

Abstract
Lung epithelial cells are considered important sources of inflammatory molecules and extracellular matrix proteins that contribute to diseases such as asthma. Understanding the factors that stimulate epithelial cells may lead to new insights into controlling lung inflammation. This study sought to investigate the responsiveness of human lung epithelial cells to the TNF family molecules LIGHT and lymphotoxin αβ (LTαβ). Bronchial and alveolar epithelial cell lines, and primary human bronchial epithelial cells, were stimulated with LIGHT and LTαβ, and expression of inflammatory cytokines and chemokines, and markers of epithelial-mesenchymal transition and fibrosis/remodeling, were measured. LTβR, the receptor shared by LIGHT and LTαβ, was constitutively expressed on all epithelial cells. Correspondingly, LIGHT and LTαβ strongly induced a limited but highly distinct set of inflammatory genes in all epithelial cells tested, namely the adhesion molecules ICAM-1 and VCAM-1; the chemokines CCL5, CCL20, CXCL1, CXCL3, CXCL5 and CXCL11; the cytokines IL-6, activin A, and GM-CSF; and metalloproteinases MMP-9 and ADAM-8. Importantly, induction of the majority of these inflammatory molecules was insensitive to the suppressive effects of the corticosteroid budesonide. LIGHT and LTαβ also moderately downregulated E-cadherin, a protein associated with maintaining epithelial integrity, but did not significantly drive production of extracellular matrix proteins or alpha-smooth muscle actin. Thus, LIGHT and LTαβ induce a distinct steroid-resistant inflammatory signature in airway epithelial cells via constitutively expressed LTβR. These findings support our prior murine studies that suggested the receptors for LIGHT and LTαβ contribute to development of lung inflammation characteristic of asthma and idiopathic pulmonary fibrosis.
INTRODUCTION
Chronic lung disorders, such as asthma, are the leading cause of morbidity and mortality throughout the world and are characterized by recurrent, reversible airflow obstruction, airway inflammation, bronchial hyperresponsiveness, and a progressive decline in lung function (1–3). In many patients, disease is poorly controlled, so the need to define new targets for intervention is important.
The inflammatory response in asthma involves infiltration of the lung by a variety of cells, including eosinophils, T lymphocytes, mast cells, neutrophils, and dendritic cells (4, 5). Furthermore, severe asthma has been associated with fibrotic activity encompassing deposition of extracellular matrix proteins (ECM), as well as hyperplasia of airway smooth muscle (6, 7). Current thinking suggests that airway epithelial cells, of bronchial and alveolar origin, are key players in orchestrating and maintaining many of the pathological features of asthma (8–11), and many asthma-susceptibility genes are expressed in the airway epithelium (12). A large array of cytokines and chemokines can be produced by these cells, and an intrinsically altered baseline release has been reported in airway epithelial cells derived from asthmatic patients (13–15). However, it remains to be understood what are the key molecules that stimulate lung epithelial cells to display inflammatory activities and to undergo changes associated with airway remodeling.
We previously suggested that the TNF superfamily molecule, LIGHT (TNFSF14, CD258), might be a primary driver of inflammatory activity as well as tissue remodeling that is characteristic of severe asthmatic disease and idiopathic pulmonary fibrosis (16–18). LIGHT can be membrane bound but might largely act as a soluble molecule, and T cells are regarded as the main source of LIGHT, although other cells may also contribute including dendritic cells, macrophages, and NKT cells. LIGHT can bind to the lymphotoxin β receptor (LTβR), a shared receptor for another TNF family molecule lymphotoxin αβ (LTαβ). LIGHT also binds a second distinct receptor, HVEM (19). We showed that T cell expressed LIGHT and HVEM regulated Th2 cell survival during acute allergen-induced responses in the mouse, and that LIGHT exerted a separate action in driving lung remodeling during chronic exposure to allergen (16, 17). Part of the activity of LIGHT in promoting remodeling was explained by lung macrophage-expressed LTβR driving TGF-β production, and eosinophil-expressed HVEM driving IL-13 production (17). Moreover, we recently showed that LIGHT can drive TSLP production in bronchial epithelial cells in vivo during the development of pulmonary fibrosis in mice, and LIGHT is required for lung fibrosis triggered by the antibiotic bleomycin (18). Given the potential of the lung epithelium to respond strongly to LIGHT, here we examined whether LIGHT or the related molecule LTαβ may exert activities on epithelial cells that could further amplify other processes associated with asthma or pulmonary fibrosis. We show that LIGHT stimulation of lung epithelial cells induced a limited but distinct set of genes with critical importance for inflammation including chemokines and matrix metalloproteinases. Cells of both bronchial and alveolar origin expressed LTβR, and correspondingly the activities elicited by LIGHT were also induced by LTαβ. Moreover, induction of many of the inflammatory genes by LIGHT/LTαβ was resistant to the effects of budesonide. Thus, LIGHT and LTαβ signaling through the LTβR can promote a very specific inflammatory signature in lung epithelial cells that can be refractory to corticosteroid treatment.
MATERIALS AND METHODS
Reagents and antibodies
Trypsin/EDTA and antibiotic/antimycotic solution (Penicillin/Streptomycin, thereafter P/S) were from Life Technologies (Carlsbad, CA). Fetal bovine serum (FBS) was from Omega Scientific (Tarzana, CA). DMEM-F12 was from ATCC (Mannasas, VA) and DMEM from Corning (Manassas, VA). BEBM basal medium and BEGM supplements and growth factors were purchased from Lonza (Walkersville, MD). Anti-human fibronectin (mouse ant-human ascites fluid, clone IST-4), α-smooth muscle actin (mouse anti-human clone 1A4), β-tubulin (mouse anti-human ascites, clone TUB 2.1), and budesonide were from Sigma (St. Louis, MO). Recombinant human LIGHT/TNFSF14, lymphotoxin α1/β2, TNF and TGF-β1, human antibodies to MMP-9 and Vimentin, and Human active MMP-9 Flurokine E kit, TGF-β1 DuoSet ELISA, and CCL5/RANTES Quantikine ELISA were obtained from R&D Systems (Minneapolis, MN). Recombinant human LIGHT FLAG-tagged was from Enzo Life Sciences (Farmingdale, NY). Anti-human CD270 (HVEM clone eBioHVEM-122), CD54 (ICAM-1, clone HA58), and CD324 (E-Cadherin, clone DECMA-1) were from eBioscience (San Diego, CA). Anti-human CD106 (VCAM-1, clone STA) and LTβR (clone 31G4D8) were from BioLegend (San Diego, CA). Human anti-E-Cadherin (rabbit anti-human, H-108) was from Santa Cruz Biotecnhology, Inc (Dallas, TX).
Primary cells and cell lines
Primary human bronchial epithelial (HBE) cells were from Lonza (Walkersville, MD) and maintained in bronchial epithelial basal medium (BEBM) supplemented with cytokines and growth factors (BEGM), also from Lonza. Human epithelial cell lines of bronchial and alveolar origin, BEAS-2B and A549, were from the American Type Culture Collection (ATCC, Manassas, USA). They were maintained in DMEM-F12 and DMEM (1X) respectively, supplemented with 10% FBS and 1% P/S. Cells (0.5–1×106) were grown as monolayers, then treated with 0.05% solution of Trypsin/EDTA, washed with HBSS (Corning, Manassas, VA), and resuspended in medium for experiments. Cells were used between passages 2–3 (HBE) and 3–15 (BEAS-2B and A549).
Culture conditions
Epithelial cells were seeded a day prior to starting the treatment in 6 well plates (Becton Dickinson, Mountain View, CA) at a density of 0.25×106/ml in 2ml. Cells were cultured alone, or with either recombinant LIGHT/TNFSF14, recombinant lymphotoxin α1/β2 (1–100ng/ml), recombinant TNF (10ng/ml) or TGF-β1 (5ng/ml). All cells were stimulated in submerged cultures based on initial experiments showing similar results in air-liquid interface cultures. In some experiments, budesonide (10−7 M) was added 16 hours before stimulation. Increasing the concentration of budesonide did not alter the conclusions (data not shown). Additional assessment of LIGHT functional activity in lung ECs were performed by comparing recombinant soluble LIGHT with either a membrane form of LIGHT present in the surface of a transfected cell line (EL4-LIGHT) or by cross-linking a soluble Flag-tagged version of LIGHT with an anti-Flag mAb. ECs stimulation by membrane-bound or cross-linked LIGHT did not further enhance any of the depicted biological activities compared to soluble LIGHT (not shown).
Flow cytometry
Cell staining was performed at 4°C for 30 min in staining buffer (PBS, 0.2% BSA, 0.1% NaN3) in 96-well round-bottom plates (Sarstedt). Irrelevant isotype matched mAb were used as negative controls to define background staining. In two-step staining protocols, conjugated Abs were used. Cell death and survival were determined by Annexin V and PI staining, using Ca2+-based staining buffer (10nM HEPES/140mM NaCl/2.5mM CaCl2). No difference was observed after LIGHT stimulation in any of the cells examined (data not shown). After staining, cells were washed and acquired with a FACSCalibur or FACSCanto (both from Becton Dickinson, Mountain View, CA). For each sample, 50,000 events were acquired using FCS/SSC characteristics and analyzed using FlowJo software.
Protein isolation and immunoblotting
After culture, cells were washed and lysed in complete Lysis-M supplemented with a protease inhibitor cocktail (Roche, Mannheim, Germany) for 10 min on ice. The lysates were centrifuged at 10,000Xg to remove cell debris. Alternatively, supernatants of treated cells were precipitated with 10% tricloroacetic acid (TCA) for 30 min on ice, centrifuged at 11,000Xg for 15 min and washed in cold acetone overnight at −20°C. Samples were centrifuged again at 11,000Xg for 15 min and resuspended in sample buffer (25%Tris pH 6.8, 10% SDS, 0.5% bromophenol blue, 10% glycerol) heated at 96°C for 10 min and quantified by the BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA). Aliquots corresponding to 20–25µl of protein were resolved in NuPAGE Novex 4–12% Bis-Tris Gels pre-cast polyacrylamide gels (Life Technologies) under reducing conditions and transferred to PVDF membranes (Life Technologies). Membranes were incubated with 5% non-fat dry milk (Santa Cruz) in TBS-T and then incubated with primary Abs followed by horseradish peroxidase (HRP)-conjugated secondary Abs. Proteins were visualized after exposure to Kodak Biomax (Sigma) using the enhanced chemiluminescense Super Signal West Pico detection reagent (Thermo Scientific, Rockford, IL). Immunoblotting for β-tubulin (mouse anti-human ascites, clone TUB 2.1, Sigma) was used as loading control.
ELISA, Fluorokine E Matrix Metalloproteinase Activity Assay, and Multiplex assay
Supernatants were centrifuged at 13,000Xg for 15 min and stored at −80°C until used. The levels of CCL5, Activin A and TGF-β1 were determined by ELISA-based kits (R&D Systems), according to the manufacturer’s instructions. The activity of MMP-9 was assessed using Flurokine E enzymatic activity assay (R&D Systems). Briefly, samples were pipetted into wells pre-coated with a monoclonal antibody specific for MMP-9. After incubation and washing of unbound substances, an activation reagent (APMA) was added to standards and samples. Following washes, a fluorogenic substrate linked to a quencher molecule was added into the well and after 20 hours of incubation the relative fluorescence units (RFU) were determined using a fluorescence plate reader. MSD multiplex human cytokine 30-plex panel (Meso Scale Discovery, Rockville, MD) was used to assess the protein levels of CCL2, CCL3, CCL13, CCL11, CCL17, CCL22, CCL24, CCL26, CXCL8, CXCL10, GM-CSF, VEGF, IL-1α, IL-1β, IL-6, IL-7 and IL-10.
RNA isolation, reverse transcription, and qRT-PCR
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA) following standard protocols. Total RNA was converted into cDNA using Transcriptor First Strand cDNA kit (Roche). The resultant cDNA was further diluted and 50ng used in 20µl quantitative real-time PCR reactions for amplification of genes in LIGHT cycler 480 DNA SYRB Green I master mix (Roche) and a LightCycler 480 Real-Time PCR System (Roche) using the specific primers (IDT, Coralville, IA) listed in Table I. PCR products were generated in triplicate and normalized to housekeeping genes, β-actin or GAPDH. Relative quantification (RQ) was derived from the difference in cycle threshold (Ct) between the gene of interest and the housekeeping genes β-actin or GAPDH using the equation RQ=2−Δ Δct. PCR product specificity was confirmed by performing a melt analysis at the end of each experiment, or alternatively by running the amplicon product in an agarose gel to confirm size. Data in each experiment are displayed as the average RQ and representative of at least three independent experiments/donors.
Table I.
Sets of primers used for mRNA amplication by qRT-PCR
| Molecule | Sense Primer (5′⇒ 3′) | Anti-Sense Primer (5′⇒ 3′) | Product (bp) |
|---|---|---|---|
| ICAM-1 | CCTGATGGGCAGTCAACAGCTA | ACAGCTGGCTCCCGTTTCA | 200 |
| VCAM-1 | CCGGATTGCTGCTCAGATTGGA | AGCGTGGAATTGGTCCCCTCA | 141 |
| CCL2 (MCP-1) | GCTCAAGCAGCCACCTTCATTC | GGACACTTGCTGCTGGTGATTC | 146 |
| CCL3 (MIP-1α) | TCCGTCACCTGCTCAGAATCA | ACTGGCTGCTCGTCTCA | 189 |
| CCL5 (RANTES) | ACCAGTGGCAAGTGCTCCAAC | CTCCCAAGCTAGGACAAGAGCAAG | 191 |
| CCL13 (MCP-4) | CAAACTGGGCAAGGAGATCTG | GGCCCAGGTGTTTCATATAATTCT | 71 |
| CCL11 (Eotaxin-1) | CTCGCTGGGCCAGCTTCTGTC | GGCTTTGGAGTTGGAGATTTTTGG | 227 |
| CCL17 (TARC) | CTCCTCCTGGGGGCTTCTCT | GTTGGGGTCCGAACAGATGG | 198 |
| CCL20 | CTGGCTGCTTTGATGTCAGT | CGTGTGAAGCCCACAATAAA | 128 |
| CCL21 | CAAGCTTAGGCTGCTCCATC | TCAGTCCTCTTGCAGCCTTT | 220 |
| CCL22 (MDC) | TGCCGTGATTACGTCCGTTAC | AAGGCCACGGTCATCAGAGTAG | 201 |
| CCL24 (Eotaxin-2) | CACATCATCCCTACGGGCTCT | GGTTGCCAGGATATCTCTGGACAGGG | 288 |
| CCL26 (Eotaxin-3) | GGAACTGCCACACGTGGGAGTGAC | CTCTGGGAGGAAACACCCTCTCC | 354 |
| CXCL1 | GAAAGCTTGCCTCAATCCTG | CTTCCTCCTCCCTTCTGGTC | 97 |
| CXCL2 | GGGCAGAAAGCTTGTCTCAA | GCTTCCTCCTTCCTTCTGGT | 103 |
| CXCL3 | CGCCCAAACCGAAGTCATAG | GCTCCCCTTGTTCAGTATCTTTT | 259 |
| CXCL5 | GGAAAGATTTTGTTGTTGTT | AGTCACCTACAATTCAAGAC | 260 |
| CXCL8 (IL-8) | CTGGCCGTGGCTCTCTTG | TTAGCACTCCTTGGCAAAACTG | 77 |
| CXCL10 (IP-10) | GGAACCTCCAGTCTCAGCACC | GCGTACGGTTCTAGAGAGAGGTAC | 108 |
| CXCL11 | ATGAGTGTGAAGGGCATGGC | TCACTGCTTTTACCCCAGGG | 121 |
| CXCL12 | ATGCCCATGCCGATTCTTCG | GCCGGGCTACAATCTGAAGG | 102 |
| CXCL13 | CTCTGCTTCTCATGCTGCTG | TGAGGGTCCACACACACAAT | 220 |
| CX3CL1 (Fractalkine) | GCTGAGGAACCCATCCAT | GAGGCTCTGGTAGGTGAACA | 165 |
| GM-CSF | CATGATGGCCAGCCACTACAA | ACTGGCTCCCAGCAGTCAAAG | 141 |
| VEGF | ATCTTCAAGCCATCCTGTGTGC | CAAGGCCCACAGGGATTTTC | 224 |
| IL-1β | ATGGCCCTAAACAGATGAAGTGCT | TCCCATGTGTCGAAGAAGATAGGT | 247 |
| IL-6 | AAGCCAGAGCTGTGCAGATGAGTA | TGTCCTGCAGCCACTGGTTC | 150 |
| TGF-β1 | TACGTCAGACATTCGGGAAGCA | AGGTAACGCCAGGAATTGTTGC | 138 |
| Activin-A | GAAAAGGAGCAGTCGCACAGA | GGCGATGAGGGTGGTCTTC | 70 |
| MMP-2 | CGTCTGTCCCAGGATGACATC | TGTCAGGAGAGGCCCCATAG | 98 |
| MMP-9 | TGGGCAGATTCCAAACCTTT | TCTTCCGAGTAGTTTTGGATCCA | 82 |
| MMP-13 | CAGACTTCACGATGGCATTGC | AGGAAAAGCATGAGCCAGCA | 109 |
| TIMP-1 | CTTCTGGCATCCTGTTGTTG | GGTATAAGGTGGTCTGGTTG | 156 |
| ADAM-8 | CACAGAGGATGGCACTGCGTATGA | CGTGCACCTCAGTCAGCAGCTT | 221 |
| E-Cadherin | CCACCAAAGTCACGCTGAATAC | GGAGTTGGGAAATGTGAGCAA | 104 |
| α-SMA | CTGGCATCGTGCTGGACTCT | GATCTCGGCCAGCCAGATC | 108 |
| Vimentin | GGAAGAGAACTTTGCCGTTGAA | GTGACGAGCCATTTCCTCCTT | 97 |
| Collagen I | CCTCAAGGGCTCCAAC | GGTTTTGTATTCAATCACTGTCTTGC | 71 |
| Fibronectin-EDA | GAGCTATTCCCTGCACCTGATG | CGTGCAAGGCAACCACACT | 97 |
| Tenascin C | CAGAAGCCGAACCGGAAGTT | TTCATCAGCTGTCCAGGACAGA | 83 |
| HVEM | AGCAGCTCCCACTGGGTATG | GATTAGGCCAACTGTGGAGCA | 78 |
| LTβR | CCGACACAACCTGCAAAAAT | GAGCAGAAAGAAGGCCAGTG | 101 |
| GILZ | GGCCATAGACAACAAGATCG | ACTTACACCGCAGAACCACCA | 264 |
| FKBP51 | CAGCTGCTCATGAACGAGTTTG | GCTTTATTGGCCTCTTCCTTGG | 203 |
| GRα | GAAGGAAACTCCAGCCAGAACTG | GATGATTTCAGCTAACATCTCG | 144 |
| GRβ | GAAGGAAACTCCAGCCAGAACTG | TGAGCGCCAAGATTGTTGG | 140 |
| β-Actin | TGCGTGACATTAAGGAGAAG | GTCAGGCAGCTCGTAGCTCT | 107 |
| GAPDH | AGCCAAAAGGGTCATCATCTCT | AGGGGCCATCCACAGTCTT | 230 |
Transfection of siRNA
ON-TARGETplus siRNA were purchased from Dharmacon (Pittsburg, PA). A 5nM-25nM concentration range of Ctrl or LTβR siRNA were transfected into ECs using HiPerFect transfection reagent (Qiagen, Venlo, Netherlands) according to the manufacturer’s instructions. Efficiency of LTβR knockdown was monitored over-time by both mRNA and surface protein levels and was stable during the 24–72hr culture period when ECs where stimulated with recombinant LIGHT.
Chemotaxis assay
Experiments were performed in 3.0µM pore size transwell permeable polycarbonate membranes (Corning). Briefly, freshly isolated peripheral blood mononuclear cells (PBMCs) were isolated by using Histopaque-1077 (Sigma) cell separation medium and loaded in the upper chamber of transwells. The supernatants from ECs were placed in the lower chamber and cells allowed to migrate for 120 minutes at 37°C. The medium in the lower chamber was then recovered and acquired for 1 min in a FACScalibur. Both lymphocyte (PBL) and monocyte populations were gated accordingly and the number of cells counted and compared to controls. All conditions were performed in triplicate wells.
Statistical analysis
Statistical analyses were performed using Excel or GraphPad Prism 6 software. A two-tail unpaired Student’s t-test between two groups, or one-way ANOVA with Greenhouse-Geisser correction followed by Dunnett’s multiple comparisons test, was used to test the significance of the differences between group means. All data are representative of at least three independent experiments and expressed as mean ± SEM. Statistical significance was defined as *(P<0.05) or ***(P<0.001).
RESULTS
Lung epithelial cells express LTβR and HVEM, the receptors for LIGHT and LTαβ
We examined the expression of the shared receptor for LIGHT and LTαβ, namely LTβR, and the second receptor for LIGHT, namely HVEM, in one human bronchial epithelial cell line (BEAS-2B), one human alveolar epithelial cell line (A549), and six primary human bronchial epithelial (HBE) cell cultures obtained from individual donors. All ECs expressed constitutive high levels of LTβR without stimulation. HVEM was detected in BEAS-2B and HBE cells, but not in A549 cells (Figure 1A and 1B). LTβR expression in HBE cells was similar between donors, whereas HVEM expression varied (Figure 1C). Thus, bronchial and alveolar epithelial cells have the potential to respond to LIGHT and LTαβ. In line with this, addition of soluble recombinant LIGHT resulted in downregulation of surface LTβR in all ECs within 24hrs, but did not affect mRNA levels (Figure 1D–F). HVEM expression was unaffected. This indirectly suggested that LIGHT could be active and LTβR was the primary receptor responsive to LIGHT.
Figure 1. Expression of LTβR and HVEM in lung epithelial cells.

The human bronchial epithelial cell line, BEAS-2B, the human alveolar cell line, A549, and human primary bronchial epithelial (HBE) cells were assessed by flow cytometry (A, C, D, E) and mRNA analysis (B, F) for expression of LTβR and HVEM. (A) LTβR and HVEM, black histograms, on representative samples of all cell populations. Isotype control, gray histograms. (B) Mean mRNA expression in all cell populations, n=3 independent experiments. (C) Staining of HBE cells from 6 individual donors. (D–F) HBE cells were cultured in the absence (Unst) or presence of recombinant LIGHT. (D) Representative LTβR and HVEM expression assessed by flow at 48hr. Dashed line indicates peak receptor expression in unstimulated cells. Data representative of three independent experiments. (E) MFI of LTβR expression by flow from 3 donors over time; (F) Mean LTβR mRNA levels of HBE over time, n=3 independent experiments.
We then assessed the biological response to LIGHT in an attempt to define a gene signature of potential relevance to future analyses of human patient samples. We primarily measured mRNA expression of the genes indicated, but where possible, we also confirmed production or upregulation of the relevant protein (see Table II). Arbitrarily, we considered a significant effect of LIGHT to be upregulation of mRNA by greater than 2-fold (see below and Table II). All activities described were dose dependent, with maximal activity at 50–100 ng/ml of LIGHT, and LIGHT did not affect the viability of any ECs in culture over 5 days (not shown). In general, mRNA for the indicated proteins was induced or upregulated by LIGHT within 24–48hrs and progressively increased over 5 days. Where LIGHT had no effect on gene transcription, this applied to both early and late time points analyzed (Table II and data not shown).
Table II.
LIGHT induction of mRNA in human lung epithelial cells
| LIGHT Induction of mRNA | |||||
|---|---|---|---|---|---|
| Molecule | BEAS-2B | A549 | NHBE | ||
| ICAM-1 | +++ | +++ | ++ | * | Adhesion molecules |
| VCAM-1 | +++ | +++ | − | * | |
| CCL2 (MCP-1) | − | − | − | * | CC Chemokines |
| CCL3 (MIP-1α) | − | − | − | * | |
| CCL5 (RANTES) | +++ | +++ | +++ | * | |
| CCL13 (MCP-4) | − | − | − | * | |
| CCL11 (Eotaxin-1) | − | − | − | * | |
| CCL17 (TARC) | + | + | − | * | |
| CCL20 | ++ | +++ | + | ||
| CCL21 | − | − | − | ||
| CCL22 (MDC) | − | − | − | * | |
| CCL24 (Eotaxin-2) | − | − | − | * | |
| CCL26 (Eotaxin-3) | − | − | − | * | |
| CXCL1 | ++ | + | + | CXC Chemokines | |
| CXCL2 | + | + | + | ||
| CXCL3 | ++ | + | ++ | ||
| CXCL5 | − | ++ | +++ | ||
| CXCL8 (IL-8) | + | + | + | * | |
| CXCL10 (IP-10) | − | − | − | * | |
| CXCL11 | ++ | ++ | ++ | ||
| CXCL12 | − | − | − | ||
| CXCL13 | − | − | − | ||
| GM-CSF | ++ | ++ | +++ | * | Growth factors and cytokines |
| VEGF | − | − | + | * | |
| IL-1α | − | − | + | * | |
| IL-1β | − | − | − | * | |
| IL-6 | ++ | ++ | ++ | * | |
| IL-7 | − | − | − | * | |
| IL-10 | − | − | − | * | |
| TGF-beta | − | − | − | * | |
| Activin-A | + | + | ++ | * | |
| MMP-2 | − | − | − | * | Remodeling factors |
| MMP-9 | +++ | +++ | +++ | * | |
| MMP-13 | − | − | − | ||
| TIMP-1 | − | − | − | ||
| ADAM-8 | +++ | − | ++ | ||
| α-SMA | + | − | − | * | |
| Vimentin | + | + | + | * | |
| Collagen I | − | − | − | ||
| Fibronectin | − | − | ++ | * | |
| Tenascin C | + | − | − | ||
confirmed at the protein level
(−) no fold increase
(+) ≤ 2 fold increase
(++) 2–5 fold increase
(+++) > 5 fold increase
LIGHT upregulates the adhesion molecules ICAM-1 and VCAM-1
We first assessed whether LIGHT could induce the expression of the adhesion molecules ICAM-1 and VCAM-1 that play important roles in enabling leukocyte recruitment and infiltration to the lungs. All ECs constitutively expressed ICAM-1, but their levels differed strongly. Importantly, LIGHT significantly upregulated ICAM-1 in both bronchial and alveolar epithelial cells. Induction was evident within 24hr and increased progressively over 5 days (Figure 2A–C). VCAM-1 was weakly detected on resting lung ECs. However, induction of VCAM-1 was observed in BEAS-2B and A549 cells when stimulated with LIGHT, again progressively increasing from 24hr (Figure 2D–E). In contrast, primary HBE cells failed to express VCAM-1 after LIGHT stimulation (Figure 2E). A summary of the relative magnitude of increase in mRNA for these molecules induced by LIGHT is in Table II.
Figure 2. LIGHT upregulates adhesion molecules in lung epithelial cells.

ECs were cultured in the absence or presence of recombinant LIGHT. (A) ICAM-1 expression in representative cells assessed by flow cytometry at 72hr. Unstimulated (gray); LIGHT (black). Isotype control, light gray. (B) Mean ICAM-1 mRNA expression after 5 days. Unstimulated (gray bar); LIGHT (black bar). n=5 independent experiments. (C) Fold increase over unstimulated cells in ICAM-1 mRNA in HBE from 6 individual donors over time. (D) VCAM-1 expression in representative cultures of unstimulated (unst) versus LIGHT stimulated BEAS-2B and A549 cells at 48hr. Data representative of three independent experiments. (E) Mean VCAM-1 mRNA expression over time in ECs. Unstimulated (gray bar); LIGHT (black bar). n=3 independent experiments. n.d. – not detected.
LIGHT induces a specific profile of chemokines, growth factors, and cytokines
Next, we examined chemokine production by ECs. LIGHT strongly enhanced production of CCL5 (RANTES), a major chemoattractant for T cells, eosinophils, and basophils (Figure 3A, and Table II). An increase in mRNA for CCL20, a chemotactic factor for lymphocytes, was also observed in all ECs, even though the base mRNA levels of this chemokine varied (Figure 3B). In contrast, other CC chemokines, like CCL2, CCL3, CCL17, CCL21 and CCL22, that promote migration of monocytes/macrophages, as well as the chemokines CCL11, CCL24, and CCL26, that are chemoattractants for eosinophils, were not upregulated at all, or to any great extent, after LIGHT stimulation, regardless of the lung EC population studied (see Table II). From the CXC chemokines studied, CXCL1, CXCL3 and CXCL5, which can act on neutrophils, were induced by LIGHT, although this varied between the EC types (Figure 3C). In contrast, CXCL11, a chemotactic factor for T cells, was strongly upregulated in all cells (Figure 3C). Also, CXCL8 (IL-8) and CXCL2, additional chemoattractants for neutrophils, which were constitutively secreted by all EC populations, were only modestly upregulated after LIGHT stimulation (Table II). CXCL10, CXCL12, CXCL13, and CX3CL1, chemotactic factors for monocytes, DC, and B cells, were not induced by LIGHT (Table II). This suggests that LIGHT might selectively contribute to chemotaxis of inflammatory cells, and in line with this conclusion, we observed increased migration of lymphocytes, including T cells, but not monocytes, in a double chamber chemotaxis assay, toward supernatants of LIGHT-treated epithelial cells (Supplementary Figure 1).
Figure 3. LIGHT induces the secretion of a specific group of chemokines and cytokines by lung epithelial cells.

ECs were left unstimulated or stimulated with recombinant LIGHT. (A) Mean CCL5 mRNA (left) and protein (right) after 5 days of culture. n=3 independent experiments. (B–C) Mean CCL20 (B), CXCL1, CXCL3, CXCL5 and CXCL11 (C) mRNA after 5 days. n=3 independent experiments. (D–F) Mean GM-CSF (D), IL-6 (E), and activin A (F) mRNA and protein levels after 5 days. n=3–6 independent experiments. Unstimulated (gray bars); LIGHT (black bars).
ECs were also analyzed for the production of growth factors and pro-inflammatory cytokines. GM-CSF, an immune modulator of the growth of myeloid cells such as dendritic cells, was strongly promoted after LIGHT stimulation in all EC cultures (Figure 3D). LIGHT additionally promoted the production of two pro-inflammatory cytokines in a selective manner, with IL-6 consistently enhanced to a strong degree in all cell populations analyzed, and activin A robustly targeted in HBE and A549 cells although more modestly in BEAS-2B (Figure 3E and 3F). VEGF and IL-1α were only weakly induced in HBE cells, but not in BEAS2-B or A549 cells, and other cytokines, TGF-β, IL-1β, IL-6, IL-7, and IL-10, were not induced after LIGHT stimulation in any ECs (Table II, data not shown). Thus, LIGHT promotes only select chemokines and cytokines suggesting a precise signature of action.
LIGHT has a selective effect in promoting metalloproteinases
To further this conclusion, we assessed molecules implicated in lung remodeling activities associated with severe asthma. Metalloproteinases and their inhibitors could play a pivotal role in the pathogenesis of asthma via regulation and fine-tuning of the balance between matrix deposition and degradation, as well as the migration of inflammatory cells into the airways (20, 21). LIGHT strongly upregulated matrix metalloproteinase-9 (MMP-9) mRNA in all ECs, even though the base levels differed between HBE, BEAS-2B, and A549 cells (Figure 4A and Table II). In contrast, mRNA levels of MMP-2, MMP-13, and tissue inhibitor of metalloproteinase-1 (TIMP-1) were not altered (Figure 4B). MMP-9 protein was found in a secreted form, as shown by western analysis of supernatants, being produced after 1 day and accumulating thereafter (Figure 4C). With a fluorometric assay, we confirmed strong MMP-9 enzymatic activity in LIGHT-stimulated HBE cell cultures (Figure 4D), and found that the majority, if not all, of MMP-9 produced was in an active form since no statistical difference was observed in enzymatic activity after addition of APMA, a reagent that converts latent into active enzymes (Figure 4D).
Figure 4. LIGHT promotes selective expression of MMP-9 and ADAM-8 in lung epithelial cells.
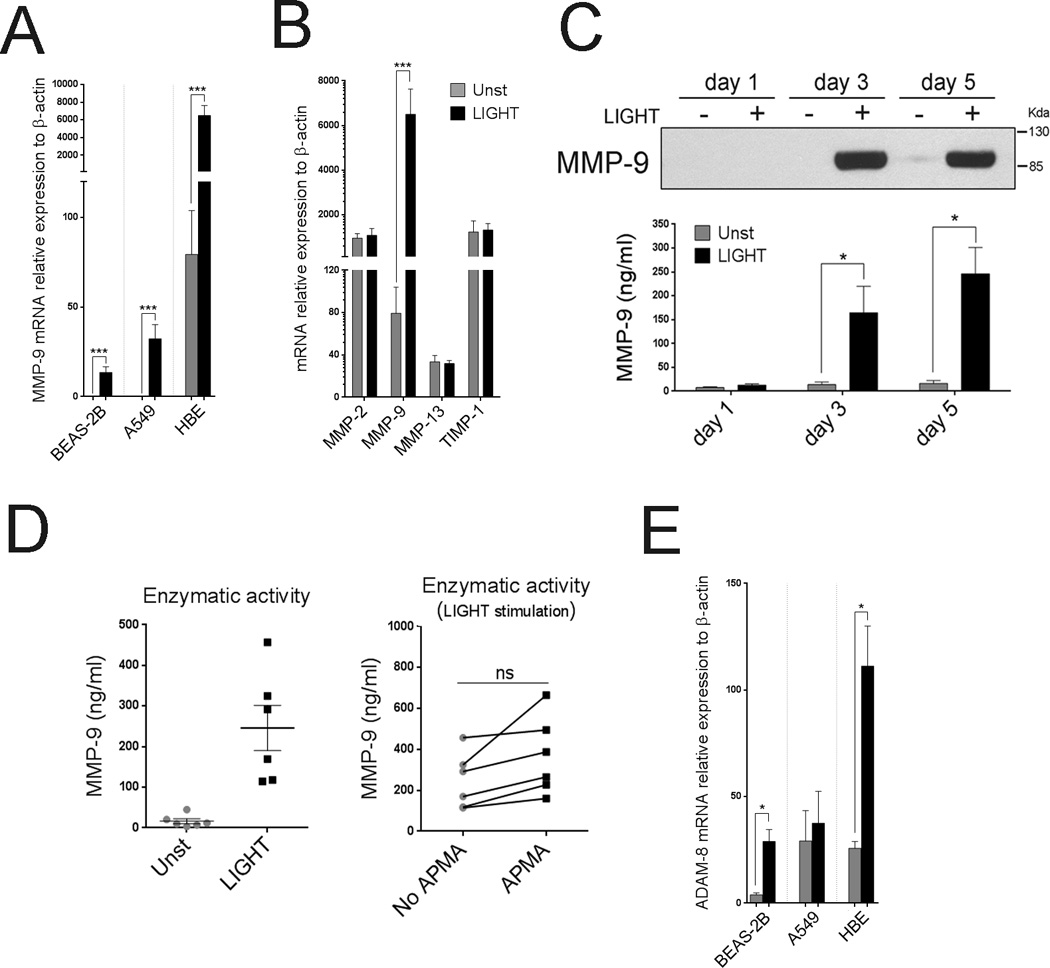
(A) Mean MMP-9 mRNA levels after 5 days in ECs cultured with (black bars) or without (gray bars) rLIGHT. n=3 independent experiments. (B) Mean MMP-2, MMP-9, MMP-13, and TIMP-1 mRNA relative expression, in HBE cells after 5 days of culture. n=6 independent experiments. (C) MMP-9 protein in supernatants of cultured HBE cells over time, by immunoblotting (top). Enzymatic activity of secreted MMP-9 over time measured by enzymatic fluorometric assay (bottom). Data representative of three independent experiments. (D) Enzymatic activity of secreted MMP-9 in HBE culture supernatants after 5 days, from 6 individual donors, measured by enzymatic fluorometric assay. Endogenous versus potential activity shown on the right after APMA activation. (E). Mean ADAM-8 mRNA levels after 5 days of culture in the absence (gray bars) or presence of LIGHT (black bars). n=3 independent experiments.
Aminopeptidase N (CD13), a membrane-bound metalloproteinase, was detected on the surface of all ECs, but levels did not change after LIGHT stimulation (data not shown). In contrast, ADAM-8, a disintegrin and metalloproteinase domain family member, that is increased in airways of asthmatic patients (22), was also strongly upregulated at the mRNA level by LIGHT in HBE and BEAS-2B cells, although not in A549 cells (Figure 4E). These data again suggest that LIGHT has very specific activities rather than being broadly stimulatory for any epithelial product.
LIGHT has little effect in promoting collagen, fibronectin, or alpha smooth muscle actin expression, and a moderate activity in EMT
We additionally assessed molecules that have been associated with epithelial-mesenchymal transition (EMT) that may contribute to decreased lung function in severe asthmatics (23, 24). In this case, we directly compared LIGHT to TGF-β as this has been described to be the primary factor contributing to EMT (25). As shown in Figure 5A–C, TGF-β strongly reduced expression of E-cadherin, a molecule thought to regulate epithelial integrity and tight junctions, although this was more pronounced in BEAS-2B and A549 cells. LIGHT stimulation reproduced this activity in BEAS-2B and A549 cells, but to a lower extent, and had no significant effect on HBE cells. Concurrently, TGF-β had some activity, albeit not strong, on enhancing expression of α-Smooth Muscle Actin (SMA) and Vimentin, although again this varied between the three cell populations. In contrast, LIGHT had only a modest effect compared to TGF-β in situations where TGF-β activity was strong (Figure 5D). Most importantly, LIGHT did not exert strong activity in promoting what are arguably the hallmarks of remodeling in epithelial cells, namely induction of Fibronectin and Collagen I, all targets of TGF-β as confirmed in our experiments (Figure 5E–F). Fibronectin was induced by LIGHT in HBE cells at the mRNA and protein levels (Figure 5E), but substantially less than seen with TGF-β, and this was not seen in BEAS-2B and A549 cells (Figure 5F). Tenascin C was also moderately induced by LIGHT in BEAS-2B cells but again less than by TGF-β, and was not significantly upregulated by either molecule in HBE or A549 cells.
Figure 5. LIGHT does not strongly promote epithelial-mesenchymal transition in lung epithelial cells.

ECs were cultured unstimulated or with TGF-β versus LIGHT. (A) Mean E-Cadherin mRNA fold decrease over unstimulated cells after 5 days. n=3 independent experiments. (B) Representative E-Cadherin expression by flow cytometry in A549 cells after 72hr. (C) E-Cadherin protein in cell lysates of BEAS-2B, A549, and HBE cells by immunoblotting over time. (D) Mean α-SMA and Vimentin mRNA fold increase after 5 days in ECs (top, n=3), and representative protein in cell lysates by immunoblotting over time (bottom) in BEAS-2B (α-SMA) and HBE (Vimentin) cells. (E–F) Mean fold increase in mRNA for Fibronectin, Collagen I, and Tenascin C after 5 days (n=3), and representative Fibronectin protein expression in HBE cell lysates over time by immunoblotting (bottom left).
LTαβ replicates the activity of LIGHT
As the alveolar epithelial cell line, A549, did not express HVEM, and we found largely similar activities of LIGHT in these cells, this suggested that LIGHT primarily signaled through LTβR. To further this conclusion, ECs were stimulated with LTαβ, the other ligand for LTβR. LTαβ treatment resulted in decreased expression of LTβR similar to LIGHT treatment (Figure 6A). Moreover, LTαβ increased the expression of ICAM-1 similar to LIGHT, and the combination of both ligands did not further impact ICAM-1 upregulation (Figure 6B and not shown). Similar results were observed for VCAM-1 (data not shown). Likewise, LTαβ treatment promoted the secretion of inflammatory mediators in the same fashion and extent as LIGHT, including GM-CSF, CCL5, MMP-9, and IL-6, and LTαβ had no activity in modulating the genes that LIGHT failed to induce (Figure 6C–E, and data not shown). This suggested that the LTβR was indeed the primary receptor active on lung epithelial cells, and that LIGHT exerted its effects through the LTβR. Moreover, siRNA knockdown of LTβR in A549 and BEAS-2B cells confirmed that this receptor was required for the activity of LIGHT (Supplementary Figure 2). Thus, LIGHT-LTβR or LTαβ-LTβR signaling generates a specific inflammatory signature in lung ECs.
Figure 6. LTαβ has similar activities as LIGHT in lung epithelial cells.

ECs were left unstimulated or stimulated with recombinant LIGHT compared to LTαβ. (A) LTβR surface expression in HBE cells by flow cytometry after 48hr of culture. (B) Mean fold increase in ICAM-1 mRNA over unstimulated cells after 5 days. n=3 independent experiments. (C) Mean fold increase in CCL5 and MMP-9 mRNA after 5 days. n=3 independent experiments. (D) Mean fold increase in GM-CSF mRNA (left) and protein levels (right) in all ECs or HBE cells, respectively, after 5 days. n=3 independent experiments. (F) Mean fold increase in IL-6 mRNA (left) and protein levels (right) in all ECs or HBE cells, respectively, after 5 days. n=3 independent experiments.
Many of the genes induced by LIGHT and LTαβ are steroid insensitive
Lastly, we evaluated whether the action of LIGHT and LTαβ could be dampened by corticosteroids. Basal expression of glucocorticoid receptor (GR) isoform α and β mRNA were assessed in the lung epithelial cells, as well as the glucocorticoid-induced genes GILZ and FKBP51, to confirm biologic activity of budesonide (Supplementary Figure 3). The cell lines expressed high levels of GRα and were strongly responsive, whereas HBE cells expressed much lower levels and were less responsive based on upregulation of GILZ and FKBP51. Differential effects of budesonide were observed dependent on the gene analyzed and the cell population. Steroid sensitivity was arbitrarily defined as greater than 75% inhibition of the induced activity. In spite of the weak expression of GRα and induction of GILZ/FKBP51 in HBE cells, budesonide completely suppressed induction of IL-6 by LIGHT. However, most interestingly, budesonide had no inhibitory action on any of the other genes driven by LIGHT in HBE cells regardless of the time point analyzed (Figure 7A, and Supplementary Figure 4 in the online data supplement, and data not shown). These results were reproduced with doses as high as 10−5 M (10−7 M shown). Similar steroid resistance was seen in BEAS-2B and A549 cells on the effect of LIGHT in promoting CCL5, CCL20, CXCL5, VCAM-1, CXCL1, CXCL11, and activin A, even though GRα expression and induction of GILZ/FKBP51 was pronounced in these cells (Figure 7B–C, Supplementary Figure 4). Induction of IL-6 by LIGHT was again steroid sensitive in BEAS-2B and A549 cells, and additionally budesonide suppression of LIGHT activity was seen for MMP-9, GM-CSF, ADAM-8 and ICAM-1 in these cell lines. For select genes analyzed, we also obtained similar data when LTαβ was used as the stimulus (data not shown). Thus, much of the effects of LIGHT/LTαβ were steroid resistant, partially dependent on the cell type analyzed and apparently related to the responsiveness of these cells to steroids, but certain gene products, in particular IL-6, were sensitive to the effects of steroids regardless of the former variables.
Figure 7. The action of LIGHT on multiple genes is steroid insensitive.

ECs were left unstimulated or stimulated with LIGHT in the absence (black bars) or presence of budesonide (grey bars). (A) HBE cells, (B) BEAS-2B cells, and (C) A549 cells. Data are mean fold increase in mRNA over unstimulated cells for the indicated genes after 3 days of culture. Steroid sensitivity was arbitrarily set at >75% inhibition of the LIGHT-induced response, indicated to the left of the thick dotted lines. Where applicable % inhibition indicated above histograms. n=3 independent experiments for all groups. Partial inhibition was seen in some cases (left of thin dotted lines) but was not statistically significant or >75% from controls.
DISCUSSION
Our previous studies in murine models of allergen challenge highlighted a strong role for LIGHT in driving acute lung inflammation and primary features of lung remodeling (16, 17). We proposed that T cells, macrophages, and eosinophils were targets of LIGHT to perpetuate these activities. However, other cell types in the lungs may be equally important for the overall action of LIGHT. In this regard, we recently found that recombinant LIGHT additionally promoted TSLP expression in lung epithelial cells when injected intratracheally in mice, and that TSLP was required for the remodeling activities of LIGHT (18). Furthermore, because LIGHT binds a shared receptor with LTαβ, the latter molecule may also be important. Here, we now show that LIGHT as well as LTαβ has additional strong inflammatory activities on human lung epithelial cells, promoting genes implicated in orchestrating several aspects of asthmatic reactions.
We found that LIGHT and LTαβ strongly up-regulated the expression of the adhesion molecule ICAM-1, and to a lesser extent VCAM-1. LIGHT or LTαβ may then contribute to enhancing the infiltration or retention of effector cells into the lung by regulating cell-cell adhesion. Additionally, ICAM-1 acts as a receptor for rhinoviruses, which can contribute to asthma exacerbations in children and adults (26, 27), raising the intriguing possibility that LIGHT or LTαβ may also perpetuate asthmatic activity in an indirect as well as direct manner. LIGHT and LTαβ additionally induced the expression of several chemokines, however a strong action was only seen on the CC chemokines CCL5 and 20, and the CXC chemokines, CXCL1, 3, 5 and 11. Thus, LIGHT/LTαβ modulation of genes related with chemotaxis appears to be quite specific. Exactly which cell type might be affected by LIGHT/LTαβ activity in vivo remains to be determined, but our results predict that migration of T lymphocytes and neutrophils are likely to be primarily impacted, based on the reported targets of these chemokines.
The other inflammatory mediators strongly and consistently upregulated by LIGHT and LTαβ in all ECs examined were GM-CSF, IL-6, activin A, and MMP-9. Again, the action was quite specific in that a number of other inflammatory cytokines/growth factors and other metalloproteinases were not induced. This draws another parallel with a prior study of LIGHT in human synovial fibroblasts where IL-6 and MMP-9, but not other MMPs, were promoted (28). GM-CSF and IL-6 can be growth and differentiation factors for a range of circulating leukocytes, including dendritic cells, macrophages, and T cells (29, 30). MMP-9 is elevated in asthmatic patients and may positively correlate with disease progression (31–33). However, the activity of MMP-9 as an inflammatory mediator is not fully understood. MMP-9-deficient mice, in an allergen-challenge model, displayed lower levels of CCL5 and eosinophilia and slightly reduced collagen deposition, suggesting a specific role of this molecule (34). MMP-9 may also promote the degradation of extracellular matrix proteins, modulate epithelium integrity, or release latent/inactive inflammatory proteins from their chaperones (35, 36). Similarly, activin A has been found elevated in patients with asthma and COPD (37, 38) and mice with allergen-induced asthma (37, 39), but its role, although implicated in several mechanisms of inflammation and remodeling, has not been defined fully (39–41). Lastly, LIGHT/LTαβ also strongly induced another proteinase, ADAM-8, that has increasingly been implicated in asthma (42, 43), although this was confined to bronchial-origin epithelial cells and was not seen in the alveolar epithelial cell line. This further promotes the notion that LIGHT or LTαβ may modulate the proteolytic environment and an imbalance between deposition of ECM components and their degradation.
A recent study also partially addressed an inflammatory role for LIGHT in primary bronchial epithelial cells (44). The authors found induction of MMP-9, activin A, CXCL5, and CXCL8, and moderate induction of mRNA for CXCL1, 2, and 3, and ICAM-1, which is largely in line with our results. However, our more broad analyses highlight the very selective nature of the activity of LIGHT, suggesting a specific functional role in modulating the epithelial environment rather than LIGHT being a broad-spectrum inflammatory factor. Understanding this signature may be important for future clinical studies aimed at identifying when LIGHT might be active during human asthma. Another aspect of LIGHT action implied in the aforementioned study of bronchial epithelial cells (44), and also suggested in a separate study of the A549 alveolar epithelial cell line (45), was that LIGHT has a major effect in promoting EMT. These authors showed that LIGHT down-regulated E-cadherin and moderately induced expression of vimentin, again in line with our results, but either did not assess (44), or did not convincingly demonstrate an action on (45), what we consider the cardinal features of EMT, namely induction of collagen, fibronectin, and alpha smooth muscle actin. These were all strongly upregulated by TGF-β, but were weakly, if at all, induced by LIGHT. Thus, although the collective findings suggest that LIGHT and LTαβ may contribute to loss of epithelial integrity via reducing E-cadherin expression, their action is again quite specific and selective in not directly promoting extracellular matrix protein production in lung epithelial cells. This suggests LIGHT and/or LTαβ will primarily be upstream drivers of pro-fibrotic factors rather than being pro-fibrotic factors per se. Our recent results in mouse models in vivo, showing that LIGHT controls TSLP, TGF-β, and IL-13 production, and that these cytokines were required for LIGHT to induce collagen and alpha smooth muscle actin expression, is in line with this conclusion (18).
Our finding that the LTβR was primarily active in all human epithelial cells, regardless of HVEM expression, are consistent with a report that found that synovial fibroblasts expressed both LTβR and HVEM, but only knockdown of LTβR prevented the effects of LIGHT in promoting fibroblast proliferation and upregulating several chemokines (33). Also Braun et al. (46) had previously found that LTαβ induced expression of ICAM-1, several chemokines including CCL5, and several MMPs, in synovial fibroblasts, further in line with the idea that LTβR may play a distinct and specific role in controlling the response of structural cells. Interestingly, this may be a general property of several TNF family molecules that can act on structural cells as a brief comparison of LIGHT to TNF itself showed induction of a similar profile of inflammatory mediators, albeit the kinetics of response were different for some gene products likely related to the nature of the signals through LTβR versus TNFR (data not shown). All TNFR family molecules can activate canonical and/or non-canonical NF-κB. TNF may elicit the fast canonical NF-κB pathway as well as the non-canonical pathway, whereas LIGHT through LTβR may primarily exert its effects through the slower acting non-canonical pathway. However, much would need to be done in this area before taking such a simplistic outlook on the signaling pathways that are operative in lung epithelial cells.
Lastly, we found that much of the action of LIGHT and LTαβ in vitro was not suppressed by budesonide, although the exact targets of LIGHT that were resistant to steroid treatment varied between the cell populations studied. Corticosteroids are the first line of therapy for asthmatics and are effective in reducing many of the symptoms of disease (47). However, it is of great interest to understand which inflammatory factors are not affected by steroid treatment, or whether certain activities of these factors are steroid sensitive and other activities are resistant. IL-6 was the only molecule promoted by LIGHT/LTαβ that was consistently suppressed by budesonide in all epithelial cells, whereas induction of other molecules like MMP-9, GM-CSF, and ICAM-1 was blocked in the cell lines but not in primary bronchial epithelial cells. The reason for this is not clear, and may reflect both the nature of the signaling pathways required for eliciting these molecules as well as intrinsic regulatory activities that are promoted by budesonide that might vary in each cell population. Indeed, we found that A549 and BEAS-2B cells have higher expression of GRα and consequently are more responsive to corticosteroids when compared to HBE cells. However, this could not fully explain the differential sensitivity of IL-6 and other molecules to budesonide that was observed across the alternate epithelial cell populations. Similarly, why many targets such as CCL5, CCL20, CXCL1, CXCL11, and activin A, were not affected by budesonide, regardless of the EC population studied, again is likely reflective of how LIGHT/LTαβ signal through the LTβR to turn on the genes for these molecules and that budesonide cannot block these specific pathways. Whether our data here will mean that the in vivo effects of LIGHT in the lung are steroid insensitive remains to be determined. Although we did find that certain activities were blocked by budesonide, one caveat is that the submerged epithelial cultures used in our experiments might not truly reflect the differentiation state of an epithelial cell in vivo. Thus, future studies will be required to correlate the in vivo activity of LIGHT with lung inflammatory activity that is resistant to steroids.
In conclusion, LIGHT and LTαβ can promote a distinct gene signature in human lung epithelial cells that express their shared receptor, LTβR. Together with our prior data suggesting that LIGHT via LTβR can drive TGF-β production by macrophages and TSLP production by epithelial cells, and LIGHT via HVEM can enhance IL-13 production by eosinophils (17,18), these findings indicate that LIGHT signaling may play a role as a pleiotropic inflammatory factor in the pathogenesis of asthma and other lung diseases, and that LTαβ signaling may further contribute to development or maintenance of the lung inflammatory response.
Supplementary Material
Acknowledgments
Sources of Support:
This work was supported by NIH grant AI070535 to M.C, and by Janssen Research and Development.
REFERENCES
- 1.Bochner BS, Undem BJ, Lichtenstein LM. Immunological aspects of allergic asthma. Annu Rev Immunol. 1994;12:295–335. doi: 10.1146/annurev.iy.12.040194.001455. [DOI] [PubMed] [Google Scholar]
- 2.Bousquet J, Clark TJ, Hurd S, Khaltaev N, Lenfant C, O'Byrne P, Sheffer A. GINA guidelines on asthma and beyond. Allergy. 2007;62:102–112. doi: 10.1111/j.1398-9995.2006.01305.x. [DOI] [PubMed] [Google Scholar]
- 3.Lemanske RF, Jr, Busse WW. Asthma: clinical expression and molecular mechanisms. J Allergy Clin Immunol. 2010;125:S95–S102. doi: 10.1016/j.jaci.2009.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hasenberg M, Stegemann-Koniszewski S, Gunzer M. Cellular immune reactions in the lung. Immunol Rev. 2013;251:189–214. doi: 10.1111/imr.12020. [DOI] [PubMed] [Google Scholar]
- 5.Nakagome K, Nagata M. Pathogenesis of airway inflammation in bronchial asthma. Auris Nasus Larynx. 2011;38:555–563. doi: 10.1016/j.anl.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 6.James AL, Elliot JG, Jones RL, Carroll ML, Mauad T, Bai TR, Abramson MJ, McKay KO, Green FH. Airway smooth muscle hypertrophy and hyperplasia in asthma. Am J Respir Crit Care Med. 2012;185:1058–1064. doi: 10.1164/rccm.201110-1849OC. [DOI] [PubMed] [Google Scholar]
- 7.Kranenburg AR, Willems-Widyastuti A, Moori WJ, Sterk PJ, Alagappan VK, de Boer WI, Sharma HS. Enhanced bronchial expression of extracellular matrix proteins in chronic obstructive pulmonary disease. Am J Clin Pathol. 2006;126:725–735. doi: 10.1309/jc477fael1ykv54w. [DOI] [PubMed] [Google Scholar]
- 8.Camelo A, Dunmore R, Sleeman MA, Clarke DL. The epithelium in idiopathic pulmonary fibrosis: breaking the barrier. Front Pharmacol. 2014;4:173. doi: 10.3389/fphar.2013.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Georas SN, Rezaee F. Epithelial barrier function: at the front line of asthma immunology and allergic airway inflammation. J Allergy Clin Immunol. 2014;134:509–520. doi: 10.1016/j.jaci.2014.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holtzman MJ, Byers DE, Alexander-Brett J, Wang X. The role of airway epithelial cells and innate immune cells in chronic respiratory disease. Nat Rev Immunol. 2014;14:686–698. doi: 10.1038/nri3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loxham M, Davies DE, Blume C. Epithelial function and dysfunction in asthma. Clin Exp Allergy. 2014;44:1299–1313. doi: 10.1111/cea.12309. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Moffatt MF, Cookson WO. Genetic and genomic approaches to asthma: new insights for the origins. Curr Opin Pulm Med. 2012;18:6–13. doi: 10.1097/MCP.0b013e32834dc532. [DOI] [PubMed] [Google Scholar]
- 13.Blume C, Swindle EJ, Dennison P, Jayasekera NP, Dudley S, Monk P, Behrendt H, Schmidt-Weber CB, Holgate ST, Howarth PH, Traidl-Hoffmann C, Davies DE. Barrier responses of human bronchial epithelial cells to grass pollen exposure. Eur Respir J. 2013;42:87–97. doi: 10.1183/09031936.00075612. [DOI] [PubMed] [Google Scholar]
- 14.Hackett TL, Singhera GK, Shaheen F, Hayden P, Jackson GR, Hegele RG, Van Eeden S, Bai TR, Dorscheid DR, Knight DA. Intrinsic phenotypic differences of asthmatic epithelium and its inflammatory responses to respiratory syncytial virus and air pollution. Am J Respir Cell Mol Biol. 2011;45:1090–1100. doi: 10.1165/rcmb.2011-0031OC. [DOI] [PubMed] [Google Scholar]
- 15.Uller L, Leino M, Bedke N, Sammut D, Green B, Lau L, Howarth PH, Holgate ST, Davies DE. Double-stranded RNA induces disproportionate expression of thymic stromal lymphopoietin versus interferon-beta in bronchial epithelial cells from donors with asthma. Thorax. 2010;65:626–632. doi: 10.1136/thx.2009.125930. [DOI] [PubMed] [Google Scholar]
- 16.Soroosh P, Doherty TA, So T, Mehta AK, Khorram N, Norris PS, Scheu S, Pfeffer K, Ware C, Croft M. Herpesvirus entry mediator (TNFRSF14) regulates the persistence of T helper memory cell populations. J Exp Med. 2011;208:797–809. doi: 10.1084/jem.20101562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doherty TA, Soroosh P, Khorram N, Fukuyama S, Rosenthal P, Cho JY, Norris PS, Choi H, Scheu S, Pfeffer K, Zuraw BL, Ware CF, Broide DH, Croft M. The tumor necrosis factor family member LIGHT is a target for asthmatic airway remodeling. Nat Med. 2011;17:596–603. doi: 10.1038/nm.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herro R, Da Silva Antunes R, Aguilera AR, Tamada K, Croft M. Tumor necrosis factor superfamily 14 (LIGHT) controls thymic stromal lymphopoietin to drive pulmonary fibrosis. J Allergy Clin Immunol. 2015 doi: 10.1016/j.jaci.2014.12.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ware CF. Targeting lymphocyte activation through the lymphotoxin and LIGHT pathways. Immunol Rev. 2008;223:186–201. doi: 10.1111/j.1600-065X.2008.00629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demedts IK, Brusselle GG, Bracke KR, Vermaelen KY, Pauwels RA. Matrix metalloproteinases in asthma and COPD. Curr Opin Pharmacol. 2005;5:257–263. doi: 10.1016/j.coph.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki R, Miyazaki Y, Takagi K, Torii K, Taniguchi H. Matrix metalloproteinases in the pathogenesis of asthma and COPD: implications for therapy. Treat Respir Med. 2004;3:17–27. doi: 10.2165/00151829-200403010-00003. [DOI] [PubMed] [Google Scholar]
- 22.Paulissen G, Rocks N, Quesada-Calvo F, Gosset P, Foidart JM, Noel A, Louis R, Cataldo DD. Expression of ADAMs and their inhibitors in sputum from patients with asthma. Mol Med. 2006;12:171–179. doi: 10.2119/2006-00028.Paulissen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hackett TL. Epithelial-mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr Opin Allergy Clin Immunol. 2012;12:53–59. doi: 10.1097/ACI.0b013e32834ec6eb. [DOI] [PubMed] [Google Scholar]
- 24.Pain M, Bermudez O, Lacoste P, Royer PJ, Botturi K, Tissot A, Brouard S, Eickelberg O, Magnan A. Tissue remodelling in chronic bronchial diseases: from the epithelial to mesenchymal phenotype. Eur Respir Rev. 2014;23:118–130. doi: 10.1183/09059180.00004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, Argentieri R, Kicic A, Stick SM, Bai TR, Knight DA. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am J Respir Crit Care Med. 2009;180:122–133. doi: 10.1164/rccm.200811-1730OC. [DOI] [PubMed] [Google Scholar]
- 26.Friedlander SL, Busse WW. The role of rhinovirus in asthma exacerbations. J Allergy Clin Immunol. 2005;116:267–273. doi: 10.1016/j.jaci.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 27.Traub S, Nikonova A, Carruthers A, Dunmore R, Vousden KA, Gogsadze L, Hao W, Zhu Q, Bernard K, Zhu J, Dymond M, McLean GR, Walton RP, Glanville N, Humbles A, Khaitov M, Wells T, Kolbeck R, Leishman AJ, Sleeman MA, Bartlett NW, Johnston SL. An anti-human ICAM-1 antibody inhibits rhinovirus-induced exacerbations of lung inflammation. PLoS Pathog. 2013;9:e1003520. doi: 10.1371/journal.ppat.1003520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pierer M, Brentano F, Rethage J, Wagner U, Hantzschel H, Gay RE, Gay S, Kyburz D. The TNF superfamily member LIGHT contributes to survival and activation of synovial fibroblasts in rheumatoid arthritis. Rheumatology (Oxford) 2007;46:1063–1070. doi: 10.1093/rheumatology/kem063. [DOI] [PubMed] [Google Scholar]
- 29.Rincon M, Irvin CG. Role of IL-6 in asthma and other inflammatory pulmonary diseases. Int J Biol Sci. 2012;8:1281–1290. doi: 10.7150/ijbs.4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, Zhang Y, Zhang L, Yuan ZR, Tan HS, Das G, Devadas S. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don't know. Cell Res. 2006;16:126–133. doi: 10.1038/sj.cr.7310017. [DOI] [PubMed] [Google Scholar]
- 31.Lee KS, Min KH, Kim SR, Park SJ, Park HS, Jin GY, Lee YC. Vascular endothelial growth factor modulates matrix metalloproteinase-9 expression in asthma. Am J Respir Crit Care Med. 2006;174:161–170. doi: 10.1164/rccm.200510-1558OC. [DOI] [PubMed] [Google Scholar]
- 32.Mautino G, Oliver N, Chanez P, Bousquet J, Capony F. Increased release of matrix metalloproteinase-9 in bronchoalveolar lavage fluid and by alveolar macrophages of asthmatics. Am J Respir Cell Mol Biol. 1997;17:583–591. doi: 10.1165/ajrcmb.17.5.2562. [DOI] [PubMed] [Google Scholar]
- 33.Oshita Y, Koga T, Kamimura T, Matsuo K, Rikimaru T, Aizawa H. Increased circulating 92 kDa matrix metalloproteinase (MMP-9) activity in exacerbations of asthma. Thorax. 2003;58:757–760. doi: 10.1136/thorax.58.9.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim DH, Cho JY, Miller M, McElwain K, McElwain S, Broide DH. Reduced peribronchial fibrosis in allergen-challenged MMP-9-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2006;291:L265–L271. doi: 10.1152/ajplung.00305.2005. [DOI] [PubMed] [Google Scholar]
- 35.Perng DW, Chang KT, Su KC, Wu YC, Chen CS, Hsu WH, Tsai CM, Lee YC. Matrix metalloprotease-9 induces transforming growth factor-beta(1) production in airway epithelium via activation of epidermal growth factor receptors. Life Sci. 2011;89:204–212. doi: 10.1016/j.lfs.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 36.Vermeer PD, Denker J, Estin M, Moninger TO, Keshavjee S, Karp P, Kline JN, Zabner J. MMP9 modulates tight junction integrity and cell viability in human airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2009;296:L751–L762. doi: 10.1152/ajplung.90578.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karagiannidis C, Hense G, Martin C, Epstein M, Ruckert B, Mantel PY, Menz G, Uhlig S, Blaser K, Schmidt-Weber CB. Activin A is an acute allergen-responsive cytokine and provides a link to TGF-beta-mediated airway remodeling in asthma. J Allergy Clin Immunol. 2006;117:111–118. doi: 10.1016/j.jaci.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 38.Verhamme FM, Bracke KR, Amatngalim GD, Verleden GM, Van Pottelberge GR, Hiemstra PS, Joos GF, Brusselle GG. Role of activin-A in cigarette smoke-induced inflammation and COPD. Eur Respir J. 2014;43:1028–1041. doi: 10.1183/09031936.00082413. [DOI] [PubMed] [Google Scholar]
- 39.Cho SH, Yao Z, Wang SW, Alban RF, Barbers RG, French SW, Oh CK. Regulation of activin A expression in mast cells and asthma: its effect on the proliferation of human airway smooth muscle cells. J Immunol. 2003;170:4045–4052. doi: 10.4049/jimmunol.170.8.4045. [DOI] [PubMed] [Google Scholar]
- 40.Gregory LG, Mathie SA, Walker SA, Pegorier S, Jones CP, Lloyd CM. Overexpression of Smad2 drives house dust mite-mediated airway remodeling and airway hyperresponsiveness via activin and IL-25. Am J Respir Crit Care Med. 2010;182:143–154. doi: 10.1164/rccm.200905-0725OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kariyawasam HH, Semitekolou M, Robinson DS, Xanthou G. Activin-A: a novel critical regulator of allergic asthma. Clin Exp Allergy. 2011;41:1505–1514. doi: 10.1111/j.1365-2222.2011.03784.x. [DOI] [PubMed] [Google Scholar]
- 42.Naus S, Blanchet MR, Gossens K, Zaph C, Bartsch JW, McNagny KM, Ziltener HJ. The metalloprotease-disintegrin ADAM8 is essential for the development of experimental asthma. Am J Respir Crit Care Med. 2010;181:1318–1328. doi: 10.1164/rccm.200909-1396OC. [DOI] [PubMed] [Google Scholar]
- 43.Oreo KM, Gibson PG, Simpson JL, Wood LG, McDonald VM, Baines KJ. Sputum ADAM8 expression is increased in severe asthma and COPD. Clin Exp Allergy. 2014;44:342–352. doi: 10.1111/cea.12223. [DOI] [PubMed] [Google Scholar]
- 44.Hung JY, Chiang SR, Tsai MJ, Tsai YM, Chong IW, Shieh JM, Hsu YL. LIGHT is a crucial mediator of airway remodeling. J Cell Physiol. 2015;230:1042–1053. doi: 10.1002/jcp.24832. [DOI] [PubMed] [Google Scholar]
- 45.Mikami Y, Yamauchi Y, Horie M, Kase M, Jo T, Takizawa H, Kohyama T, Nagase T. Tumor necrosis factor superfamily member LIGHT induces epithelial-mesenchymal transition in A549 human alveolar epithelial cells. Biochem Biophys Res Commun. 2012;428:451–457. doi: 10.1016/j.bbrc.2012.10.097. [DOI] [PubMed] [Google Scholar]
- 46.Braun A, Takemura S, Vallejo AN, Goronzy JJ, Weyand CM. Lymphotoxin beta-mediated stimulation of synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2004;50:2140–2150. doi: 10.1002/art.20356. [DOI] [PubMed] [Google Scholar]
- 47.Alangari AA. Corticosteroids in the treatment of acute asthma. Ann Thorac Med. 2014;9:187–192. doi: 10.4103/1817-1737.140120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.