

Abstract
Energy dense “Western” diets (WD) are known to cause obesity as well as learning and memory impairments, blood-brain barrier damage, and psychological disturbances. Impaired glucose (GLUT1) and monocarboxylate (MCT1) transport may play a role in diet-induced dementia development. In contrast, ketogenic diets (KD) have been shown to be neuroprotective. We assessed the effect of 10, 40 and 90 days WD, KD and Chow maintenance on spontaneous alternation (SA) and vicarious trial and error (VTE) behaviors in male rats, then analyzed blood glucose, insulin, and ketone levels; and hippocampal GLUT1 and MCT1 mRNA. Compared to Chow and KD, rats fed WD had increased 90 day insulin levels. SA was decreased in WD rats at 10, but not 40 or 90 days. VTE was perturbed in WD-fed rats, particularly at 10 and 90 days, indicating hippocampal deficits. WD rats had lower hippocampal GLUT1 and MCT1 expression compared to Chow and KD, and KD rats had increased 90 day MCT1 expression compared to Chow and WD. These data suggest that WD reduces glucose and monocarboxylate transport at the hippocampus, which may result in learning and memory deficits. Further, KD consumption may be useful for MCT1 transporter recovery, which may benefit cognition
Keywords: Obesity, GLUT1, MCT1, Vicarious Trial and Error, Spontaneous Alternation, Hippocampus
Introduction
In the United States more than one in three adults and one in six children can be classified as obese (Flegal, Carroll, Ogden, & Curtin, 2010; Ogden, Carroll, Kit, & Flegal, 2014). Obesity during childhood typically persists into adulthood (Deshmukh-Taskar et al., 2005; Wang, Beydoun, Liang, Caballero, & Kumanyika, 2008), and is associated with increased disease risk as well as neurological and psychological deficits (Agranat-Meged et al., 2005; Cserjési, Molnár, Luminet, & Lénárd, 2007; Kamijo et al., 2014; Yau, Castro, Tagani, Tsui, & Convit, 2012).
Though obesity and the metabolic syndrome can develop via multiple routes, there is evidence that “Western” diets (WD) high in fat and sugar play a substantial role in their etiologies, as well as the often comorbid learning and memory deficits. In adult humans, WD not only induces weight gain, but also increases risk for dementia (de la Monte, 2009; Whitmer, Gunderson, Barrett-Connor, Quesenberry, & Yaffe, 2005). Hippocampal function appears to be particularly vulnerable to dietary insult. For example, adult rats fed WD diet show impairments on radial maze learning (Kanoski, Zhang, Zheng, & Davidson, 2010), Morris water maze performance (Molteni, Barnard, Ying, Roberts, & Gomez-Pinilla, 2002; Pistell et al., 2010; Stranahan et al., 2008; A. Wu, Ying, & Gomez-Pinilla, 2004), spontaneous alternation (Kaczmarczyk et al., 2013), deprivation-discrimination learning (Sample, this issue), reversal learning (Kanoski, Meisel, Mullins, & Davidson, 2007); and feature negative discrimination (Davidson et al., 2013; Davidson et al., 2012; Kanoski et al., 2010) but are not impaired on tasks such as simple discrimination and non-spatial maze learning, which do not depend on the hippocampus (Davidson et al., 2013; Kanoski et al., 2007; Kanoski et al., 2010). WD also reduces hippocampal BDNF (Molteni et al., 2002; A. Wu et al., 2004) and increases hippocampal blood-brain barrier (BBB) permeability (Davidson et al., 2013; Davidson et al., 2012; Kanoski et al., 2010).
These deficits do not appear to be specific to adult animals. In juvenile rats, two months WD exposure is associated with impairments in spatial reference memory on the Morris water maze, delayed spatial reference learning, and an enhancement of lipopolysaccharide (LPS)-induced inflammation in the hippocampus (Boitard et al., 2014; Boitard et al., 2012). Compared to their lean counterparts, obese human children also show impairments in academic performance and deficits in the inhibitory “NoGo” task (Kamijo et al., 2012), while adolescents with the metabolic syndrome have lower overall IQ, impaired attentional abilities, reduced mental flexibility, smaller hippocampal volumes, and damaged white matter tracts (Yau et al., 2012), suggesting that the hippocampus is vulnerable to dietary insults throughout development.
We have noted previously (Davidson et al., 2013; Kanoski et al., 2007) that diet composition plays a significant role in the development of these cognitive deficits. We have also noted that rats fed a very high fat ketogenic diet (KD) did not show hippocampal impairments when circulating ketone bodies were high, despite markedly increased body adiposity levels (Davidson et al., 2013).
KD is known to be an effective treatment for many types of epilepsy (Barañano & Hartman, 2008; Kwon, Jeong, Kim, Choi, & Son, 2008; Lutas & Yellen, 2012), and has been shown to exert numerous other neurological benefits. For instance, peripheral inflammation was significantly reduced in obese men after 12 weeks maintenance on KD (Forsythe et al., 2008), even prior to the onset of weight loss (Sharman et al., 2002; Sharman & Volek, 2004; Volek et al., 2004). Adults with Alzheimer’s disease, probable Alzheimer’s disease, mild cognitive impairment, and age-associated memory impairments have demonstrated improvements after consuming AC-1202, a medical food known to increase circulating ketone levels (Costantini, Vogel, Barr, & Henderson, 2007; Henderson et al., 2009).
Ketones act as an alternate energy source for neurons, particularly when there are problems with glucose transport. In DeVivo syndrome, which is characterized by a mutation to the SLC2A-1 gene, GLUT1 transporter levels are reduced or abolished, thereby preventing glucose from crossing the BBB. Individuals with DeVivo syndrome have a cluster of neurological and behavioral symptoms, including seizures, developmental delays, and cognitive deficits that include spatial deficits, suggesting that the hippocampus may be particularly affected by this condition (Akman et al., 2010; De Vivo & Wang, 2008). The majority of these symptoms can be improved or averted via maintenance on KD (Friedman et al., 2006; Ramm-Pettersen, Stabell, Nakken, & Selmer, 2014). This suggests that many of the deficits in De Vivo syndrome are not due to reduced glucose transport per se, but a general energy deficit within the brain which can be overcome by switching to a ketone metabolism.
Under normal circumstances, GLUT1 expression increases in response to low, and decreases in response to high, circulating glucose levels, thereby maintaining glucose homeostasis within the brain interstitial fluid (Kumagai, Kang, Boado, & Pardridge, 1995; Pardridge, Triguero, & Farrell, 1990). When glucose levels are low, the primary source of energy for neuronal tissues becomes monocarboxylates, which include lactate, pyruvate, leucine, and ketone bodies such as beta-hydroxybutyrate (BHB), all of which are escorted across the BBB via the MCT1 transporter, which unlike GLUT1 is typically regulated via positive feedback mechanisms in proportion to monocarboxylates (Cortes-Campos et al., 2011; Nehlig, 2004; Vannucci & Simpson, 2003). Therefore, under conditions of starvation (where glucose levels are low, but ketone bodies are relatively high), expression of both GLUT1 and MCT1 are increased. Low levels of both transporters have been associated with cognitive deficits (De Vivo & Wang, 2008; Ding, Yao, Rettberg, Chen, & Brinton, 2013). Low GLUT1 levels have been measured in patients with AD (Bailey, Rivara, Rocher, & Hof, 2004; Mooradian, Chung, & Shah, 1997; Z. Wu et al., 2005), particularly in regions such as the hippocampus and neocortex (Kalaria & Harik, 1989); MCT1 levels are significantly lower in mouse models of AD (Ding, Yao, Rettberg, et al., 2013).
While it is unknown whether this is the result of degeneration at the neurovascular unit or an adaptive response to chronic hyperglycemia, it is clear that AD and related syndromes are associated with progressive reductions in glucose metabolism at the hippocampus and cortex (de la Monte, 2009; Mosconi, 2005; Mosconi et al., 2009), which suggests that the dementia-afflicted neurons may be deprived of energy for extended periods of time. Because GLUT1 and MCT1 transporters can be regulated quickly and play a role in cognitive function, we hypothesized that they may play a role in WD-induced cognitive deficits.
These experiments were conducted with three objectives in mind. First, we assessed the role of short- (10 day), moderate- (40 day) and long-term (90 day) exposure to WD and KD on body weight, markers of the metabolic syndrome, and circulating BHB levels. Our second goal was to determine the relationship between diet and exposure duration on behavior. We accomplished this by analyzing spontaneous alternation (SA) behavior, a task which is noninvasive and, importantly, requires no food restriction. Because we wished to investigate changes in hippocampal functioning as a result of diet, we assessed vicarious trial and error (VTE) behaviors during the SA task.
VTE is measured by recording the total number of head turns an animal performs at choice points during a maze task. There is evidence (Schmidt, Papale, Redish, & Markus, 2013) that these behaviors are analogous to the “mental time travel” humans engage in during task acquisition (Suddendorf & Corballis, 2007), whereby an individual accesses declarative memories of past events in order to predict future outcomes. During planning and goal-directed learning, the hippocampus is strongly activated (Addis, Moscovitch, & McAndrews, 2007; Addis & Schacter, 2008; Buckner & Carroll, 2007), and animals with hippocampal lesions do not engage in VTE behaviors (Hu & Amsel, 1995; Voss et al., 2011). Therefore, this measure served as an index of hippocampal involvement.
Finally, because we hypothesized that high circulating glucose levels could decrease expression of GLUT1 transporter at the hippocampus, and because we were interested in the effects of WD and KD on the MCT1 transporter, we performed qPCR on hippocampal homogenates for GLUT1 and MCT1 mRNA expression.
Materials and Methods
Animals and Diets
Male Sprague-Dawley rats (n = 93; Harlan Laboratories, Indianapolis, IN) weighing between 275-300g were housed individually in hanging-wire cages in a temperature- and humidity-controlled room, and maintained on a 12h light/dark cycle. Rats were provided with ad libitum tap water. All procedures were approved by the Purdue University Animal Care and Use Committee.
Animals were handled for two weeks prior to experimentation, and regular body weight measurements began six days prior to experimental diet administration. During the acclimation period, rats were maintained on standard chow (2018, Harlan Teklad, Indianapolis, IN), after which they were weight-matched into diet treatment groups. Diets included Chow, a high-fat, high-dextrose “Western” diet (WD; Harlan TD.10768), and a low-carbohydrate, high-fat ketogenic diet (KD; Research Diets, New Brunswick, NJ; D06040601); for macronutrient composition, see Table 1. Rats from each diet group were then matched into groups based on exposure duration, and were fed their diets ad libitum for 10 (n = 10 per diet), 40 (n = 10 per diet) or 90 (n = 11 per diet) days, except for the two hours immediately prior to glucose testing and sacrifice. For Chow and WD rats, food was delivered in stainless steel hoppers. KD rats received their food in small, enameled cups, which were changed daily to prevent spoilage. Each week, daily food intake was estimated by weighing food containers and spillage on two consecutive days. Data are expressed in kCal.
Table 1.
Macronutrient composition of diets.
| Chow Harlan 2018 |
WD Harlan TD.10768 |
KD Research Diets D06040601 |
|
|---|---|---|---|
| % kCal Fat | 18 | 38 | 80 |
| % kCal Dextrose | - | 20 | - |
| % kCal Other Carb. | 58 | 18 | 5 |
| % kCal Protein | 24 | 24 | 15 |
| Caloric Density | 3.4 kCal/g | 4.5 kCal/g | 6.1 kCal/g |
Rats were weighed 6 hours into the light cycle, twice per week. Body composition was analyzed approximately once every 10 days using an EchoMRI whole-body composition analyzer (EchoMRI-900, Echo Medical Systems, LLC, Houston, TX), and expressed as percent body fat (grams fat mas / grams total body weight × 100).
Behavioral Assessment – Spontaneous Alternation Task
Spontaneous alternation (SA) behavior was analyzed in a Y maze apparatus as an assessment of general cognitive function. During SA trials, vicarious trial and error (VTE) behaviors were quantified as an index of hippocampal involvement in the task. The Y maze consisted of three black, wooden arms radiating 120° from a midpoint. Each arm was 1m long and 20cm wide, and was surrounded by a 20cm high wall on all sides. Lightly soiled bedding was placed inside the maze in order to neutralize the effect of odor cues; bedding was tossed thoroughly between trials.
During testing, rats were placed into the center of the apparatus and allowed to explore the maze for 8 min while an overhead digital camera recorded the rat’s behaviors. After 8 min, the rat was returned to its home cage.
The sequence of arm entries was scored by hand according to the following criteria: a rat was deemed to have entered an arm if all four of its paws had crossed the threshold between the center triangle and the rectangular arm space. Animals were considered to have re-entered the same arm if all four paws were withdrawn from and returned to the same arm without simultaneously penetrating another branch. To facilitate scoring, arms were numbered. Number sequences were tabulated, and the number of triplets in which the rat entered exactly three arms were quantified and expressed as a ratio of the total possible number of triplets (the number of arm entries minus two).
During spontaneous alternation performance, VTE, or the total number of lateral head movements at choice points, was quantified and expressed as a ratio of total arm entries. Choice points were defined as occasions prior to the entry of a new arm when the rat’s head was within 5 cm of the new arm opening. Lateral head movements were the total number of head turns toward either of the arm choices. Lateral head movements could be accompanied by limb movements, but quantification was paused if the animal turned 180° within the same arm or moved backward such that the head was no longer within 5 cm of the arm opening.
Blood Glucose Quantification
On the day of sacrifice, a tail nick was performed, and blood glucose was analyzed in duplicate via handheld glucometer (Novamax Plus, Nova Diabetes Care, Inc., Billerica, MA). Because brains were to be dissected for fasting-sensitive gene expression analyses immediately following blood collection, rats were exposed to a mild two hour fast in lieu of a more typical overnight food deprivation. If values were disparate by > 20 mg/dL, a third reading was obtained, and the two closest values were averaged.
Sacrifice and Tissue Processing
Rats were anesthetized via isoflurane inhalation and rapidly decapitated with a guillotine. Trunk blood was collected into K3EDTA+ tubes and temporarily stored on ice. Brains were then rapidly rinsed in 1× phosphate buffered saline (PBS), hippocampi were dissected on ice, placed into nuclease-free microcentrifuge tubes, and stored on dry ice for the duration of sacrifice. Blood samples were centrifuged for 15m at 4°C, at 2500 RPM, after which plasma was aspirated and stored in microcentrifuge tubes. Samples were stored at −80°C until further analysis could be performed.
Plasma Analyses
Plasma insulin was assessed in duplicate via radioimmunoassay (Rat Insulin RIA Kit, Millipore, St. Charles, MO) according to the instructions provided by the manufacturer. Sample values were interpolated from a standard curve that was generated using enclosed known insulin quantities. The range of sensitivity for the kit was from 0.081-10 ng/mL.
Plasma beta-hydroxybutyrate (BHB) was measured in duplicate using the beta-hydroxybutyrate LiquiColor® Test (Stanbio Laboratory, Boerne, TX) with a microplate, using instructions provided by the manufacturer. Data were expressed in mg/dL as a function of a known standard value.
RNA Extraction, Quantification, Purification and cDNA Synthesis
Dissected brain tissues were homogenized in TRI Reagent (Molecular Research Center, Inc., Cincinnati, OH), after which RNA was extracted by separating the RNA-containing aqueous phase from the interphase and organic phase (which contain DNA and proteins) using bromochloropropane (Molecular Research Center). RNA was then isolated via alcohol precipitation, and analyzed via spectrophotometry. Prior to cDNA synthesis, RNA was incubated with RNAse-free DNase I and MnCl2 buffer (Fermentas Maxima, Thermo Scientific, Waltham, MA). A First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA) was used to synthesize cDNA in 20 μL PCR reactions according to instructions provided by the manufacturer. A negative control (RT-) was also created by substituting nuclease-free water for the Enzyme Mix.
Quantitative Polymerase Chain Reaction
In order to quantify the hippocampal expression of GLUT1 and MCT1 (relative to GAPDH), SYBR green qPCR was performed using an iCycler iQ Real-Time PCR Detection System with MyIQ software (Bio-Rad Laboratories, Hercules, CA). For each primer set (Table 2), both amplification and melt curve analyses of serially diluted cDNA were conducted to verify the effectiveness of the primer, confirm the absence of primer dimers, and determine the reaction efficiency. Primers were optimized such that the correlation coefficient was 0.98-1.0 and the PCR efficiency was within the range of 98-107%. The RT- control was also compared to a matched cDNA sample for the GAPDH primer. As amplification did not occur in the RT- sample, it was concluded that RNA samples used in cDNA synthesis were void of residual DNA contamination. Amplification of sample cDNA was performed in triplicate. The thermal profile began with a 10 min initiation phase at 95°C, followed by 45 cycles of 95°C (30 s), 62°C (30 s) and 72°C (60 s). Primers are listed in Table 2. Efficiency of primers were 98.2%, 98.5% and 96.7% for GLUT1, MCT1 and GAPDH, respectively. Water controls were run on each plate to assess contamination; there were no cases of water control amplification. Threshold values were standardized across plates for a given primer, such that all plate thresholds were within the exponential region of amplification. Changes in mRNA expression were determined by comparing the CT values of the genes of interest to that of GAPDH using the Pfaffl method (Pfaffl, 2001).
Table 2.
Primer sequences and reference numbers.
| Target | FWD sequence | REV sequence | NCBI Reference |
|---|---|---|---|
| GLUT1 | TGG CCC CTA CGT CTT CAT CAT CTT CA |
TCG CCC GAG ATC TGT CAG TTT GGA AG |
NM_138827.1 |
| MCT1 | TGT GGA GCA TGA AGA GAG CAG GTG TG |
CCC CAT ATT CTT TGT CAA CCA CTC CC |
NM_012716.2 |
| GAPDH | ACA GCA ACA GGG TGG TGG AC |
TTT GAG GGT GCA GCG AAC TT |
NM_017008 |
Statistical Analyses
Differences in behavioral and physiological measures between diet groups were analyzed via two-way ANOVA, with Bonferroni posttests where appropriate. Correlations were also performed to analyze the relationship between behavioral tests, with significance set to p < 0.05.
Results
Effect of Diet Type on Food Intake, Body Weight, and Adiposity
Caloric intake was measured during the first day of diet presentation, then weekly by weighing hopper on and off weights during a 24h period. Data from each exposure duration group were analyzed separately via two-way, repeated measures ANOVA (Figure 1A), all three of which revealed significant interactions (F2,27 = 7.228, p < 0.001 at 10 days; F8,108 = 10.27, p < 0.0001 at 40 days; F20,300 = 7.813, p < 0.0001 at 90 days) and main effects of both diet (F2,27 = 19.88, p < 0.0001 at 10 days; F2,108 = 22.08, p < 0.0001 at 40 days; F2,300 = 12.14, p < 0.0001 at 90 days) and exposure time (F1,27 = 36.66, p < 0.0001 at 10 days; F4,108 = 51.17, p < 0.0001 at 40 days; F10,300 = 27.53, p < 0.0001 at 90 days). All three cohorts showed the same pattern of WD-induced hyperphagia at day 1 (p < 0.001 for all cohorts, compared to both Chow and KD). On day 7, WD rats from the 40 day group consumed more than KD rats (p < 0.05); though there was a trend towards increased WD intake compared to both Chow and KD in all cohorts at this time.
Figure 1.

Caloric intake, body weight, and adiposity data for rats fed Chow, Western diet (WD) and ketogenic diet (KD). A) Amount consumed (kCal) for rats belonging to the 10, 40, and 90 day cohorts throughout the experiment. There was an overall effect of diet on caloric consumption, with WD rats consuming significantly more than Chow (p < 0.05) and KD-fed (p < 0.01) rats. According to post-hoc tests, WD-associated hyperphagia was present for only one week, with WD rats consuming more than Chow and KD at day 1 (p < 0.001, for all cohorts) and compared to KD at day 7 in the 40 day cohort (p < 0.05). No differences were detected at any other time points. (B) Change in body weight at sacrifice for Chow, WD, and KD rats. There were no differences in terminal weight gain between Chow, WD, and KD rats at any time point. (C) Body adiposity at sacrifice. Chow-fed rats had lower adiposity than WD (p < 0.001 at days 10 and 90; p < 0.05 at day 40), and KD (p < 0.01 at day 10; p < 0.001 at days 40 and 90) rats at the time of sacrifice. (*) indicates significant difference between WD and Chow; (o) indicates significant difference between WD and KD; (^) indicates significant difference between Chow and KD.
Terminal body weight gain was analyzed via two-way ANOVA and is expressed in Figure 1B. At the time of sacrifice, there were no interactions or effects of diet; however, a main effect of exposure duration was observed (F2,84 = 565.7, p < 0.0001).
Terminal body fat was also analyzed via NMR for all exposure duration cohorts via two-way ANOVA (Figure 1C). There were no interactions, but significant main effects of both diet (F2,84 = 43.46, p < 0.0001) and exposure time (F2,84 = 56.01, p < 0.0001) were observed. Compared to Chow, adiposity was significantly higher at sacrifice for WD and KD rats at all times (for WD, p < 0.001 at days 10 and 90, p < 0.05 at day 40. For KD, p < 0.01 at day 10, and p < 0.001 at days 40 and 90). These data suggest that while rats fed WD or KD were not markedly heavier than Chow rats, they carried a significantly greater proportion of their body weight in adipose tissue.
Effect of Diet on Behavior
Because previous analyses revealed that WD is capable of inducing cognitive deficits, we analyzed SA and VTE performance in rats after 10, 40, or 90 days access to Chow, WD, or KD. Data were analyzed via two-way ANOVA with Bonferroni post-hoc tests, and are depicted in Figure 2. Correlations were calculated for each group for % Sequential Arm Entries × Number of Arm Entries and % Sequential Arm Entries × VTE Episodes per Alternation.
Figure 2.
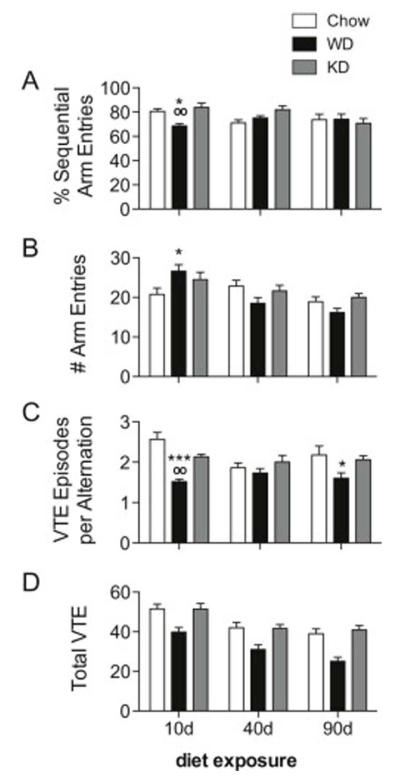
Spontaneous alternation (SA) and vicarious trial and error (VTE) behaviors for Chow, WD and KD rats. (A) An interaction between diet and exposure duration was detected for SA performance (expressed as percent sequential arm entries; p = 0.0218). After 10 days, WD rats had fewer sequential arm entries than Chow (p < 0.05) and KD (p < 0.01), but no differences were detected at later time points. (B) A similar interaction (p = 0.007) was noted for the total number of arms explored. At 10 days, WD rats entered significantly more arms than Chow (p < 0.05). (C) VTE was measured by the total number of head turns per alternation. An interaction between diet and time was noted (p = 0.0367), with WD rats engaging in fewer VTE behaviors than Chow rats at 10 (p < 0.001) and 90 days (p < 0.05), and than KD rats after 10 days (p < 0.01). (D) The total number of head turns per 8 minute session were also analyzed. There was no interaction, but main effects of diet and time (p < 0.0001) were observed, with WD rats engaging in fewer VTE behaviors than Chow and KD rats. (*) indicates significant difference compared to Chow; (o) indicates significant difference compared to KD.
Spontaneous Alternation
The total percentage of arm entry triplets without redundant entries were quantified as % Sequential Arm Entries (Figure 2A). A significant interaction between diet treatment and duration was noted (F4,84 = 3.039, p = 0.0218), though no main effects of diet or treatment duration were detected. After 10 days diet access, rats fed WD had significantly fewer sequential arm entries than either Chow (p < 0.05) or KD (p < 0.01). The total number of arm entries was also analyzed (Figure 2B), and an interaction (F4,84 = 3.788, p = 0.007) and main effect of time (F2,84 = 11.90, p = 0.0001) were found. After 10 days, WD rats had entered significantly more arms than Chow (p < 0.05). A significant and robust negative correlation between the total number of arm entries and SA performance was noted for WD rats (r = −0.6402; p = 0.0002). This correlation was absent in KD and Chow-fed rats.
Vicarious Trial and Error
We then analyzed VTE behaviors by counting the total number of head turns at choice points. Data are expressed as the total number of head turns per alternation (Figure 2C). A significant interaction between diet and duration was observed (F4,84 = 2.687, p = 0.0367), along with a main effect of diet (F2,84 = 14.48, p < 0.0001). After 10 days diet access WD rats engaged in fewer VTE behaviors than Chow (p < 0.001) and KD (p < 0.01). There were no differences observed at 40 days, but after 90 days diet access, WD rats engaged in fewer VTE behaviors compared to Chow (p < 0.05). To be sure that these data were not an artifact of increases in total arm entries, total number of VTE behaviors over the entire 8 minute session were analyzed via two-way ANOVA (Figure 2D). There was no interaction, but main effects of both diet (F2,84 = 27.36, p < 0.0001) and time (F2,84 = 22.95, p < 0.0001) were observed, with rats engaging in progressively fewer VTE behaviors over time, and WD rats engaging in fewer VTE movements than Chow and KD rats. This suggests that rats fed WD were less inclined to engage in these behaviors, which may indicate diminished hippocampal involvement. There were no correlations between VTE Episodes per Alternation and SA performance for any group.
Effect of Diet on Glucose, Insulin, and BHB
Blood was collected for analysis after a two-hour fast on the day of sacrifice (10, 40 or 90 days maintenance on Chow, WD or KD), and levels of glucose, insulin and BHB were measured (Figure 3). Data were analyzed via two-way ANOVA with Bonferroni posttests. For glucose (Figure 3A), no interactions between diet and time were detected; however, main effects of diet (F2,84 = 4.023, p = 0.0215) and duration (F2,84 = 3.925, p = 0.0235) were observed. At all time points, glucose levels were marginally higher in WD rats compared to Chow and KD.
Figure 3.
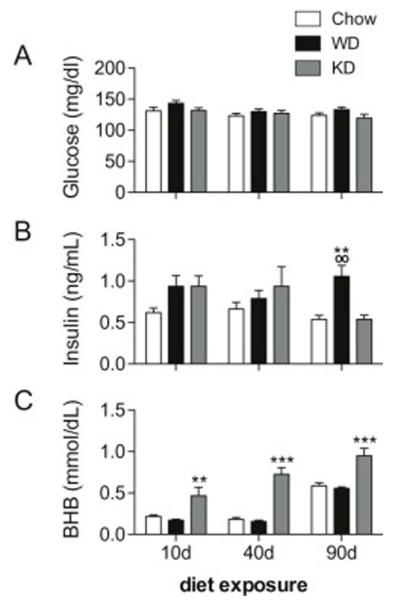
Blood glucose, plasma insulin, and plasma beta-hydroxybutyrate (BHB) from rats fed Chow, WD, and KD for 10, 40 and 90 days. (A) Main effects of diet (p = 0.0215) and time (p = 0.0235) on blood glucose were detected. Glucose levels were higher in WD-fed rats, compared to Chow and KD. (B) An interaction between diet and time was observed for plasma insulin (p = 0.0484), and post-hoc tests revealed that WD-fed rats had higher insulin levels at 90 days, compared to Chow and KD (p < 0.01). (C) A main effect of diet was observed (p < 0.0001), and KD rats had higher BHB levels at all time points. A marginally significant interaction was also present (p = 0.0926); post-hoc tests revealed that KD rats had elevated BHB than Chow and WD rats at all time points (p < 0.01 at 10 days, p < 0.001 at 40 and 90 days). (*) indicates significant difference compared to Chow; (o) indicates significant difference compared to KD.
Plasma insulin (Figure 3B) was measured in duplicate using a commercially available radioimmunoassay kit. A significant interaction between diet and exposure time was present (F4,84 = 2.516, p = 0.0484), along with a main effect of diet treatment (F2,84 = 6.101, p = 0.0035). Post-hoc tests revealed that 90 day insulin levels were significantly higher in WD rats, compared to both Chow and KD rats (p < 0.01). There were no differences between other groups or time points.
Plasma BHB (Figure 3C) was quantified in duplicate using a microtiter assay. An interaction approached significance (F4,84 = 2.065, p = 0.0926), and robust main effects of diet (F2,84 = 50.55, p < 0.0001) and time (F2,84 = 47.24, p < 0.0001) were observed. At all time points, KD rats had significantly higher levels of BHB compared to both Chow and WD rats (p < 0.01 at 10 days, and p < 0.001 at 40 and 90 days), indicating that, as expected, maintenance on KD led to increases in circulating ketone bodies. The lack of BHB differences between Chow and WD rats suggests that despite its high fat content, WD was not ketogenic.
Effects of Diet on Hippocampal mRNA Expression
Hippocampal glucose (GLUT1) and monocarboxylate (MCT1) transporter expression was measured via qPCR in rats fed Chow, WD or KD relative to GAPDH, and analyzed via two-way ANOVA with post-hoc Bonferroni contrasts. Two samples were excluded due to poor cDNA quality, one from the 10 day KD group, and one from the 90 day Chow group. Data are presented in Figure 4.
Figure 4.
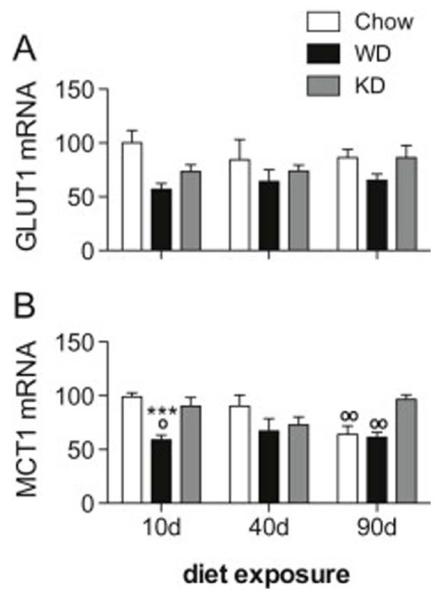
Hippocampal GLUT1 and MCT1 mRNA expression for rats fed Chow, WD and KD. (A) A main effect of diet on GLUT1 was observed, and expression was lower in WD rats compared to Chow and KD, particularly after 10 days diet access. (B) An interaction between diet and time (p = 0.0037), and a main effect of diet (p < 0.0001) on MCT1 mRNA expression were observed. Post-hoc tests revealed that MCT1 levels were significantly lower in WD rats compared to Chow (p < 0.001) and KD (p < 0.05) rats at 10 days, and higher in KD rats, compared to Chow rats at 90 days (p < 0.01). (*) indicates significant difference compared to Chow; (o) indicates significant difference compared to KD.
Though no interaction was detected, a main effect of diet on hippocampal GLUT1 (Figure 4A) expression was observed (F2,82 = 5.822, p = 0.0043). At all time points, GLUT1 mRNA was lower in WD rats compared to Chow and, to a lesser extent, KD; however, the magnitude of this difference was most pronounced at the 10 day time point.
We then analyzed hippocampal MCT1 mRNA (Figure 4B), and noted a significant interaction between diet and time (F4,82 = 4.223, p = 0.0037) and a main effect of diet (F2,82 = 10.23, p < 0.0001). Bonferroni post-hoc revealed that MCT1 levels were significantly lower in WD rats at 10 days compared to both Chow (p < 0.001) and KD (p < 0.05) rats. At 90 days, KD rats had MCT1 mRNA expression levels significantly higher than both Chow and WD (p < 0.01).
Discussion
These data are in agreement with previous reports that hippocampal-dependent cognitive performance is affected by exposure to WD (Davidson et al., 2013; Davidson et al., 2012; Kanoski et al., 2007; Kanoski et al., 2010) but not KD, despite KD-fed rats’ propensity to maintain extremely high levels of body fat (Davidson et al., 2013). We also replicated previous findings suggesting maintenance on KD does not induce excess weight gain, but does lead to increases in overall fat mass (Davidson et al., 2013; Kinzig, Hargrave, Hyun, & Moran, 2007; Kinzig, Honors, & Hargrave, 2010).
We now report significant decreases in hippocampal GLUT1 and MCT1 mRNA in after 10 days WD access, and higher levels of MCT1 mRNA in KD-fed rats after 90 days. The increased 90 day MCT1 levels in KD rats were not surprising given the marked increase in BHB expression at this time. However, BHB levels were also higher in KD rats at 10 and 40 days without an accompanying MCT1 up-regulation. Whether MCT1 up-regulation depends on duration of exposure to ketones, ketone levels, or both, is currently unknown.
The 10 day decrease in GLUT1 and MCT1 mRNA expression exhibited by WD rats (which appear to be associated with hyperphagia and diet change, rather than weight or body composition) cannot be explained by blood glucose or BHB levels, which did not differ significantly from Chow rats. It is possible that an increased fasting duration prior to blood collection would have revealed significant differences in glucose expression. Further, glucose levels in the brain interstitial fluid represent only a small fraction of the levels in circulation, and could therefore differ from blood glucose levels measured. Given the hyperphagia that was present throughout the first week of WD access, but not during the testing phases, it is plausible that transient, intake-induced hyperglycemia precipitated a reduction in transporter levels, which may not have normalized by 10 days. Typically, glucose-induced changes in GLUT1 mRNA peak approximately 24 hours following experimental metabolic manipulations (Ruben J. Boado & Pardridge, 1993; Kumagai et al., 1995), but other insults can lead to more sustained GLUT1 changes (McCall et al., 1996). Whether the present reduction in GLUT1 was sustained across days 7 through 10 despite normalized glucose levels, or was in response to brain interstitial hyperglycemia or high fasting glucose levels is not known.
GLUT1 levels may also be decreased in response to another factor, such as neurotrophins, which are decreased following WD exposure (Cordeira & Rios, 2011; Kanoski et al., 2007; Molteni et al., 2002), and are positively correlated with GLUT1 expression (Rubén J. Boado, 1996). Alternately, since GLUT1 is a critical component of the BBB (Maher, Vannucci, & Simpson, 1994; Maurer, Canis, Kuschinsky, & Duelli, 2004; Roberts et al., 2008), which is compromised after extended WD exposure (Davidson et al., 2013; Kanoski et al., 2010), GLUT1 may also be decreased in response to BBB damage.
The regulation of the MCT1 transporter has not been fully elucidated; however, it is likely that factors beyond BHB are capable of altering its expression patterns. For instance, PPARα has been shown to up-regulate MCT1 in peripheral tissues (König et al., 2008) Another possibility is that other monocarboxylates (e.g., lactate and pyruvate) are capable of regulating neuronal MCT1 expression and were altered following maintenance on WD, though this relationship has not yet been established. However, peripheral administration of pyruvate and lactate have been shown to reduce the hyperphagia associated with WD intake (Nagase, Bray, & York, 1996). While glucose and ketone bodies have generally been considered the primary energy substrates within the brain, there is evidence that lactate is also an effective energy source for neurons (Pellerin et al., 2007; Schurr, West, & Rigor, 1988; Suzuki et al., 2011; Zilberter, Zilberter, & Bregestovski, 2010). Further investigation should be conducted to determine whether WD has the potential to alter pyruvate and/or lactate levels and the effect this has on central MCT1 expression.
Disruptions to astrocytic MCT1 transporter expression in the hippocampus have been shown to induce amnesia, which is restored following lactate, but not glucose, administration (Suzuki et al., 2011). Very low GLUT1 levels limit the rate of glucose transport into the brain interstitial fluid (Ding, Yao, Zhao, et al., 2013; Qutub & Hunt, 2005), which is associated with cognitive deficits (de la Monte, 2009; De Vivo et al., 1991; De Vivo & Wang, 2008; Ding, Yao, Rettberg, et al., 2013). The point at which GLUT1 and MCT1 levels are low enough to limit the rate at which glucose or monocarboxylates enter the interstitial fluid is not currently known. Future research should analyze the composition of the brain interstitial fluid of WD-fed rats to assess whether this down-regulation of GLUT1 and MCT1 simply normalized glucose concentration levels or induced brain-specific reductions in glucose, ketone bodies, or lactate. If paradoxical reductions in glucose, BHB, or lactate are indeed present in the hippocampus of rats fed WD, energy-deprived neurons could sustain damage that manifests as cognitive deficits.
In addition to the novel transporter data, we have observed several WD-induced behavioral outcomes, including slight reductions in SA after 10 days exposure, significant decreases in VTE episodes per alternation after 10 and 90 days exposure, and reductions in total VTE behaviors after all WD exposure durations. The total number of arm entries was also significantly higher in WD rats after 10 days, and was negatively correlated with % sequential arm entries, suggesting SA performance may be influenced by the animal’s activity patterns.
Notably, we observed performance deficits in a non-appetitive task, suggesting that WD does not merely influence cognitive behaviors directly related to ingestion. Furthermore, while there was a significant main effect of diet on body weight over the course of the study, no such difference was present between groups at the time of behavioral testing or sacrifice, suggesting WD can induce behavioral deficits even in normal weight animals. Throughout the study we observed reductions in VTE behaviors in WD-fed rats, which remained significant after accounting for the total number of alternations at both 10 and 90 days. VTE depends on the hippocampus (Addis et al., 2007; Addis & Schacter, 2008; Buckner & Carroll, 2007; van der Meer, Kurth-Nelson, & Redish, 2012), and reduced VTE behaviors may indicate diminished behavioral flexibility. Reductions in VTE following WD maintenance may make food consumption more mindless and less malleable via cognitive inputs (Blass et al., 2006; Cserjési et al., 2007; Delgado-Rico, Río-Valle, González-Jiménez, Campoy, & Verdejo-García, 2012). This may impair an animal’s ability to exert inhibitory control over their feeding patterns, thereby increasing inappropriate responding to food cues and impairing adherence to dietary regimens, ultimately exacerbating any innately obesogenic properties of WD and leading to a vicious cycle of obesity and cognitive decline (Benoit, Davis, & Davidson, 2010; Davidson, Kanoski, Schier, Clegg, & Benoit, 2007; Kanoski & Davidson, 2011).
A consideration in interpreting the VTE data within this experiment is that VTE behaviors were observed during the SA task, rather than during a traditional, appetitive spatial learning paradigm. While SA behaviors are quite robustly expressed, the underlying behavioral and neural mechanisms have not yet been elucidated. SA is often touted as a “manifestation of curiosity” (Gerlai, 1998), but it is not known if SA behaviors are due to an animal’s “lose-shift” foraging strategy (Estes & Schoeffler, 1955), preference for novelty (Dember & Fowler, 1958), or “obsessive-compulsive”-like behaviors (Albelda & Joel, 2012; Yadin, Friedman, & Bridger, 1991). Further, mazes such as that used in the SA task can be navigated using either place or response strategies, which (respectively) rely on extra-maze spatial cues and habitual motor responses (such as turning in the same direction at each choice point).
Unlike place strategies, response strategies do not require an intact hippocampus (Packard & McGaugh, 1996), and are associated with reductions in VTE (Schmidt et al., 2013). Though alternation can be reduced following elimination of external cues (Gerlai, 1998), these cues are not required for SA behaviors to occur (Lennartz, 2008). If maintenance on WD induces hippocampal deficits and biases rats toward a response strategy, it would be expected that SA, but not VTE, performance would remain intact.
The neural correlates of SA are predominantly (but not exclusively) limbic structures (Lalonde, 2002). Many have noted SA changes in response to hippocampal manipulations. For example, alternation was reduced in mice with ibotenic lesions to the hippocampus, or with genetically-induced LTP deficits (Gerlai, 1998). SA performance is also enhanced via administration of glucose to the septum (Stefani, Nicholson, & Gold, 1999), a region with numerous connections to the hippocampus. This suggests that this task may depend on adequate nutrient transport to these brain regions, and may be sensitive to reductions in hippocampal GLUT1 or MCT1 expression.
However, several studies (e.g., Asin & Fibiger, 1984; Caston, Vasseur, Delhaye-Bouchaud, & Mariani, 1997; Divac, Wikmark, & Gade, 1975) suggest that SA is not hippocampal-dependent. Since alternation can be influenced by behavioral strategy, learning, and environmental factors (Lalonde, 2002), it is understandable that lesions induce divergent effects.
Whether WD-induced GLUT1 and MCT1 reductions are due to an immediate effect of the diet which is compensated for later, or as a result of the hyperphagia present during the first week of WD access is currently unknown. Whatever the mechanism, changes in transporter levels may be one mechanism by which short-term, diet-induced cognitive impairments develop. In adults, diet-induced changes in hippocampal function could facilitate overeating, whereas in juvenile organisms, perturbations in neuronal glucose homeostasis could lead not only to cognitive deficits (Boitard et al., 2014), but also to atypical hippocampal development (Boitard et al., 2012; Ekdahl, Kokaia, & Lindvall, 2009) which may in turn influence the development of other neurological structures (Lipska, Aultman, Verma, Weinberger, & Moghaddam, 2002) and result in psychiatric illness (Agranat-Meged et al., 2005; Schaefer et al., 2000).
Fortunately, isolated deficits in GLUT1 and MCT1 are likely to be modifiable via dietary changes such as maintenance on a ketogenic diet, which has been demonstrated to up-regulate both GLUT1 and MCT1 (Leino, Gerhart, Duelli, Enerson, & Drewes, 2001) and ameliorate the cognitive deficits associated with GLUT1 deficiency syndrome (Akman et al., 2010; De Vivo et al., 1991; De Vivo & Wang, 2008), Alzheimer’s disesase (Van der Auwera, Wera, Van Leuven, & Henderson, 2005) and other conditions. Ketone bodies, or other monocarboxylates such as lactate, may represent safe, effective supplements for diet-induced cognitive deficits. Further investigation may be useful for identifying the mechanism behind the purported therapeutic properties of these compounds and identify pharmaceutical targets for more focused treatments.
Highlights.
Short-term western diet access impairs spontaneous alternation performance.
Vicarious trial and error is disturbed throughout the course of western diet treatment.
Hippocampal GLUT1 and MCT1 mRNA levels decreased following western diet maintenance.
Hippocampal MCT1 mRNA levels increased following long-term access to ketogenic diet.
Acknowledgments
The authors would like to thank Melissa McCurley, Arielle Zawadski-Weist, and Joshua Stephenson for assistance with animal care and handling. These studies were supported by the US National Institute of Health grants DK078654 (KPK) and HD29792 (TLD), and the Bilsland Dissertation Fellowship (SLH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Addis DR, Moscovitch M, McAndrews MP. Consequences of hippocampal damage across the autobiographical memory network in left temporal lobe epilepsy. Brain: a journal of neurology. 2007;130:2327–2342. doi: 10.1093/brain/awm166. doi: 10.1093/brain/awm166. [DOI] [PubMed] [Google Scholar]
- Addis DR, Schacter DL. Constructive episodic simulation: temporal distance and detail of past and future events modulate hippocampal engagement. Hippocampus. 2008;18:227–237. doi: 10.1002/hipo.20405. doi: 10.1002/hipo.20405. [DOI] [PubMed] [Google Scholar]
- Agranat-Meged AN, Deitcher C, Goldzweig G, Leibenson L, Stein M, Galili-Weisstub E. Childhood obesity and attention deficit/hyperactivity disorder: a newly described comorbidity in obese hospitalized children. International Journal of Eating Disorders. 2005;37(4):357–359. doi: 10.1002/eat.20096. [DOI] [PubMed] [Google Scholar]
- Akman CI, Engelstad K, Hinton VJ, Ullner P, Koenigsberger D, Leary L, De Vivo DC. Acute hyperglycemia produces transient improvement in glucose transporter type 1 deficiency. Annals of Neurology. 2010;67:31–40. doi: 10.1002/ana.21797. doi: 10.1002/ana.21797. [DOI] [PubMed] [Google Scholar]
- Albelda N, Joel D. Current animal models of obsessive compulsive disorder: an update. Neuroscience. 2012;211:83–106. doi: 10.1016/j.neuroscience.2011.08.070. doi: 10.1016/j.neuroscience.2011.08.070. [DOI] [PubMed] [Google Scholar]
- Asin KE, Fibiger HC. Spontaneous and delayed spatial alternation following damage to specific neuronal elements within the nucleus medianus raphe. Behavioural Brain Research. 1984;13:241–250. doi: 10.1016/0166-4328(84)90166-9. [DOI] [PubMed] [Google Scholar]
- Bailey T, Rivara C, Rocher A, Hof P. The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurological Research. 2004;26:573–578. doi: 10.1179/016164104225016272. [DOI] [PubMed] [Google Scholar]
- Barañano KW, Hartman AL. The ketogenic diet: uses in epilepsy and other neurologic illnesses. Current treatment options in neurology. 2008;10:410–419. doi: 10.1007/s11940-008-0043-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit SC, Davis JF, Davidson TL. Learned and cognitive controls of food intake. Brain Research. 2010;1350:71–76. doi: 10.1016/j.brainres.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blass EM, Anderson DR, Kirkorian HL, Pempek TA, Price I, Koleini MF. On the road to obesity: Television viewing increases intake of high-density foods. Physiology & Behavior. 2006;88:597–604. doi: 10.1016/j.physbeh.2006.05.035. doi: 10.1016/j.physbeh.2006.05.035. [DOI] [PubMed] [Google Scholar]
- Boado RJ. Brain-derived peptides increase the expression of a blood-brain barrier GLUT1 glucose transporter reporter gene. Neuroscience Letters. 1996;220(1):53–56. doi: 10.1016/s0304-3940(96)13237-7. doi: 10.1016/S0304-3940(96)13237-7. [DOI] [PubMed] [Google Scholar]
- Boado RJ, Pardridge WM. Glucose Deprivation Causes Posttranscriptional Enhancement of Brain Capillary Endothelial Glucose Transporter Gene Expression via GLUT1 mRNA Stabilization. Journal of Neurochemistry. 1993;60(6):2290–2296. doi: 10.1111/j.1471-4159.1993.tb03516.x. doi: 10.1111/j.1471-4159.1993.tb03516.x. [DOI] [PubMed] [Google Scholar]
- Boitard C, Cavaroc A, Sauvant J, Aubert A, Castanon N, Layé S, Ferreira G. Impairment of hippocampal-dependent memory induced by juvenile high-fat diet intake is associated with enhanced hippocampal inflammation in rats. Brain, Behavior, and Immunity. 2014 doi: 10.1016/j.bbi.2014.03.005. [DOI] [PubMed] [Google Scholar]
- Boitard C, Etchamendy N, Sauvant J, Aubert A, Tronel S, Marighetto A, Ferreira G. Juvenile, but not adult exposure to high-fat diet impairs relational memory and hippocampal neurogenesis in mice. Hippocampus. 2012;22(11):2095–2100. doi: 10.1002/hipo.22032. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Carroll DC. Self-projection and the brain. Trends in cognitive sciences. 2007;11:49–57. doi: 10.1016/j.tics.2006.11.004. doi: 10.1016/j.tics.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Caston J, Vasseur F, Delhaye-Bouchaud N, Mariani J. Delayed spontaneous alternation in intact and cerebellectomized control and lurcher mutant mice: differential role of cerebellar cortex and deep cerebellar nuclei. Behavioral Neuroscience. 1997;111:214–218. doi: 10.1037//0735-7044.111.1.214. [DOI] [PubMed] [Google Scholar]
- Cordeira J, Rios M. Weighing in the Role of BDNF in the Central Control of Eating Behavior. Molecular Neurobiology. 2011;44:441–448. doi: 10.1007/s12035-011-8212-2. doi: 10.1007/s12035-011-8212-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Campos C, Elizondo R, Llanos P, Uranga RM, Nualart F, Garcia MA. MCT Expression and Lactate Influx/Efflux in Tanycytes Involved in Glia-Neuron Metabolic Interaction. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0016411. doi: 10.1371/journal.pone.0016411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini LC, Vogel JL, Barr LJ, Henderson ST. Clinical Efficacy of AC-1202 (AC-1202TM) in mild to moderate Alzheimer’s disease; Proceedings of the 59th Annual Meeting of the American Academy of Neurology Conference; Boston, MA. 28 April to 5 May 2007.2007. [Google Scholar]
- Cserjési R, Molnár D, Luminet O, Lénárd L. Is there any relationship between obesity and mental flexibility in children? Appetite. 2007;49:675–678. doi: 10.1016/j.appet.2007.04.001. doi: 10.1016/j.appet.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Davidson TL, Hargrave SL, Swithers SE, Sample CH, Fu X, Kinzig KP, Zheng W. Inter-relationships among diet, obesity and hippocampal-dependent cognitive function. Neuroscience. 2013;253:110–122. doi: 10.1016/j.neuroscience.2013.08.044. doi: 10.1016/j.neuroscience.2013.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson TL, Kanoski SE, Schier LA, Clegg DJ, Benoit SC. A potential role for the hippocampus in energy intake and body weight regulation. Current opinion in pharmacology. 2007;7:613–616. doi: 10.1016/j.coph.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson TL, Monnot A, Neal AU, Martin AA, Horton JJ, Zheng W. The effects of a high-energy diet on hippocampal-dependent discrimination performance and blood–brain barrier integrity differ for diet-induced obese and diet-resistant rats. Physiology & Behavior. 2012;107:26–33. doi: 10.1016/j.physbeh.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM. Insulin resistance and Alzheimer’s disease. BMB reports. 2009;42:475. doi: 10.5483/bmbrep.2009.42.8.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. The New England journal of medicine. 1991;325:703–709. doi: 10.1056/NEJM199109053251006. doi: 10.1056/NEJM199109053251006. [DOI] [PubMed] [Google Scholar]
- De Vivo DC, Wang D. Glut1 deficiency: CSF glucose. How low is too low? Revue neurologique. 2008;164:877–880. doi: 10.1016/j.neurol.2008.10.001. doi: 10.1016/j.neurol.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Delgado-Rico E, Río-Valle JS, González-Jiménez E, Campoy C, Verdejo-García A. BMI Predicts Emotion-Driven Impulsivity and Cognitive Inflexibility in Adolescents With Excess Weight. Obesity. 2012;20:1604–1610. doi: 10.1038/oby.2012.47. doi: 10.1038/oby.2012.47. [DOI] [PubMed] [Google Scholar]
- Dember WN, Fowler H. Spontaneous alternation behavior. Psychological Bulletin. 1958;55:412–428. doi: 10.1037/h0045446. doi: 10.1037/h0045446. [DOI] [PubMed] [Google Scholar]
- Deshmukh-Taskar P, Nicklas TA, Morales M, Yang SJ, Zakeri I, Berenson GS. Tracking of overweight status from childhood to young adulthood: the Bogalusa Heart Study. European Journal of Clinical Nutrition. 2005;60(1):48–57. doi: 10.1038/sj.ejcn.1602266. [DOI] [PubMed] [Google Scholar]
- Ding F, Yao J, Rettberg JR, Chen S, Brinton RD. Early Decline in Glucose Transport and Metabolism Precedes Shift to Ketogenic System in Female Aging and Alzheimer’s Mouse Brain: Implication for Bioenergetic Intervention. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0079977. doi: 10.1371/journal.pone.0079977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, Yao J, Zhao L, Mao Z, Chen S, Brinton RD. Ovariectomy induces a shift in fuel availability and metabolism in the hippocampus of the female transgenic model of familial Alzheimer’s. PLoS ONE. 2013;8:e59825. doi: 10.1371/journal.pone.0059825. doi: 10.1371/journal.pone.0059825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divac I, Wikmark RGE, Gade A. Spontaneous alternation in rats with lesions in the frontal lobes: An extension of the frontal lobe syndrome. Physiological Psychology. 1975;3:39–42. doi: 10.3758/BF03326820. [Google Scholar]
- Ekdahl CT, Kokaia Z, Lindvall O. Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience. 2009;158(3):1021–1029. doi: 10.1016/j.neuroscience.2008.06.052. [DOI] [PubMed] [Google Scholar]
- Estes WK, Schoeffler MS. Analysis of variables influencing alternation after forced trials. Journal of Comparative and Physiological Psychology. 1955;48:357–362. doi: 10.1037/h0043836. doi: 10.1037/h0043836. [DOI] [PubMed] [Google Scholar]
- Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999-2008. JAMA: the journal of the American Medical Association. 2010;303:235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- Forsythe CE, Phinney SD, Fernandez ML, Quann EE, Wood RJ, Bibus DM, Volek JS. Comparison of low fat and low carbohydrate diets on circulating fatty acid composition and markers of inflammation. Lipids. 2008;43:65–77. doi: 10.1007/s11745-007-3132-7. [DOI] [PubMed] [Google Scholar]
- Friedman JRL, Thiele EA, Wang D, Levine KB, Cloherty EK, Pfeifer HH, Natowicz MR. Atypical GLUT1 deficiency with prominent movement disorder responsive to ketogenic diet. Movement disorders: official journal of the Movement Disorder Society. 2006;21:241–245. doi: 10.1002/mds.20660. doi: 10.1002/mds.20660. [DOI] [PubMed] [Google Scholar]
- Gerlai R. A new continuous alternation task in T-maze detects hippocampal dysfunction in mice: A strain comparison and lesion study. Behavioural Brain Research. 1998;95:91–101. doi: 10.1016/s0166-4328(97)00214-3. doi: 10.1016/S0166-4328(97)00214-3. [DOI] [PubMed] [Google Scholar]
- Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab (Lond) 2009;6:31. doi: 10.1186/1743-7075-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Amsel A. A simple test of the vicarious trial-and-error hypothesis of hippocampal function. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:5506–5509. doi: 10.1073/pnas.92.12.5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarczyk MM, Machaj AS, Chiu GS, Lawson MA, Gainey SJ, York JM, Freund GG. Methylphenidate prevents high-fat diet (HFD)-induced learning/memory impairment in juvenile mice. Psychoneuroendocrinology. 2013;38:1553–1564. doi: 10.1016/j.psyneuen.2013.01.004. doi: 10.1016/j.psyneuen.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaria RHN, Harik SI. Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. Journal of Neurochemistry. 1989;53:1083–1088. doi: 10.1111/j.1471-4159.1989.tb07399.x. [DOI] [PubMed] [Google Scholar]
- Kamijo K, Khan NA, Pontifex MB, Scudder MR, Drollette ES, Raine LB, Hillman CH. The relation of adiposity to cognitive control and scholastic achievement in preadolescent children. Obesity. 2012;20(12):2406–2411. doi: 10.1038/oby.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo K, Pontifex MB, Khan NA, Raine LB, Scudder MR, Drollette ES, Hillman CH. The negative association of childhood obesity to cognitive control of action monitoring. Cerebral Cortex. 2014;24(3):654–662. doi: 10.1093/cercor/bhs349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanoski SE, Davidson TL. Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiology & Behavior. 2011;103:59–68. doi: 10.1016/j.physbeh.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanoski SE, Meisel RL, Mullins AJ, Davidson TL. The effects of energy-rich diets on discrimination reversal learning and on BDNF in the hippocampus and prefrontal cortex of the rat. Behavioural Brain Research. 2007;182:57–66. doi: 10.1016/j.bbr.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanoski SE, Zhang Y, Zheng W, Davidson TL. The effects of a high-energy diet on hippocampal function and blood-brain barrier integrity in the rat. Journal of Alzheimer’s Disease. 2010;21:207–219. doi: 10.3233/JAD-2010-091414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzig KP, Hargrave SL, Hyun J, Moran TH. Energy balance and hypothalamic effects of a high-protein/low-carbohydrate diet. Physiology & Behavior. 2007;92(3):454–460. doi: 10.1016/j.physbeh.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzig KP, Honors MA, Hargrave SL. Insulin sensitivity and glucose tolerance are altered by maintenance on a ketogenic diet. Endocrinology. 2010;151(7):3105–3114. doi: 10.1210/en.2010-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König B, Koch A, Giggel K, Dordschbal B, Eder K, Stangl GI. Monocarboxylate transporter (MCT)-1 is up-regulated by PPARα. Biochimica et Biophysica Acta (BBA) - General Subjects. 2008;1780(6):899–904. doi: 10.1016/j.bbagen.2008.03.002. doi: http://dx.doi.org/10.1016/j.bbagen.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Kumagai AK, Kang Y-S, Boado RJ, Pardridge WM. Upregulation of Blood-Brain Barrier GLUT1 Glucose Transporter Protein and mRNA in Experimental Chronic Hypoglycemia. Diabetes. 1995;44:1399–1404. doi: 10.2337/diab.44.12.1399. doi: 10.2337/diab.44.12.1399. [DOI] [PubMed] [Google Scholar]
- Kwon YS, Jeong S-W, Kim DW, Choi ES, Son BK. Effects of the ketogenic diet on neurogenesis after kainic acid-induced seizures in mice. Epilepsy research. 2008;78:186–194. doi: 10.1016/j.eplepsyres.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Lalonde R. The neurobiological basis of spontaneous alternation. Neuroscience & Biobehavioral Reviews. 2002;26:91–104. doi: 10.1016/s0149-7634(01)00041-0. doi: 10.1016/S0149-7634(01)00041-0. [DOI] [PubMed] [Google Scholar]
- Leino RL, Gerhart DZ, Duelli R, Enerson BE, Drewes LR. Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain. Neurochemistry International. 2001;38:519–527. doi: 10.1016/s0197-0186(00)00102-9. doi: 10.1016/S0197-0186(00)00102-9. [DOI] [PubMed] [Google Scholar]
- Lennartz R. The role of extramaze cues in spontaneous alternation in a plus-maze. Learning & behavior. 2008;36(2):138–144. doi: 10.3758/lb.36.2.138. doi: 10.3758/LB.36.2.138. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Aultman JM, Verma A, Weinberger DR, Moghaddam B. Neonatal Damage of the Ventral Hippocampus Impairs Working Memory in the Rat. Neuropsychopharmacology. 2002;27:47–54. doi: 10.1016/S0893-133X(02)00282-8. doi: 10.1016/S0893-133X(02)00282-8. [DOI] [PubMed] [Google Scholar]
- Lutas A, Yellen G. The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends in Neurosciences. 2012 doi: 10.1016/j.tins.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher F, Vannucci SJ, Simpson IA. Glucose transporter proteins in brain. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1994;8:1003–1011. doi: 10.1096/fasebj.8.13.7926364. [DOI] [PubMed] [Google Scholar]
- Maurer MH, Canis M, Kuschinsky W, Duelli R. Correlation between local monocarboxylate transporter 1 (MCT1) and glucose transporter 1 (GLUT1) densities in the adult rat brain. Neuroscience Letters. 2004;355:105–108. doi: 10.1016/j.neulet.2003.10.056. doi: 10.1016/j.neulet.2003.10.056. [DOI] [PubMed] [Google Scholar]
- McCall AL, Van Bueren AM, Nipper V, Moholt-Siebert M, Downes H, Lessov N. Forebrain Ischemia Increases Glut1 Protein in Brain Microvessels and Parenchyma. J Cereb Blood Flow Metab. 1996;16(1):69–76. doi: 10.1097/00004647-199601000-00008. [DOI] [PubMed] [Google Scholar]
- Molteni R, Barnard RJ, Ying Z, Roberts CK, Gomez-Pinilla F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience. 2002;112:803–814. doi: 10.1016/s0306-4522(02)00123-9. [DOI] [PubMed] [Google Scholar]
- Mooradian A, Chung H, Shah G. GLUT-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiology of Aging. 1997;18:469–474. doi: 10.1016/s0197-4580(97)00111-5. [DOI] [PubMed] [Google Scholar]
- Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. European Journal of Nuclear Medicine and Molecular Imaging. 2005;32:486–510. doi: 10.1007/s00259-005-1762-7. doi: 10.1007/s00259-005-1762-7. [DOI] [PubMed] [Google Scholar]
- Mosconi L, Mistur R, Switalski R, Tsui WH, Glodzik L, Li Y, Leon M. J. d. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. European Journal of Nuclear Medicine and Molecular Imaging. 2009;36:811–822. doi: 10.1007/s00259-008-1039-z. doi: 10.1007/s00259-008-1039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H, Bray GA, York DA. Pyruvate and hepatic pyruvate dehydrogenase levels in rat strains sensitive and resistant to dietary obesity. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 1996;270(3):R489–R495. doi: 10.1152/ajpregu.1996.270.3.R489. [DOI] [PubMed] [Google Scholar]
- Nehlig A. Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins, Leukotrienes and Essential Fatty Acids. 2004;70:265–275. doi: 10.1016/j.plefa.2003.07.006. doi: 10.1016/j.plefa.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA. 2014;311(8):806–814. doi: 10.1001/jama.2014.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard MG, McGaugh JL. Inactivation of Hippocampus or Caudate Nucleus with Lidocaine Differentially Affects Expression of Place and Response Learning. Neurobiology of Learning and Memory. 1996;65:65–72. doi: 10.1006/nlme.1996.0007. doi: 10.1006/nlme.1996.0007. [DOI] [PubMed] [Google Scholar]
- Pardridge WM, Triguero D, Farrell CR. Downregulation of blood-brain barrier glucose transporter in experimental diabetes. Diabetes. 1990;39:1040–1044. doi: 10.2337/diab.39.9.1040. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Bouzier-Sore A-K, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity-dependent regulation of energy metabolism by astrocytes: An update. Glia. 2007;55:1251–1262. doi: 10.1002/glia.20528. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic acids research. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, Bruce-Keller AJ. Cognitive impairment following high fat diet consumption is associated with brain inflammation. Journal of Neuroimmunology. 2010;219:25–32. doi: 10.1016/j.jneuroim.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qutub AA, Hunt CA. Glucose transport to the brain: A systems model. Brain Research Reviews. 2005;49:595–617. doi: 10.1016/j.brainresrev.2005.03.002. doi: 10.1016/j.brainresrev.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Ramm-Pettersen A, Stabell KE, Nakken KO, Selmer KK. Does ketogenic diet improve cognitive function in patients with GLUT1-DS? A 6- to 17-month follow-up study. Epilepsy & Behavior. 2014;39:111–115. doi: 10.1016/j.yebeh.2014.08.015. doi: 10.1016/j.yebeh.2014.08.015. [DOI] [PubMed] [Google Scholar]
- Roberts LM, Black DS, Raman C, Woodford K, Zhou M, Haggerty JE, Grindstaff KK. Subcellular localization of transporters along the rat blood–brain barrier and blood–cerebral-spinal fluid barrier by in vivo biotinylation. Neuroscience. 2008;155:423–438. doi: 10.1016/j.neuroscience.2008.06.015. doi: 10.1016/j.neuroscience.2008.06.015. [DOI] [PubMed] [Google Scholar]
- Schaefer CA, Brown AS, Wyatt RJ, Kline J, Begg MD, Bresnahan MA, Susser ES. Maternal prepregnant body mass and risk of schizophrenia in adult offspring. Schizophrenia bulletin. 2000;26(2):275–286. doi: 10.1093/oxfordjournals.schbul.a033452. [DOI] [PubMed] [Google Scholar]
- Schmidt B, Papale A, Redish AD, Markus EJ. Conflict between place and response navigation strategies: effects on vicarious trial and error (VTE) behaviors. Learning & memory (Cold Spring Harbor, N.Y.) 2013;20:130–138. doi: 10.1101/lm.028753.112. doi: 10.1101/lm.028753.112. [DOI] [PubMed] [Google Scholar]
- Schurr A, West CA, Rigor BM. Lactate-supported synaptic function in the rat hippocampal slice preparation. Science. 1988;240(4857):1326–1328. doi: 10.1126/science.3375817. doi: 10.1126/science.3375817. [DOI] [PubMed] [Google Scholar]
- Sharman MJ, Kraemer WJ, Love DM, Avery NG, Gómez AL, Scheett TP, Volek JS. A ketogenic diet favorably affects serum biomarkers for cardiovascular disease in normal-weight men. J Nutr. 2002;132:1879–1885. doi: 10.1093/jn/132.7.1879. [DOI] [PubMed] [Google Scholar]
- Sharman MJ, Volek JS. Weight loss leads to reductions in inflammatory biomarkers after a very-low-carbohydrate diet and a low-fat diet in overweight men. Clinical Science. 2004;107:365–370. doi: 10.1042/CS20040111. [DOI] [PubMed] [Google Scholar]
- Stefani MR, Nicholson GM, Gold PE. ATP-sensitive potassium channel blockade enhances spontaneous alternation performance in the rat: a potential mechanism for glucose-mediated memory enhancement. Neuroscience. 1999;93:557–563. doi: 10.1016/s0306-4522(99)00128-1. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, Mattson MP. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18:1085–1088. doi: 10.1002/hipo.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suddendorf T, Corballis MC. The evolution of foresight: What is mental time travel, and is it unique to humans? Behavioral and Brain Sciences. 2007;30:299–313. doi: 10.1017/S0140525X07001975. doi: 10.1017/S0140525X07001975. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. Astrocyte-Neuron Lactate Transport Is Required for Long-Term Memory Formation. Cell. 2011;144:810–823. doi: 10.1016/j.cell.2011.02.018. doi: 10.1016/j.cell.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera I, Wera S, Van Leuven F, Henderson ST. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutrition & metabolism. 2005;2:28. doi: 10.1186/1743-7075-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer M, Kurth-Nelson Z, Redish AD. Information processing in decision-making systems. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2012;18:342–359. doi: 10.1177/1073858411435128. doi: 10.1177/1073858411435128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannucci SJ, Simpson IA. Developmental switch in brain nutrient transporter expression in the rat. American Journal of Physiology - Endocrinology and Metabolism. 2003;285:E1127–E1134. doi: 10.1152/ajpendo.00187.2003. doi: 10.1152/ajpendo.00187.2003. [DOI] [PubMed] [Google Scholar]
- Volek J, Sharman M, Gomez A, Judelson DA, Rubin MR, Watson G, Kraemer WJ. Comparison of energy-restricted very low-carbohydrate and low-fat diets on weight loss and body composition in overweight men and women. Nutr Metab (Lond) 2004;1:13. doi: 10.1186/1743-7075-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss JL, Warren DE, Gonsalves BD, Federmeier KD, Tranel D, Cohen NJ. Spontaneous revisitation during visual exploration as a link among strategic behavior, learning, and the hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:E402–409. doi: 10.1073/pnas.1100225108. doi: 10.1073/pnas.1100225108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Beydoun MA, Liang L, Caballero B, Kumanyika SK. Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity. 2008;16:2323–2330. doi: 10.1038/oby.2008.351. [DOI] [PubMed] [Google Scholar]
- Whitmer RA, Gunderson EP, Barrett-Connor E, Quesenberry CP, Yaffe K. Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. Bmj. 2005;330:1360. doi: 10.1136/bmj.38446.466238.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F. The interplay between oxidative stress and brain-derived neurotrophic factor modulates the outcome of a saturated fat diet on synaptic plasticity and cognition. European Journal of Neuroscience. 2004;19:1699–1707. doi: 10.1111/j.1460-9568.2004.03246.x. [DOI] [PubMed] [Google Scholar]
- Wu Z, Guo H, Chow N, Sallstrom J, Bell RD, Deane R, Sagare A. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat Med. 2005;11:959–965. doi: 10.1038/nm1287. [DOI] [PubMed] [Google Scholar]
- Yadin E, Friedman E, Bridger WH. Spontaneous alternation behavior: An animal model for obsessive-compulsive disorder? Pharmacology Biochemistry and Behavior. 1991;40:311–315. doi: 10.1016/0091-3057(91)90559-k. doi: 10.1016/0091-3057(91)90559-K. [DOI] [PubMed] [Google Scholar]
- Yau PL, Castro MG, Tagani A, Tsui WH, Convit A. Obesity and metabolic syndrome and functional and structural brain impairments in adolescence. Pediatrics. 2012;130(4):e856–e864. doi: 10.1542/peds.2012-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberter Y, Zilberter T, Bregestovski P. Neuronal activity in vitro and the in vivo reality: the role of energy homeostasis. Trends in Pharmacological Sciences. 2010;31:394–401. doi: 10.1016/j.tips.2010.06.005. doi: 10.1016/j.tips.2010.06.005. [DOI] [PubMed] [Google Scholar]