phosphate buffer (1 mL, 5 mM, pH 6) and then centrifuged at 21,000×g for 30 min at 4°C. The supernatants were discarded, and the pellets were washed again as described above. To extract MPO, the pellets were suspended in 0.5% hexadecyltrimethylammonium bromide (Sigma Aldrich) in potassium phosphate buffer (50 mM, pH 6) at a tissue w/v ratio of 1/10 and frozen at −40°C. Three freeze/thaw/sonication cycles were performed. Samples were incubated at 4°C for 10 min and then centrifuged at 12,500×g for 15 min at 4°C. Supernatants were collected and 7 μL were allowed to react with 200 μL of a solution containing 167 mg L−1 o-dianisidine dihydrochloride (Sigma Aldrich) and 0.0005% hydrogen peroxide (Sigma Aldrich) in potassium phosphate buffer (50 mM, pH 6). The change in absorbance was recorded at intervals of 30 s for 2 min at 460 nm using a microplate reader. One unit of MPO activity was defined as the quantity of enzyme degrading 1 μmol of hydrogen peroxide per minute at 25°C.
TNF-α ELISA
ELISA kits were used to measure TNF-α levels on mouse pleural exudate (BioLegend) and human macrophages cell culture supernatant (eBioscience). The assays were performed according to the manufacturer's instructions.
Flow cytometry
Whole blood was obtained from adult healthy donors by venous puncture after informed consent was obtained. Total leukocytes were analyzed following removal of red blood cells with Lysing Buffer (BD Biosciences). For NAAA expression, 1×106 cells were washed twice with PBS and stained with fluorescence-conjugated antibodies against specific markers of immune cells for 15 min in staining buffer (PBS supplemented with 0.5% FCS and 0.02% NaN3) at 4°C (CD15-APC and CD68-FITC from Miltenyi Biotec; CD3 e780 from eBioscience; CD19 PE-Cy7 and CD56 ECD from Beckman Coulter). Cells were then fixed with 4% formaldehyde for 15 min, stained intracellularly with anti-NAAA antibody (1:100) for 20 min, washed twice with cold PBS and stained with host-specific Alexa-488 secondary antibody (1:200) for additional 20 min. For cytokine production, monocyte-derived macrophages were pretreated with various concentrations of 6 or vehicle (DMSO, 0.1%) for 30 min and then challenged with 100 ng mL−1 LPS for 6 h in the presence of 1 μg ml−1 brefeldin A (Sigma Aldrich) added to prevent cytokine-containing vesicles exocytosis. At the end of the incubation, cells were stained with FITC-conjugated anti-CD68, fixed with 4% formaldehyde for 15 min and intracellularly stained with PE-conjugated anti-TNF-α (1:100) for 20 min (TNF-α-PE from Miltenyi Biotec). Intracellular expression of NAAA in the different immune cells and of cytokine within macrophages was analyzed by flow cytometry in a FACS-Cyan ADP (Beckman Coulter), as reported.26 For each analysis, at least 300,000 events were acquired and viable cells were analyzed by using the FlowJo software version 7.6 (TreeStar).
Isolation of human monocytes and differentiation into macrophages
Monocytes were isolated from human whole blood using positive selection with CD14 microbeads and sorted on an AutoMACSPro Separator (Miltenyi Biotech). Cells were constantly controlled for their purity (always >90%) by means of flow cytometry. For monocyte-to-macrophage differentiation, monocytes were cultured in RPMI with 10 ng mL−1 human GM–CSF plus 10 ng ml−1 M-CSF (R&D Systems) at 37 °C for 7 days.
Supplementary Material
Acknowledgments
We thank S. Mandrup-Bertozzi, S. Venzano, M. Summa, T. De Vita, and L. Goldoni for experimental help; and the American Asthma Foundation and National Institutes of Health (grant DA012413 to D.P.) for financial support.
Footnotes
Note: Authors declare the following financial interest. AF, AN, GT, MM, FB, TB and DP are inventors in patent applications filed by the University of California and the Fondazione Istituto Italiano di Tecnologia, which protect composition and use of chemicals described in the present study.
Supporting information: Supporting information includes methods, Supplementary Figures 1–5, and Supplementary Tables 1–3. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Piomelli D, Sasso O. Peripheral gating of pain signals by endogenous lipid mediators. Nat Neurosci. 2014;17:164–74. doi: 10.1038/nn.3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodríguez De Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 3.Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, Piomelli D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67:15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- 4.D'Agostino G, La Rana G, Russo R, Sasso O, Iacono A, Esposito E, Raso GM, Cuzzocrea S, Lo Verme J, Piomelli D, Meli R, Calignano A. Acute intracerebroventricular administration of palmitoylethanolamide, an endogenous peroxisome proliferator-activated receptor-alpha agonist, modulates carrageenan-induced paw edema in mice. J Pharmacol Exp Ther. 2007;322:1137–1143. doi: 10.1124/jpet.107.123265. [DOI] [PubMed] [Google Scholar]
- 5.Blankman JL, Cravatt BF. Chemical probes of endocannabinoid metabolism. Pharmacol Rev. 2013;65:849–71. doi: 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ueda N, Yamanaka K, Yamamoto S. Purification and Characterization of an Acid Amidase Selective for N-Palmitoylethanolamine, a Putative Endogenous Anti-inflammatory Substance. J Biol Chem. 2001;276:35552–35557. doi: 10.1074/jbc.M106261200. [DOI] [PubMed] [Google Scholar]
- 7.Sasso O, Moreno-Sanz G, Martucci C, Realini N, Dionisi M, Mengatto L, Duranti A, Tarozzo G, Tarzia G, Mor M, Bertorelli R, Reggiani A, Piomelli D. Antinociceptive effects of the N-acylethanolamine acid amidase inhibitor ARN077 in rodent pain models. Pain. 2013;154:350–360. doi: 10.1016/j.pain.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khasabova IA, Xiong Y, Coicou LG, Piomelli D, Seybold V. Peroxisome proliferator-activated receptor alpha mediates acute effects of palmitoylethanolamide on sensory neurons. J Neurosci. 2012;32:12735–12743. doi: 10.1523/JNEUROSCI.0130-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solorzano C, Zhu C, Battista N, Astarita G, Lodola A, Rivara S, Mor M, Russo R, Maccarrone M, Antonietti F, Duranti A, Tontini A, Cuzzocrea S, Tarzia G, Piomelli D. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc Natl Acad Sci U S A. 2009;106:20966–20971. doi: 10.1073/pnas.0907417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrosino S, Ahmad A, Marcolongo G, Esposito E, Allarà M, Verde R, Cuzzocrea S, Di Marzo V. Diacerein is a potent and selective inhibitor of palmitoylethanolamide inactivation with analgesic activity in a rat model of acute inflammatory pain. Pharmacol Res. 2014;91C:9–14. doi: 10.1016/j.phrs.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Yang L, Chen L, Zhu C, Huang R, Zheng X, Qiu Y, Fu J. Design and synthesis of potent N-acylethanolamine-hydrolyzing acid amidase (NAAA) inhibitor as anti-inflammatory compounds. PLoS One. 2012;7 doi: 10.1371/journal.pone.0043023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuboi K, Hilligsmann C, Vandevoorde S, Lambert DM, Ueda N. N-cyclohexanecarbonylpentadecylamine: a selective inhibitor of the acid amidase hydrolysing N-acylethanolamines, as a tool to distinguish acid amidase from fatty acid amide hydrolase. Biochem J. 2004;379:99–106. doi: 10.1042/BJ20031695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bandiera T, Ponzano S, Piomelli D. Advances in the discovery of N-acylethanolamine acid amidase inhibitors. Pharmacol Res. 2014 doi: 10.1016/j.phrs.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ponzano S, Bertozzi F, Mengatto L, Dionisi M, Armirotti A, Romeo E, Berteotti A, Fiorelli C, Tarozzo G, Reggiani A, Duranti A, Tarzia G, Mor M, Cavalli A, Piomelli D, Bandiera T. Synthesis and structure-activity relationship (SAR) of 2-methyl-4-oxo-3- oxetanylcarbamic acid esters, a class of potent N-acylethanolamine acid amidase (NAAA) inhibitors. J Med Chem. 2013;56:6917–6934. doi: 10.1021/jm400739u. [DOI] [PubMed] [Google Scholar]
- 15.Armirotti A, Romeo E, Ponzano S, Mengatto L, Dionisi M, Karacsonyi C, Bertozzi F, Garau G, Tarozzo G, Reggiani A, Bandiera T, Tarzia G, Mor M, Piomelli D. β-Lactones inhibit N -acylethanolamine acid amidase by S-acylation of the catalytic N-terminal cysteine. ACS Med Chem Lett. 2012;3:422–426. doi: 10.1021/ml300056y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fiasella A, Nuzzi A, Summa M, Armirotti A, Tarozzo G, Tarzia G, Mor M, Bertozzi F, Bandiera T, Piomelli D. 3-Aminoazetidin-2-one derivatives as N-acylethanolamine acid amidase (NAAA) inhibitors suitable for systemic administration. ChemMedChem. 2014;9:1602–1614. doi: 10.1002/cmdc.201300546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuboi K, Sun YX, Okamoto Y, Araki N, Tonai T, Ueda N. Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. J Biol Chem. 2005;280:11082–11092. doi: 10.1074/jbc.M413473200. [DOI] [PubMed] [Google Scholar]
- 18.Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature. 1996;384:39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- 19.Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012;379:1341–51. doi: 10.1016/S0140-6736(11)60968-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baumgart DC, Sandborn WJ. Crohn's disease. Lancet. 2012;380:1590–605. doi: 10.1016/S0140-6736(12)60026-9. [DOI] [PubMed] [Google Scholar]
- 21.Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380:1606–19. doi: 10.1016/S0140-6736(12)60150-0. [DOI] [PubMed] [Google Scholar]
- 22.O'Shea JJ, Laurence A, McInnes IB. Back to the future: oral targeted therapy for RA and other autoimmune diseases. Nat Rev Rheumatol. 2013;9:173–82. doi: 10.1038/nrrheum.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Astarita G, Piomelli D. Lipidomic analysis of endocannabinoid metabolism in biological samples. J Chromatogr B Anal Technol Biomed Life Sci. 2009 doi: 10.1016/j.jchromb.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crisafulli C, Bruscoli S, Esposito E, Mazzon E, Di Paola R, Genovese T, Bramanti P, Migliorati G, Cuzzocrea S. PPAR-alpha contributes to the anti-inflammatory activity of 17beta-estradiol. J Pharmacol Exp Ther. 2009;331:796–807. doi: 10.1124/jpet.109.156646. [DOI] [PubMed] [Google Scholar]
- 25.Couturier JY, Ding-Zhou L, Croci N, Plotkine M, Margaill I. 3-Aminobenzamide reduces brain infarction and neutrophil infiltration after transient focal cerebral ischemia in mice. Exp Neurol. 2003;184:973–980. doi: 10.1016/S0014-4886(03)00367-4. [DOI] [PubMed] [Google Scholar]
- 26.Chiurchiù V, Cencioni MT, Bisicchia E, De Bardi M, Gasperini C, Borsellino G, Centonze D, Battistini L, Maccarrone M. Distinct modulation of human myeloid and plasmacytoid dendritic cells by anandamide in multiple sclerosis. Ann Neurol. 2013;73:626–636. doi: 10.1002/ana.23875. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.