

Abstract
Detection of intracellular DNA triggers activation of the STING-dependent interferon-stimulatory DNA (ISD) pathway, which is essential for antiviral immune responses. However, chronic activation of this pathway is implicated in autoimmunity. Mutations in TREX1, a 3’ repair exonuclease that degrades cytosolic DNA, cause Aicardi-Goutieres Syndrome (AGS) and chilblain lupus. Trex1−/− mice develop lethal, IFN-driven autoimmune disease that is dependent on activation of the ISD pathway, but the DNA sensors that detect the endogenous DNA that accumulates in Trex1−/− mice have not been defined. Multiple DNA sensors have been proposed to activate the ISD pathway, including cyclic GMP-AMP synthase (cGAS). Here we show that Trex1−/− mice lacking cGAS are completely protected from lethality, exhibit dramatically reduced tissue inflammation, and fail to develop autoantibodies. These findings implicate cGAS as a key driver of autoimmune disease and suggest that cGAS inhibitors may be useful therapeutics for AGS and related autoimmune diseases.
Introduction
Detection of intracellular nucleic acids and production of type I IFNs are essential for antiviral immunity. The RNA helicases RIG-I and MDA5 sense intracellular RNA and signal via MAVS to promote IFN production, whereas cGAS detects intracellular DNA ligands and signals via the adaptor STING to trigger the interferon-stimulatory DNA (ISD) response (1). cGAS is a nucleotidyltransferase that directly binds dsDNA in the cytosol and, upon DNA binding, catalyzes the production of cGAMP, which directly binds to and activates STING, triggering production of type I IFNs (2, 3). Studies with cGAS-deficient mice have shown that cGAS is required for the IFN response to transfected DNA ligands, infection with DNA viruses, and retrovirus infection (4-8).
Although critical for antiviral immunity, innate nucleic acid sensing pathways must be tightly regulated to avoid an IFN response against endogenous nucleic acid ligands. The 3’ repair exonuclease Trex1, which degrades cytosolic DNA, is a key regulator of the ISD pathway (9). Loss of function mutations in TREX1 cause Aicardi-Goutieres syndrome (AGS), a rare autoimmune disease that presents in neonates with clinical features that mimic congenital viral infection (10). AGS patients exhibit elevated type I IFNs, encephalopathy, psychomotor retardation, and premature death (11, 12). Currently, no ameliorative treatments exist for AGS patients (13). TREX1 mutations also cause familial chilblain lupus (FCL) and retinal vasculopathy with cerebral leukodystrophy (RVCL) and are associated with systemic lupus erythematosus (SLE) (14-18).
Trex1−/− mice recapitulate many of the key features of AGS and are a tractable model to study AGS disease mechanisms. Trex1-deficient cells accumulate endogenous DNA substrates, including reverse-transcribed retroelement cDNA, that triggers excessive production of type I IFNs resulting in lethal autoimmune disease (9). Autoimmune disease in Trex1−/− mice is dependent on activation of the ISD pathway, as Trex1−/− mice that lack STING, IRF3, the type I IFN receptor, or lymphocytes are completely protected from lethality and tissue destruction (9, 19). However, the DNA sensors that detect these endogenous DNA substrates that accumulate in Trex1−/− mice and trigger IFN production have not been defined. cGAS is a key DNA sensor that activates the ISD response, yet multiple other DNA sensors have been proposed to activate this pathway, including DAI (20), DDX41 (21), and the AIM2-like receptors, IFI16, IFI204, and IFI203 (22-24). One recent study demonstrated that depletion of cGAS within Trex1−/− mouse embryonic fibroblasts reduced the type I IFN gene signature that is present within these cells (25). However, nothing is known about the in vivo role of cGAS in autoimmune pathology. Here, we demonstrate that cGAS is essential for all aspects of the autoimmune disease in Trex1−/− mice, providing further evidence for its central role as an ISD sensor and revealing a new therapeutic target for AGS and related diseases.
Materials and Methods
Mice
We generated cGAS−/− (Mb21d1−/−) mice by in vitro fertilization of C57BL/6J oocytes with frozen sperm carrying a “knockout first” null allele of cGAS from the International Knockout Mouse Consortium (C57BL/6N-Mb21d1tm1a(EUCOMM)Hmgu, http://www.mousephenotype.org/data/alleles/MGI:2442261/tm1a(EUCOMM)Hmgu). Mice were genotyped using the following primers: WT Fwd, GCTGTCTCCACTTGGGCATCT; KO Fwd, AGATGGCGCAACGCAATTAATG; Common Rev, AAGCAGCCACATGAATAGTCTC. cGAS-deficient mice with the “knockout first” null allele of cGAS were intercrossed with Trex1−/− mice to generate cGAS−/−Trex1−/− mice. Trex1−/−, Tmem173−/−, and Rag2−/− mice have been described (19). All mouse studies were carried out in a specific-pathogen-free facility at the University of Washington with approval of the UW Institutional Animal Care and Use Committee.
Pathology
All tissues were fixed in 10% neutral buffered formalin and paraffin embedded. Tissue sections (5μm) were stained with hematoxylin and eosin and histological scores were evaluated as described (19).
Detection of autoantibodies
Heart extracts were prepared and blotted with mouse sera as described (9). Anti-nuclear antibodies were detected with ANA (HEp-2) antigen substrate slides (MBL-BION) with 1:200 diluted mouse sera and anti-mouse IgG Alexa Fluor 488 (Invitrogen) as described (19).
Cell treatments and analysis
Primary mouse bone marrow-derived macrophages and embryonic fibroblasts were cultured, transfected with nucleic acid ligands, and Ifnb mRNA and type I IFN induction were measured as described (26).
Detection of cGAMP by LC-MS/MS
Mouse hearts were homogenized in extract buffer (50mM HEPES, 2mM EDTA) at a ratio of 0.1g heart tissue per 1ml of buffer. Ice-cold methanol was added to 80% final concentration. Extracts were spiked with isotope-labeled cGAMP (cGAMP*) at 10nM, sonicated, cleared three times by centrifugation at 16,000 × g, lyophilized, and resuspended in water. Detection of cGAMP in heart lysates was performed with LC-MS/MS as described (27) with the following modifications. Isotope-labeled cGAMP (cGAMP*, 12C1013C10H2415N514N5O13P2, MW=689 g/mol), synthesized with recombinant cGAS and per labeled 13C, 15N-ATP and natural abundance GTP, was used as an internal standard. Both cGAMP and cGAMP* were eluted at 2.7 min. The following MRM transitions were detected: cGAMP: +675.1/152.1 (qualifier), +675.1/136.0 (quantifier), and cGAMP*: +690.0/146.0 (quantifier). A standard curve was constructed using standard 1 (250 nM cGAMP*, 10 nM cGAMP) and standard 2 (250 nM cGAMP*, 1000 nM cGAMP).
Results and Discussion
cGAS is required for lethal autoimmunity in Trex1-deficient mice
To explore cGAS function in vivo, we generated cGAS-deficient mice from frozen sperm carrying a null allele of cGAS (cGAS−/−, also referred to as Mb21d1−/−, Supplementary Figure 1A,B). cGAS-deficient mice were physically indistinguishable from their wild type littermates (data not shown). Moreover, consistent with recent studies (5), the IFN response to transfected DNA in cGAS−/− bone marrow macrophages (Supplementary Figure 2A,B) and embryonic fibroblasts (Supplementary Figure 2C,D) was dramatically impaired and similar to STING-deficient cells, while the response to transfected RNA remained intact.
To determine whether cGAS is required for autoimmune disease in Trex1-deficient mice, we intercrossed Trex1−/− and cGAS−/− mice and monitored survival. Trex1 and cGAS are genetically linked, separated by 16 cM on chromosome 9. For this reason, recovery of cGAS−/−Trex1−/− mice from intercrosses of cGAS+/−Trex1+/− mice occurred at a much lower frequency than would be expected with unlinked genes. In contrast to cGAS+/+Trex1−/− mice, which succumbed to lethal disease as expected, cGAS−/−Trex1−/− mice were completely protected from lethality (Figure 1). Trex1−/− mice heterozygous for cGAS were partially rescued from death similar to Irf3+/−Trex1−/− mice (9). This contrasts with Trex1−/− mice lacking a single copy of Tmem173 (which encodes STING), which are completely protected from lethal disease (19).
Figure 1. cGAS is required for lethal autoimmune disease in Trex1-deficient mice.
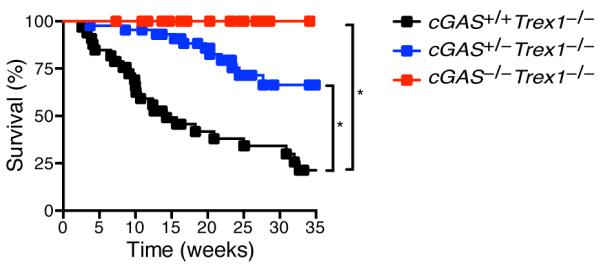
Survival curves of cGAS+/+Trex1−/− (n=33), cGAS+/−Trex1−/− (n=44), and cGAS−/−Trex1−/− (n=29) mice. Both cGAS-deficient and Trex1-deficient mice were on a pure C57BL/6 background. Statistical analysis was performed with a Log-rank (Mantel-Cox) test. *p < 0.0001.
cGAS promotes autoimmune tissue destruction in Trex1−/− mice
Trex1-deficient mice develop multiorgan inflammation, including severe inflammatory myocarditis that is dependent on type I IFNs and STING (9, 19). To determine whether cGAS is required to drive autoimmune tissue destruction, we performed a blinded histological analysis of tissues from cGAS−/−Trex1−/− mice compared to cGAS+/+Trex1−/−, cGAS+/−Trex1−/− and cGAS−/−Trex1+/+ mice. Consistent with previous studies, we found evidence of inflammation in multiple tissues from cGAS+/+Trex1−/− mice, including the heart, skin, glandular stomach, skeletal muscle, tongue, and kidney, whereas the brain was unaffected (Figure 2). Heart tissue from Trex1−/− mice was characterized by focally extensive subendocardial and perivascular lymphoplasmacytic myocarditis (Figure 2A, left panel). Skin tissue exhibited focally extensive proliferative dermatitis with lymphoplasmacytic and lesser neutrophilic cellulitis and myositis (Figure 2A, middle panel). Finally, the glandular stomach was characterized by diffuse moderate proliferative gastritis with multifocal lymphoid aggregates (Figure 2A, right panel). In contrast, analysis of heart and skin tissue from cGAS−/−Trex1−/− revealed no signs of inflammation (Figure 2A,B). Mild inflammation was observed in glandular stomach, skeletal muscle, tongue, and kidney tissue from cGAS−/−Trex1−/−mice, which was similar to cGAS−/−Trex1+/+ mice and clearly distinct from that observed in cGAS+/+Trex1−/− mice (Figure 2B and data not shown). For example, multifocal lymphoid aggregates were not detected in glandular stomach tissue from cGAS−/−Trex1−/− mice and the mild inflammation observed in these mice was in a distinct region of the stomach (margo plicatus/limiting ridge) compared to cGAS+/+Trex1−/− mice (Fig. 2A and data not shown). Thus, cGAS−/−Trex1−/− mice are protected from tissue destruction, similar to Tmem173−/−Trex1−/− mice (9, 19), indicating that the cGAS/STING ISD pathway is required to drive autoimmune tissue destruction in Trex1-deficient mice.
Figure 2. cGAS drives tissue inflammation in Trex1-deficient mice.

(A) Representative H&E-stained heart, skin, and glandular stomach tissue sections from cGAS+/+Trex1−/− and cGAS−/−Trex1−/− mice (20X original magnification). (B) Histological scores of inflammation in the indicated tissues from cGAS+/+Trex1−/−, cGAS+/−Trex1−/−, cGAS−/−Trex1−/− and cGAS−/−Trex1+/+ mice. All histological analysis was performed in a blinded manner. Data are representative of at least four mice of each genotype. Statistical analysis was performed comparing cGAS−/−Trex1−/− to cGAS+/+Trex1−/− and cGAS−/−Trex1+/+ mice using a one-way ANOVA with Tukey’s multiple comparison posttest. *p < 0.05, **p < 0.005, ****p < 0.0001.
cGAS drives the autoantibody response in Trex1−/− mice
Trex1-deficient mice develop a T cell-dependent autoantibody response against heart tissue antigens that is dependent on IRF3 and type I IFN signaling (9, 19). To determine whether cGAS is required for the development of these autoantibodies, we blotted heart tissue extracts with sera from cGAS+/+Trex1−/−, cGAS+/−Trex1−/−, and cGAS−/−Trex1−/− mice. Sera from multiple cGAS+/+Trex1−/− and cGAS+/−Trex1−/− mice exhibited strong autoreactivity against heart extracts (Figure 3A, left panels). The antigen specificities of these heart autoantibodies were variable among different mice, consistent with our previous analysis of Trex1-deficient mice (9, 19). In contrast, minimal autoreactivity, similar to that observed in Trex1+/− and Trex1+/+ mice, was observed in sera from cGAS−/−Trex1−/− mice (Figure 3A, right panels). This demonstrates that cGAS is required for the development of autoantibodies against heart tissue antigens in Trex1-deficient mice.
Figure 3. cGAS-dependent autoantibodies in Trex1-deficient mice.

(A) Autoantibodies against heart antigens evaluated by blotting neat and 1:5 diluted heart extracts from Rag2−/−Trex1−/− mice with sera from mice of the indicated genotype. Immunoblots were prepared with sera from different mice harvested at the age indicated, rather than with sera harvested repeatedly from a single mouse. Data are representative of two independent experiments. (B) Antinuclear antibodies (ANA) in sera from littermate cGAS+/+Trex1−/−, cGAS+/−Trex1−/−, and cGAS−/−Trex1−/− mice (left panels) analyzed by immunofluorescence staining of HEp-2 cell-coated slides. ANA in serum from cGAS−/−Trex1+/− mice was analyzed as a control (right panel). Data are representative of three independent experiments with a total of at least three mice of each genotype.
In addition to the heart-specific autoantibody response, Trex1-deficient mice develop several hallmarks of lupus including anti-nuclear antibodies (ANAs) (19). Whether the development of ANAs in Trex1-deficient mice requires the cGAS/STING ISD pathway has not yet been tested. To explore this question, we evaluated sera from cGAS+/+Trex1−/−, cGAS+/−Trex1−/−, and cGAS−/−Trex1−/− mice for the presence of ANAs by staining HEp-2 cell-coated slides. Consistent with previous work, ANAs with variable signal intensity were observed in sera from all cGAS+/+Trex1−/− mice (n=7/7) and the majority of cGAS+/−Trex1−/− mice (n=6/7) (Figure 3B). In contrast, ANAs were not detected in sera from age-matched cGAS−/−Trex1−/− mice (n=0/3) (Figure 3B) indicating that cGAS is required for the development of ANAs in Trex1-deficient mice.
cGAS promotes type I IFN production in Trex1−/− mice
Disease in Trex1-deficient mice is driven by type I IFN signaling, as Ifnar−/−Trex1−/− mice are protected from disease (9). To determine whether cGAS promotes type I IFN production in Trex1−/− mice, we measured a panel of IFN-stimulated genes (ISGs) in total peripheral blood cells from cGAS+/+Trex1−/−, cGAS−/−Trex1−/− and Trex1+/+ mice. We detected a significant increase in expression of two ISGs, Cxcl10 and Mx1, and a trend towards increased expression of Isg15 and Ifit2 in cells from cGAS+/+Trex1−/− mice compared to Trex1+/+ mice, while ISG expression was similar in blood cells from cGAS−/−Trex1−/− and Trex1+/+ mice (Figure 4A).
Figure 4. cGAS promotes type I IFN production in Trex1-deficient mice.
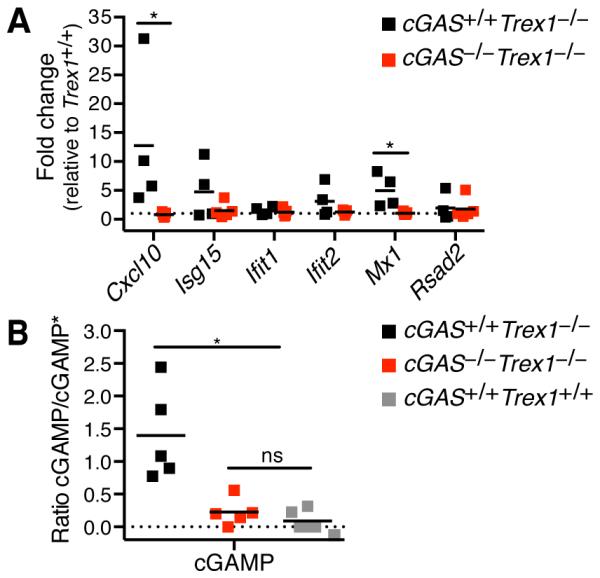
(A) Quantification by RT-PCR of a panel of IFN-stimulated gene transcripts in total peripheral blood cells from cGAS+/+Trex1−/− and cGAS−/−Trex1−/− mice relative Trex1+/+ mice. Data are representative of one independent experiment with at least four mice of each genotype. (B) Analysis of cGAMP production in whole heart extracts from cGAS+/+Trex1−/−, cGAS−/−Trex1−/−, and Trex1+/+ mice by LC-MS/MS. Data are plotted relative to the internal standard (cGAMP*). Data are representative of one independent experiment with at least five mice of each genotype. Statistical analysis was performed with a Mann-Whitney test. *p < 0.05.
In a prior study, we identified a localized focus of IFN-responsive cells at the endocardial surface of the heart in neonatal Trex1-deficient mice, suggesting that disease is initiated in this tissue. To explore the possibility that cGAS is activated in Trex1−/− hearts, we used mass spectrometry to measure cGAMP in Trex1−/− heart tissue. We reproducibly detected elevated levels of cGAMP in heart tissue extracts from cGAS+/+Trex1−/− mice, while the cGAMP was low or undetectable in extracts from cGAS−/−Trex1−/− and Trex1+/+ mice (Figure 4B). We hypothesize that cGAS activation in heart tissue drives lethal myocarditis in Trex1−/− mice. Trex1-deficient mice with a conditional allele of cGAS (Supplementary Fig. 1A) will allow for selective depletion of cGAS in future experiments aiming to define the cell types in which cGAS is required to drive disease.
We provide clear genetic evidence that cGAS is absolutely required for disease in Trex1-deficient mice. cGAS−/−Trex1−/− mice are completely protected from lethality, lack autoimmune inflammation, fail to develop autoantibodies, and lack the peripheral blood IFN signature that is present in plain Trex1−/− mice. Moreover, we show that the inflamed hearts of Trex1−/− mice contain elevated levels of cGAMP, thus demonstrating in vivo activation of cGAS during autoimmune inflammation. While we cannot rule out the possibility that other DNA sensors proposed to activate the STING-dependent IFN response may contribute to autoimmunity in Trex1-deficient mice, our data suggest that, if other sensors participate, they must do so by functioning together with cGAS, and not as independent sensors. Altogether, our studies demonstrate that cGAS is a key driver of autoimmune disease and suggest that cGAS inhibitors may be useful therapeutics for AGS patients with TREX1 mutations.
Supplementary Material
Acknowledgments
We are grateful to the University of Washington Histology and Imaging Core and B. Johnson for histology preparation and members of the Stetson lab for helpful discussions. D.B.S. is a scholar of the Rita Allen Foundation and a Burroughs Wellcome Investigator in the Pathogenesis of Infectious Disease. E.E.G. is supported by a Cancer Research Institute Irvington postdoctoral fellowship.
Footnotes
This work was supported by NIH grant AI084914.
References
- 1.Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. 2014;32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- 2.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lahaye X, Satoh T, Gentili M, Cerboni S, Conrad C, Hurbain I, El Marjou A, Lacabaratz C, Lelievre JD, Manel N. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39:1132–1142. doi: 10.1016/j.immuni.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, Towers GJ. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. 2013;503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 11.Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, A. Montalto S, Bacino CA, Barroso B, Baxter P, Benko WS, Bergmann C, Bertini E, Biancheri R, Blair EM, Blau N, Bonthron DT, Briggs T, Brueton LA, Brunner HG, Burke CJ, Carr IM, Carvalho DR, Chandler KE, Christen HJ, Corry PC, Cowan FM, Cox H, D'Arrigo S, Dean J, De Laet C, De Praeter C, Dery C, Ferrie CD, Flintoff K, Frints SG, Garcia-Cazorla A, Gener B, Goizet C, Goutieres F, Green AJ, Guet A, Hamel BC, Hayward BE, Heiberg A, Hennekam RC, Husson M, Jackson AP, Jayatunga R, Jiang YH, Kant SG, Kao A, King MD, Kingston HM, Klepper J, van der Knaap MS, Kornberg AJ, Kotzot D, Kratzer W, Lacombe D, Lagae L, Landrieu PG, Lanzi G, Leitch A, Lim MJ, Livingston JH, Lourenco CM, Lyall EG, Lynch SA, Lyons MJ, Marom D, McClure JP, McWilliam R, Melancon SB, Mewasingh LD, Moutard ML, Nischal KK, Ostergaard JR, Prendiville J, Rasmussen M, Rogers RC, Roland D, Rosser EM, Rostasy K, Roubertie A, Sanchis A, Schiffmann R, Scholl-Burgi S, Seal S, Shalev SA, Corcoles CS, Sinha GP, Soler D, Spiegel R, Stephenson JB, Tacke U, Tan TY, Till M, Tolmie JL, Tomlin P, Vagnarelli F, Valente EM, Van Coster RN, Van der Aa N, Vanderver A, Vles JS, Voit T, Wassmer E, Weschke B, Whiteford ML, Willemsen MA, Zankl A, Zuberi SM, Orcesi S, Fazzi E, Lebon P, Crow YJ. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;81:713–725. doi: 10.1086/521373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lebon P, Badoual J, Ponsot G, Goutieres F, Hemeury-Cukier F, Aicardi J. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J Neurol Sci. 1988;84:201–208. doi: 10.1016/0022-510x(88)90125-6. [DOI] [PubMed] [Google Scholar]
- 13.Crow YJ, Vanderver A, Orcesi S, Kuijpers TW, Rice GI. Therapies in Aicardi-Goutieres syndrome. Clinical and experimental immunology. 2014;175:1–8. doi: 10.1111/cei.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, Alarcon-Riquelme ME, Gallant CJ, Boackle SA, Criswell LA, Kimberly RP, Brown E, Edberg J, Stevens AM, Jacob CO, Tsao BP, Gilkeson GS, Kamen DL, Merrill JT, Petri M, Goldman RR, Vila LM, Anaya JM, Niewold TB, Martin J, Pons-Estel BA, Sabio JM, Callejas JL, Vyse TJ, Bae SC, Perrino FW, Freedman BI, Scofield RH, Moser KL, Gaffney PM, James JA, Langefeld CD, Kaufman KM, Harley JB, Atkinson JP. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 2011;12:270–279. doi: 10.1038/gene.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hubner N. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 16.Rice GI, Rodero MP, Crow YJ. Human Disease Phenotypes Associated With Mutations in TREX1. J Clin Immunol. 2015;35:235–243. doi: 10.1007/s10875-015-0147-3. [DOI] [PubMed] [Google Scholar]
- 17.Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O'Hara A, Herrick AL, Bowden AP, Perrino FW, Lindahl T, Barnes DE, Crow YJ. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;80:811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-Labarca ML, Terwindt GM, Kasai Y, McLellan M, Grand MG, Vanmolkot KR, de Vries B, Wan J, Kane MJ, Mamsa H, Schafer R, Stam AH, Haan J, de Jong PT, Storimans CW, van Schooneveld MJ, Oosterhuis JA, Gschwendter A, Dichgans M, Kotschet KE, Hodgkinson S, Hardy TA, Delatycki MB, Hajj-Ali RA, Kothari PH, Nelson SF, Frants RR, Baloh RW, Ferrari MD, Atkinson JP. C-terminal truncations in human 3'-5' DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet. 2007;39:1068–1070. doi: 10.1038/ng2082. [DOI] [PubMed] [Google Scholar]
- 19.Gall A, Treuting P, Elkon KB, Loo YM, Gale M, Jr., Barber GN, Stetson DB. Autoimmunity Initiates in Nonhematopoietic Cells and Progresses via Lymphocytes in an Interferon-Dependent Autoimmune Disease. Immunity. 2012;36:120–131. doi: 10.1016/j.immuni.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. 2011;12:959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stavrou S, Blouch K, Kotla S, Bass A, Ross SR. Nucleic Acid recognition orchestrates the anti-viral response to retroviruses. Cell Host Microbe. 2015;17:478–488. doi: 10.1016/j.chom.2015.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li T, Chen J, Cristea IM. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe. 2013;14:591–599. doi: 10.1016/j.chom.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol. 2014;192:5993–5997. doi: 10.4049/jimmunol.1400737. [DOI] [PubMed] [Google Scholar]
- 26.Brunette RL, Young JM, Whitley DG, Brodsky IE, Malik HS, Stetson DB. Extensive evolutionary and functional diversity among mammalian AIM2-like receptors. J Exp Med. 2012;209:1969–1983. doi: 10.1084/jem.20121960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huynh TN, Luo S, Pensinger D, Sauer JD, Tong L, Woodward JJ. An HD-domain phosphodiesterase mediates cooperative hydrolysis of c-di-AMP to affect bacterial growth and virulence. Proc Natl Acad Sci U S A. 2015;112:E747–756. doi: 10.1073/pnas.1416485112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.