

Abstract
Resistance to inhibitors of cholinesterase 8A (Ric-8A) is a highly evolutionarily conserved cytosolic protein initially identified in C. elegans, where it was assigned a regulatory role in asymmetric cell divisions. It functions as a guanine nucleotide exchange factor for Gαi, Gαq, and Gα12/13 and as a molecular chaperone required for the initial association of nascent Gα subunits with cellular membranes in embryonic stem cell lines. To test its role in hematopoiesis and B lymphocytes specifically, we generated ric8fl/flvav1-cre and ric8fl/flmb1-cre mice. The major hematopoietic cell lineages developed in the ric8fl/flvav1-cre mice, notwithstanding severe reduction in Gαi2/3, Gαq, and Gα13 proteins. B lymphocyte specific loss of Ric-8A did not compromise bone marrow B lymphopoiesis, but splenic marginal zone B cell development failed, and B cells underpopulated lymphoid organs. The ric8fl/flmb1-cre B cells exhibited poor responses to chemokines, abnormal trafficking, improper in situ positioning, and loss of polarity components during B cell differentiation. The ric8fl/flmb1-cre mice had a severely disrupted lymphoid architecture and poor primary and secondary antibody responses. In B lymphocytes, Ric-8A is essential for normal Gα protein levels; and is required for B cell differentiation, trafficking, and antibody responses.
Keywords: Ric-8A, heterotrimeric G-protein, marginal zone, hypogammaglobulemia, asymmetric cell division
Introduction
In canonical G-protein signaling, an agonist binds a G-protein coupled receptor (GPCR), which adopts a conformation that triggers the Gα subunit of a heterotrimeric G protein to exchange GDP for GTP resulting in the functional dissociation of Gα from its associated Gβγ. This leads to the activation of downstream intracellular effector enzymes that mediate cellular responses. For example, most chemokine receptors signal by triggering Gαi nucleotide exchange resulting in the activation of small GTPases that communicate with actin regulatory proteins to drive the cell motility needed for chemotaxis (1). In non-canonical G-protein signaling, the guanine exchange factor (GEF) activity exerted by the GPCR is replaced by the action of intracellular GEFs. One such intracellular GEF is Ric-8A. Ric-8A acts on Gαi, Gαq, and Gα12/13 while a related protein Ric-8B acts on Gαs (2).
A highly evolutionarily conserved cytosolic protein Ric-8 was initially identified in C. elegans, where its functions include a regulatory role in asymmetric cell divisions (3–5). In human cells, Ric-8A recruits to the cell cortex a signaling complex that helps orient the mitotic spindle in response to spatial clues (6). In non-canonical signaling pathways, Gα subunits are often paired with proteins containing one or more conserved Gαi/o-Loco interaction (GoLoco) motifs, also known as G-protein regulatory (GPR) motifs, which act as a guanine nucleotide dissociation inhibitor (GDI) much like Gβγ does in the canonical pathway (7). In Drosophila, a GoLoco protein Partner of Inscuteable (Pins) forms an apical protein complex essential for neuroblast asymmetric cell division (8). In humans, mutations in the GoLoco protein LGN cause brain malformations and hearing loss in Chudley-McCullough syndrome (9). Also in human cells, the non-canonical G-protein signaling proteins Ric-8A and LGN localize at the midbody during cytokinesis along with Gαi and RGS14, a GoLoco motif containing protein (10, 11). Interference with Gαi expression or with Gαi nucleotide exchange prolongs cytokinesis, the final step of the cell cycle (12). Gene targeting the GoLoco protein Ags3 in mice leads to phenotypes in the kidney, and the nervous and immune systems (13–15). Arguing for a role of AGS3 in Gαi regulation in immune cells, ags3−/− dendritic cells and lymphocytes exhibit suboptimal responds to chemokines in chemotaxis, calcium mobilization, and effector protein activation assays (16). In addition, during neutrophil chemotaxis GDP-bound Gαi accumulates at the leading ledge where it recruits the adaptor molecule Inscuteable (Insc), LGN/AGS3, and the Par3-aPKC polarity complex. Insc−/− neutrophils poorly stabilize leading edge pseudopods and that stability can be restored by the addition of wild-type Insc protein, but not by a mutant protein that doesn’t bind LGN/AGS3 (17). These studies and others implicate Ric-8A and non-canonical G-protein signaling in an array of biologic processes likely to impact immune cells (18).
Ablation of ric8 in mice results in early embryonic lethality as embryos died at E6.5-E8.5. The mice die shortly after initiation of gastrulation with a disorganized epiblast (19). Derived ric8−/− embryonic cell lines exhibited pleiotropic G protein signaling defects and an approximate 85% loss of Gαi1/2, Gαq, and Gα13 proteins. An accelerated rate of protein degradation accounted for the reduced levels of proteins. These data indicated that Ric-8A had an additional function that of a molecular chaperone helping to target newly translated Gαi, Gαq, and Gα12/13 proteins to cellular membranes (20, 21). Conditional gene targeting using a floxed ric8 allele and an hGFAP-cre that targets Ric-8A expression in neural progenitors and astroglia resulted in mice with a disorganized Bergmann glial scaffolding, defective granule cell migration, and disrupted Purkinje cell positioning (22). A synapsin I promoter driven Cre ablated Ric-8A function in most differentiated neuron populations and resulted in early post natal death due to a severe neuromuscular phenotype (23). However, whether the phenotypes that arose in these conditionally ric8 targeted mice resulted from Gα protein deficiency or due to a loss of Ric-8A function in non-canonical G-protein signaling was unexplored in these studies.
Despite increasing evidence that asymmetrical localization of proteins during lymphocyte cell division contributes to differential cell fates and the known role of Gα proteins and their partners in model organism asymmetric cell divisions relatively little attention has been paid to whether they participate in asymmetric cell divisions in lymphocytes. One study did note that interference with the Pins (LGN)/G-protein module reduced the number of dividing T cells with a mitotic axis compatible with asymmetric cell division (24). We sought to determine whether Ric-8A had chaperone like activity for Gα subunits in hematopoietic cells, to investigate the consequences of a specific loss of Ric-8A in B cells, and to determine whether the loss of Ric-8A affected B lymphocyte symmetric and asymmetric cell divisions. We found that Ric-8A has chaperone like activity for Gαi2, Gαi3, and Gαq, while steady state levels of Gαs and Gα12 were unaffected in spleen cells and bone marrow derived macrophages. A loss of Ric-8A in B cells led to a severe B cell immunodeficiency likely due to the Gαi proteins. In response to mitotic signals the Ric-8A deficient and wild type B cells divided symmetrically with an equal frequency, although on occasion the final abscission step was delayed in the absence of Ric-8A. In contrast, activated ric8fl/flmb1-cre B cells and germinal center B cells from immunized ric8fl/flmb1-cre mice underwent fewer asymmetric cell divisions when compared to control cells. The implications of our results are discussed.
Materials and Methods
Animals
C57BL/6, and B6.SJL-Ptprca Pepcb/BoyJ mice were obtained from Jackson Laboratory. The previously characterized Ric-8Afl/fl mice (22) on a mixed background were backcrossed 10 times on to C57BL/6. The C57/BL6 mb1-cre mice were kindly provided by Dr. Michael Reth (25). The C57/BL6 vav1-cre mice were obtained from Jackson Laboratory and previously characterized (26). For bone marrow reconstitution, seven weeks old B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) mice were irradiated twice with 550 rads for total of 1100 rads and received bone marrow from C57BL/6 CD45.2 control or mutant mice. The engraftment was monitored by sampling the blood 28 days later. The mice were used 6–8 weeks after reconstitution. All mice were used in this study were 6–14 weeks of age. Mice were housed under specific-pathogen-free conditions. All the animal experiments and protocols used in the study were approved by the NIAID Animal Care and Use Committee (ACUC) at the National Institutes of Health.
Cells
Splenic B cells were isolated by negative depletion using biotinylated antibodies to CD4, CD8, Gr-1 (Ly-6C and Ly 6G), and CD11c and Dynabeads M-280 Streptavidin (Invitrogen). The B cell purity was greater than 95%. When needed B cells were cultured in RPMI 1640 containing 10% FCS (Gibco), 2 mM L-glutamine, antibiotics (100 IU/mL penicillin and 100 μg/mL streptomycin), 1 mM sodium pyruvate, and 50 μM 2-mercaptoethanol. When very high purity B cells were needed they were isolated by cell sorting following immunostaining for CD19 and B220.
Flow cytometry and antibodies
Single cells were re-suspended in PBS, 2% FBS, and stained with fluorochrome-conjugated or biotinylated antibodies against B220 (RA3-6B2), IgD (11-26c-2a), IgM (R6-60.2), CD24 (M1/69), CD3 (145-2C11), CD4 (GK1.5), CD5 (53–7.3), CD8 (53–6.7), CD11c (HL3), CD11b (M1/70), CD138 (281–2), CD19 (1D3), CD38 (90), IgG1 (X56), CD93 (AA4.1), BP-1 (6C3), GL-7 (GL-7, Ly-77), CD95 (Jo2), CD21 (4E3), CD23 (B3B4), CD43 (S7), CD184 (CXCR4, 2B11), CXCR5 (2G8), CCR7 (4B12), CD11a (M17/4), PD-1 (CD279, RMP1-30), CD45.1 (A20), or CD45.2 (104) (all from eBioscience, Biolegend, or BD Pharmingen). Biotin-labeled antibodies were visualized with fluorochrome-conjugated streptavidin (eBioscience). LIVE/DEAD® Fixable Aqua Dead Cell Stain Kit (Molecular Probes) was used in all experiments to exclude dead cells. Data acquisition was done on FACSCanto II (BD) flow cytometer and analyzed with FlowJo software (Treestar). The following flow cytometry gates were done to identify bone marrow B-cells subsets: Fr. A: B220+CD43+CD24−BP1−, Fr. B: B220+CD43+CD24+BP1−, Fr. C: B220+CD43+CD24+BP1+, Fr. D: B220+CD43−IgM−IgD−, Fr. E: B220+CD43−IgM+IgD−, and Fr. F: B220+CD43−IgM+IgD+. Gating for splenic subsets were as followed: T0: B220+CD93+IgM+IgD−CD23−, T1: B220+CD93+IgM+IgD+CD23−CD21−, Follicular (FO): B220+CD23+CD21−CD24+, T2-Follicular (T2-FO): B220+CD23+CD21−CD24++, Marginal zone precursor (MZP): B220+CD23+CD21+CD24++, Marginal zone (MZ): B220+CD23−CD21+CD24+, germinal center (GC): B220+CD19+CD38−CD95+GL-7+, and Follicular helper T-cell (Tfh): CD4+CXCR5+PD-1+. The following gates were used to identify thymocytes subsets (B220−CD11c−Gr1−): Double positive (DP): CD4+CD8+,Double negative (DN)1: CD4−CD8−CD44+CD25−, DN2: CD4−CD8−CD44+CD25+, DN3: CD4−CD8−CD44−CD25+, DN4: CD4−CD8−CD44−CD25−, mature CD4 Single positive (SP): CD4+CD8−CD69−CD62L+CD24− immature CD4SP: CD4+CD8−CD69+CD62L−CD24+ mature CD8SP: CD4−CD8+CD69−CD62L+CD24- immature CD8SP: CD4−CD8+ CD69+CD62L−CD24+.
Cell proliferation
The cell proliferation studies were performed using CFSE (Molecular Probes) in a standard dye dilution assay. Purified B cells were stimulated for 96 hours with various combinations of the following reagents: 1 μg/ml CD40 (HM40-3), 1 μg/ml LPS (055:B5, Sigma-Aldrich), recombinant mouse IL-4 (10 ng/ml), or 10 μg/ml AffiniPure F(ab′)2 fragment goat anti-mouse IgM (Jackson ImmunoResearch Laboratories). Data acquisition was done on FACSCanto II flow cytometer. The proliferation index is the average number of cell divisions that a cell in the original population undergoes; the division index is the average number of cell divisions of the responding cells; and the percent cell division is defined as the proliferation index divided by the division index, and multiplying the results by 100, assuming no cell death, were calculated using FlowJo software.
Chemotaxis assays
Chemotaxis assays were performed using a transwell chamber (Costar) as previously described (27). Splenic B cells were immunostained for B cell subsets with fluorochrome-conjugated antibodies against B220, CD21, CD23, CD24, CD45.1, and CD45.2 washed twice, re-suspended in complete RPMI 1640 medium and added in a volume of 100 μl to the upper wells of a 24-well transwell plate with a 5 μm insert. Lower wells contained various doses of chemokines in 600 μl of complete RPMI 1640 medium. The numbers of cells that migrated to the lower well after 2 h incubation were counted using a MACSQuant flow cytometer (Miltenyi Biotec). The percent migration was calculated by the numbers of cells of a given subset that migrated into the bottom chamber divided by the total number of cells of that subset in the starting cell suspension, and multiplying the results by 100. CXCL13, CCL19 and CXCL12 were purchased (R&D Systems). Fatty acid free bovine serum albumin (FAF-BSA) was purchased (Sigma-Aldrich).
Intracellular calcium measurements
Cells were seeded at 105 cells per 100 μl loading medium (RPMI 1640, 10% FBS) into poly-D-lysine coated 96-well black wall, clear-bottom microtiter plates (Nalgene Nunc). An equal volume of assay loading buffer (FLIPR Calcium 4 assay kit, Molecular Devices) in Hanks’ balanced salt solution supplemented with 20 mM HEPES and 2 mM probenecid was added. Cells were incubated for 1 h at 37 °C before adding chemokine or unconjugated AffiniPure F(ab′)2 fragment goat anti-mouse IgM (Jackson ImmunoResearch Laboratories) and then the calcium flux peak was measured using a FlexStation 3 (Molecular Devices). The data was analyzed with SOFT max Pro 5.2 (Molecular Devices). Data is shown as fluorescent counts and the y-axis labeled as Lm1.
ImageStream (IS): labeling, acquisition and analysis
The splenic B cells were stained with B220 PE-Cy7 conjugated for 10 min then fixed in 2% PFA. The PKCζ distribution was visualized by indirect labeling where antibody was diluted (Santa Cruz, 1:100) in permeabilization wash buffer (PWB) consisting of 3% BSA, and 0.1% Triton X-100 in PBS. Samples were incubated for 2h at room temperature. Primary antibody was removed and 1:200 dilution of secondary Alexa488 conjugated antibody (Jackson ImmunoResearch Laboratories Inc.) was added and incubated at room temp in the dark for 1h. Secondary antibody was removed and cells resuspended in 100μL PBS. Just prior to running on the ImageStream, all samples had Draq5 (Cell Signaling) added (20nM final concentration), to visualize the nucleus. At least, 100,000 events were collected for all samples on an ImageStream MarkII using 488nm, 405nm and 642nm laser excitations. Cell populations were hierarchically gated for single cells that were in focus and were positive for both PKCζ and B220. Then, based on Draq5 intensity histogram, we gated on cells that were in G2/M phase. An additional gating for germinal center was applied for those experiments where we focused on B220+Fas+GL7+ cells. Following data acquisition, the spatial relationship between B220 and Draq5 was measured using a feature (delta centroid intensity weighted) calculated in the IDEAS software package that eliminates non- specific asymmetric distributions. The same feature was applied between PKCζ and Draq5 and asymmetry was defined when delta centroid value was greater than half the median of the B-cell radius.
Immunohistochemistry
Immunohistochemistry was performed modified method of a previously published protocol (28). Briefly, freshly isolated lymph nodes and spleens were fixed in freshly prepared 4% paraformaldehyde (Electron Microscopy Science) for overnight at 4°C on agitation stage. Lymph nodes and spleens were embedded in 4% low melting agarose (Invitrogen) in PBS and sectioned with a vibratome (Leica VT-1000 S) at a 30 μm thickness. Thick sections were blocked in PBS containing 10% FCS, 1 mg/ml anti-Fcγ receptor (BD Biosciences), and 0.1% Triton X-100 (Sigma) for 30 minutes at room temperature. Sections were stained for overnight at 4°C on agitation stage with the following antibodies: anti-B220, anti-CD3e, anti-CD4, anti-Ki67 (all from eBioscience), anti-CD169 (R&D System), anti-CD21/35 (BioLegend). Stained thick sections were microscopically analyzed by using a Leica SP5 confocal microscope (Leica Microsystem, Inc.) and images were processed with Leica LAS AF software (Leica Microsystem, Inc.) and Imaris v.7.6.1 (Bitplane).
Live cell time lapse confocal microscopy
Splenic B cells were prepared from ric8wt/wt Lifeact and ric8fl/flmb1-cre Lifeact mice. The isolated B cells were cultured at initial concentration 1 × 106 cells/mL in complete lymphocyte medium in presence of 2 μg/mL LPS (from E. coli, Serotype R515 (Re), TLR grade, ENZO Life Sciences), for 48 h. For live cell imaging, B cells were allowed to adhere to ICAM-1 + VCAM-1 (Recombinant Mouse ICAM-1/CD54 Fc Chimera, CF; Recombinant Mouse VCAM-1/CD106 Fc Chimera; R&D Systems) on coated glass-bottom dishes (No 1.5 coverglass; MatTek). Confocal imaging was performed using Leica SP8 inverted 5 channel confocal microscope (Leica Microsystems) equipped with incubation chamber (CO2, 37°C) for live cell imaging (Pecon). Argon laser was tuned to 488 nm excitation wavelength using laser power between 0.2 and 1 %. Z stacks were acquired every 10–12 sec over time period of 1 h, and single plane images used to generate video. Images were processed using Imaris (Bitplane) software.
Intravital and spleen section two-photon laser scanning microscopy (TP-LSM)
Inguinal LNs were prepared for intravital microscopy as described (29). Cell populations were labeled for 15 minutes at 37°C with 1 uM green cell tracker CMFDA, 2.5~5 uM red cell tracker CMTMR (Molecular probes). 5–10 million labeled cells of each population in 200 ml of PBS were adoptively transferred by tail vein injection into 6~10-week-old recipient mice. After anesthetizing the mice by intraperitoneal injection of Avertin (300 mg/kg, tribromoethanol, Sigma), the skin and fatty tissue over inguinal LN were removed. The mouse was placed in a pre-warmed coverglass chamber slide (Nalgene, Nunc). The chamber slide was then placed into the temperature control chamber on the Leica SP5 microscope. The temperature of air was monitored and maintained at 37.0± 0.5°C. Inguinal LN was intravitally imaged from the capsule over a range of depths (10–220 μm). All two-photon imaging was performed with a Leica SP5 inverted 5 channel confocal microscope (Leica Microsystems) equipped with 0.95 NA (immersion medium used distilled water). Two-photon excitation was provided by a Mai Tai Ti:Sapphire laser (Spectra Physics) with a 10 W pump, tuned to 810 or 910 nm. Emitted fluorescence was collected using a 4 channel non-descanned detector. Wavelength separation was through a dichroic mirror at 560 nm and then separated again through a dichroic mirror at 495 nm followed by 525/50 emission filter for CMFDA (Molecular probes); a dichroic mirror at 650 nm followed by 610/60 nm emission filter for CMTMR; and the Evans blue signal was collected by 680/50 nm emission filter. Sequences of image stacks were transformed into volume-rendered four-dimensional videos using Imaris software and tracking analysis was transformed by using autoregressive motion algorithm of Imaris software v.7.6.1 (Bitplane). Polarity measurements were performed using Imaris software.
Immunizations and ELISA
Wild-type and mutant 6- to 8-wk-old mice were immunized with either sheep RBCs, NP35-KLH, or NP40-Ficoll. For the sheep RBC immunizations 200 μl of 10% solution of sheep RBCs (Lonza Walkerville, Inc.) was given by intraperitoneal injection. NP35-KLH (Biosearch Technology) was mixed with Imject® Alum (Thermo Scientific) and introduced into mice (100 μg) via intraperitoneal injection. Mice were boosted with same dose of antigen at the indicated days along with Alum. Mice were also immunized with 25 μg NP40-Ficoll (Biosearch Technologies) via intraperitoneal injection. Serum NP specific Ig levels in these mice were analyzed by ELISA. Briefly, 96 well ELISA plates (Nunc) were coated with NP30-BSA (Biosearch Technology) overnight at 4°C, washed and blocked with 1% BSA fraction V (Sigma-Aldrich), serum titers were then added to the plates and incubated 4 h at 4°C. After washing alkaline phosphatase-labeled goat anti-mouse Ig isotype specific antibodies were added for 2 h at room temperature (SouthernBiotech). After washing, PNPP one component substrate (SouthernBiotech) was used to detect the amount of secondary antibody bound.
Western blotting
Purified splenic were lysed on ice for 10min in 50 mM HEPES, pH 7.4, 250 mM NaCl, 2 mM EDTA, 0.2% NP40, containing Complete™ protease inhibitors (Roche) and supplemented with 100 μM orthovanadate sodium 10 μM NaF and 10 μM PMSF. After centrifugation at 13000×g for 10 min, the supernatant was collected and protein concentration determined (ODλ=280nm measurement Nanodrop, ThermoScientific). 50 μg of lysates were resolved on 4–20% Tris-Glycine gels. After transfer on nitrocellulose, blots were probed with anti-actin-HRP conjugated (Sigma, 1:20,000), anti-Gαi2 mouse monoclonal (Santa Cruz, 1:500), anti-Gαi3 rabbit polyclonal (Santa Cruz, 1:500), anti-Gα12 rabbit polyclonal (Santa Cruz, S20, 1:50), anti-Gαs rabbit polyclonal (EMD Millipore, 1:100), anti-Gαq rabbit polyclonal (EMD Millipore, 1:100), anti-Gα13 mouse monoclonal (New East Biosciences); anti-Ric-8A rabbit polyclonal (kind gift from Dr. G. Tall, University of Rochester), anti-LGN rabbit polyclonal (kind gift from Dr. J. Blumer, University of South Carolina) and revealed using HRP conjugated TrueBlottm (eBioscience) secondary antibody (1:10,000). Immunoblots were revealed by luminescence (ECL, Amersham Bioscience).
Statistics
In vivo results represent samples from 3–9 mice per experimental group. Results represent mean values of at least triplicate samples. Standard errors of the mean (SEM) and p values were calculated with Student t-test or 2-way ANOVA using GraphPad Prism (GraphPad software). * p < 0.05; ** p < 0.005; *** p < 0.0005.
Results
Disrupting Ric-8A expression in mouse hematopoietic cells results in a loss of Gαi2, Gαi3 and Gαq; anemia; leukocytosis; and a loss of B lymphocytes
To assess the role of Ric-8A in mouse hematopoietic cells we crossed ric8fl/fl mice to vav1-cre transgenic mice to generate ric8fl/flvav1-cre mice (both strains on a C57/BL6 background). Ric-8A and Vav1 are expressed in hematopoietic stem cells, progenitors, and cells of the hematopoietic lineage (18, 26, 30). The ric8fl/flvav1-cre mice were born with the expected Mendelian frequency. However, we noted that they had an increased perinatal mortality, diminished vigor, and reduced longevity with many mice dying within 4 months of birth, yet their body weights at 6 weeks were similar to that of controls (C.B., unpublished observation). Pathological examination did not reveal any consistent cause of their early demise. The loss of a single allele of ric8 in hematopoietic cells was without evident consequence as the ric8fl/wtvav1-cre mice thrived similar to that of WT mice. We verified the loss of Ric-8A in the conditionally deleted hematopoietic cells by immunoblotting cell lysates prepared from splenocytes and bone marrow derived macrophages for Ric-8A expression (Fig. 1A). Next, we checked the expression levels of Gα proteins and that of LGN. We found that the loss of Ric-8A had led to a severe reduction in Gαi2, Gαi3, Gα13, and Gαq; while the levels of Gα12, Gαs, and LGN were similar to that of WT mice (Fig. 1A). Assessment of blood obtained from the ric8fl/flvav1-cre mice revealed a modest anemia, an increased in white cell numbers, and normal platelet numbers. A lymphocytosis accounted for the noted leukocytosis (Fig. 1B,C). Assessment of B and T cells in the spleen and thymus did not reveal any striking abnormalities while lymph nodes, Peyer’s Patches, and the bone marrow all had a reduction in B cell numbers (Fig. 1D,E). An analysis of B cell development in the bone marrow and spleen revealed a significant loss of T2 transitional and marginal zone B cells in the spleen (Fig. 1F). These results indicate that Ric-8A is needed for normal levels of Gαi2, Gαi3, Gα13, and Gαq in hematopoietic cells; but not for Gα12. Its loss caused a modest anemia, decreased longevity, loss of B lymphocytes particularly from lymph nodes and Peyer’s patches, and interfered with B cell development in the spleen. Surprisingly T cell development proceeded relatively normally and secondary lymphoid organs were populated with normal numbers of T cells (Fig. 1D, E, G). We did observe an increase in the number of mature single positive CD4 and CD8 T cells, a result consistent with a mild thymus egress defect likely due to the reductions in Gαi2 and Gαi3.
Figure 1.

Targeting ric8 in hematopoietic cells causes the loss of Gα proteins in spleen cells; anemia; impaired marginal zone B cell development; increased mature CD4+ T thymocytes; and reduced peripheral B cell numbers. (A) Immunoblot analysis of cell lysates prepared from spleen cells and BMDM from ric8fl/flvav1-cre, ric8fl/+vav1-cre mice and ric8+/+vav1-cre mice for the indicated proteins. Similar results in 3 separate experiments. (B,C) Complete blood counts using blood obtained from vav1-cre (n=7) and ric8fl/flvav1-cre (n=6) mice. RBC, red blood cell count; WBC, white blood count; PMN, neutrophils; Lymph, lymphocytes; Mono, monocytes; Eosin, eosinophils; and baso, basophils. (D,E) Flow cytometry characterization of cells prepared from the lymphoid organs of the mice used in part B and C. Single cell suspensions were prepared from spleen, thymus, inguinal lymph node, Peyer’s patches, and bone marrow. The number of lymphocytes, CD4+, CD8+, and B220+ cells in each preparation are shown. (F) Flow cytometry analysis of bone marrow and splenic B cell development in vav1-cre and ric8fl/flvav1-cre mice. The bone marrow development is shown as the % of cells in fractions (Fr.) A-F based on the B220+ cell gate. Spleen B cell development is shown as the % of cells in the transitional type 1 (T1), transitional type 2 (T2), follicular (FO), and marginal zone (MZ) B cell subsets. (G) Flow cytometry analysis of thymocytes prepared from in vav1-cre and ric8fl/flvav1-cre mice. The percentages of thymocytes in the double negative (DN) subsets 1–4, double positive (DP), CD4 single positive, and CD8 single positive are shown. The CD4 and CD8 single positive cells are divided into immature and mature based on expression of CD69 and CD62L. * p < 0.05; ** p < 0.005.
Disrupting Ric-8A expression in mouse B cells leads to a loss of Gαi2, Gαi3, and Gαq; a reduction in B lymphocytes; and major decreases in germinal center B cells and early switched memory B cells
Because the initial assessment of the loss of Ric-8A in hematopoietic cells had suggested that B cells had been the most severely affected and to remove the complicating issue of the loss of Ric-8A in multiple cell types we crossed the ric8fl/fl mice to mb1-cre mice and generated ric8fl/fl mb1-cre mice. The mb1-cre mice have a single normal mb1 (CD79a) allele that is sufficient for normal B cell development and function, while the other allele express the Cre recombinase at the pro-B stage of B-cell bone marrow development (25). Because both ric-8 and mb1 are located on chromosome seven, we had to screen the progeny for a cross-over event that resulted in a floxed ric-8 allele and an mb-1 Cre allele on the same chromosome. Having established that mouse line on a C57BL/6 background, we assessed Ric-8A, and Gα protein expression in B cells from WT and mice that lacked B cell specific expression of ric8. Similar to the vav1-cre mice the loss of ric8 expression in B cells led to a marked decrease in their Gαi2, Gαi3, and Gαq protein levels although Gαi3 was less affected (Fig. 2A). Attempts to discern whether the loss of Ric-8A affected Gα13 levels in B cells were stymied by our failure to detect Gα13 in wild type B cell lysates. In the blood the mice had an increased number of lymphocytes and normal numbers of other leukocytes with the exception of a modest increase in the number of neutrophils (Fig. 2B). Similar to the ric8fl/flvav1-cre mice, the ric8fl/flmb1-cre had reduced number of B cells in lymph nodes, Peyer’s patches, and also in the spleen (Fig. 2C). B cell development in the bone marrow was largely intact and, in contrast, to the ric8fl/flvav1-cre mice no reduction of B cells in the bone marrow was noted (Fig. 2D, data not shown). Similar to the ric8fl/flvav1-cre there was a marked loss of marginal zone precursors and marginal zone B cells in the spleen (Fig. 2E). The Ric-8A expression levels did not differ appreciably between the splenic B cell subsets in the wild type mice (Fig. 2E). Immunizing the ric8fl/flmb1-cre mice led to a very poor germinal center response in the spleen, lymph nodes, and Peyer’s patches; reduced plasma cell response in lymph nodes, and in Peyer’s patches, and a very poor induction of switched memory cells at all of the tested sites (Fig. 2F). These results indicate that the loss of Ric-8A in B cells would likely lead to a severe impairment in humoral immunity.
Figure 2.

B-cell specific loss of Ric-8A confirms its role as a chaperone for Gαi 2/3, Gα13, and Gαq and reveals a B cell intrinsic role for Ric-8A in B cell development and function. (A) Ric-8A, Gα proteins, and actin expressions were detected by immunoblot of lysates prepared from sorted B220+CD19+ splenic cells from ric8wt/wtmb1-cre and ric8fl/flmb1-cre. (B) Data shown are from complete blood counts from 14 ric8wt/wtmb1-cre and 12 ric8fl/flmb1-cre mice. (C) Flow cytometry characterization of lymphoid organs from control and ric8fl/flmb1-cre mice used in part B. Absolute number of cells are shown from the spleen, mesenteric and inguinal lymph nodes (mLN & iLN), and from Peyer’s Patches (PP). (D) Flow cytometry was used to characterize B cell development in the bone marrow of ric8wt/wtmb1-cre and ric8fl/flmb1-cre mice. Shown are the % of B220+ cells in bone marrow Fr. A-F. (E) Flow cytometry was used to characterize B cell development in the spleen of ric8wt/wtmb1-cre and ric8fl/flmb1-cre mice. Shown are % of B220+ cells in the T1, T2-follicular (T2-FO), T2-marginal zone precursor (T2-MZP), FO, MZ subsets. Below is an immunoblot of Ric-8A expression in the different subsets. (F) Flow cytometry was used to analyze cells isolated from the spleen, iLN, and PP 10 days following immunization of ric8wt/wtmb1-cre and ric8fl/flmb1-cre mice with sRBCs. Absolute numbers of B220+, CD4+, CD8+, germinal center (GC), T follicular helper cells (TFH), early plasma cell (B220+CD138+), switched memory cells (B220+, CD38+/−, CD138−, IgG1+). Results are from the analysis of 4 experimental and 4 control mice. Experiment repeated three times with similar results. * p < 0.05; ** p < 0.005.
The loss of Ric-8A reduces B cell chemotaxis and profoundly impairs chemokine induced intracellular calcium responses
We used splenic B cells isolated from mixed chimeric mice reconstituted with 1:1 mix of bone marrow from ric8fl/flmb1-cre and WT mice in standard chemotaxis assays using CXCL12, CCL19, and CXCL13 as chemoattractants. We could distinguish the WT and the Ric-8A deficient B cells by flow cytometry using the differential expression of CD45.1 versus CD45.2. This allowed a direct comparison between the two sources of B cells in the same assay. We examined the response of the entire population of splenic B cells as well as the splenic B cell subsets (Fig. 3A). We did not compare the marginal zone B cells as the ric8fl/flmb1-cre mice essentially lacked them. The ric8fl/flmb1-cre B cells responded only at the highest ligand concentration tested. Although the loss of Ric-8A had markedly reduced the responsiveness of the cells, sufficient Gαi remained to allow the cells to respond to the higher dose of chemoattractant (Fig. 3B). In contrast, mice lacking Gαi2 and Gαi3 exhibit little or no chemotaxis even at high ligand concentrations (27). The absence of Ric-8A in B cells did not affect their expression of CXCR4, CCR7, or CXCR5 or that of several different homing and adhesion receptors (Fig. 3B). This differs from the Gαi2/3 deficient B cells where we noted a modest reductions in CXCR4, CCR7, and CXCR5 expression (27). The B cells from the ric8fl/flmb1-cre mice exhibited a profoundly impaired intracellular calcium response to the three chemokines. Even at the highest ligand concentration little intracellular calcium was elicited (Fig. 3C). While lymphocyte chemoattractant receptor signaling including increases in intracellular calcium is absolutely dependent on Gαi, it is possible that Gαq also contributes to the calcium flux. In the context of reduced levels of Gαi proteins, the loss of Gαq in the ric8fl/flmb1-cre B cells may have resulted in a more severe loss in the mobilization of intracellular calcium than otherwise.
Figure 3.

B cell specific loss of Ric-8A impairs responses to chemokines. (A) Chemotaxis assays using splenic B cells from mixed bone marrow chimera mice. C57BL/6 CD45.1 mice were reconstituted with bone marrow from ric8fl/flmb1-cre CD45.2 mice and C57BL/6 CD45.1 mice. Eight weeks later purified B220+ cells were immunostained for B cell subset markers, CD45.1, and CD45.2; and subjected to chemotaxis assays with the indicated concentrations of chemokine. The percentages of migratory cells for each subset and each genotype are shown. The data is from 4 reconstituted mice performed in duplicate. The indicated chemokine concentrations are ng/ml. Data shown as mean +/− SEM. (B) Flow cytometry analysis of CXCR4, ICAM1, CXCR5, CCR7, CD11a, CD62L and α4β7 expression on B220+ cells from the same mice used in part A. WT and ric8fl/flmb1-cre cells were distinguished by CD45.1 versus CD45.2 immunostaining. (C) Intracellular calcium response to chemokines using B cells from mb1-cre and ric8fl/flmb1-cre mice. The cells were stimulated with indicated amount of CXCL12, CCL19 or CXCL13 and the induced change in intracellular calcium monitored over 3 minutes. Data represents the maximal calcium change plotted against the chemokine concentration (ng/ml). Data are from 3 experiments.
Lymphoid organ architecture is disrupted in the ric8fl/flmb1-cre mice
To assess the status of the lymphoid architecture in the ric8fl/flmb1-cre mice we performed high resolution multicolor confocal microscopy with spleens and lymph nodes from non-immunized and immunized mice (Fig. 4A,B). Thick sections were immunostained for B220 (B cell marker), CD21 (enriched on FDCs and marginal zone B cells), CD4 (marker for CD4+ T cells), Ki67 (expressed by activated and proliferating cells), and CD169 (marker for marginal zone macrophages). The loss of Ric-8A in B cells severely disturbed the organization of the spleen. Shown are images of the spleens from WT (Fig. 4A) and ric8fl/flmb1-cre mice (Fig. 4B). There was nearly an 80% reduction in the number of visible primary B cell follicles in the ric8fl/flmb1-cre mice spleens. There was little or no discernable marginal zone surrounding the B cell follicles and no spontaneous germinal center (GC) formation. Numerous ric8fl/flmb1-cre B cells inappropriately localized in T cell zones and an excessive number of B cells resided in the red pulp. The residual B cell follicles were poorly delineated from the T cell zone as the normal sharp B/T borders were missing in the mutant mice. Following immunization those GCs that formed were small and disorganized. No discernible light and dark zone were identifiable and Ki67 positive cells were distributed throughout the GC-like structure. The lymph node architecture in the ric8fl/flmb1-cre mice largely mirrored that of the spleen. The non-immunized mice had small, poorly delineated primary follicles and immunization led to a poor GC response with small GCs present only occasionally in the primary follicles (Fig. 4A,B). These results show that the loss of Ric-8A in B cells profoundly disturbs the normal lymphoid architecture in secondary lymphoid organs, however the abnormalities are not as extreme as that noted in mice whose B cells totally lacked both Gαi2 and Gαi3 (27).
Figure 4.

Ric8fl/flmb1-cre mice have disorganized lymphoid organs and poorly structured germinal centers. (A,B) Confocal microscopy imaging of the spleen and lymph nodes from non-immunized mice and mice 8 days post sRBC immunization. Spleen and lymph node sections from ric8fl/fl (A) and ric8fl/flmb1-cre (B) mice were immunostained for B220 (green), CD4 (blue), CD21/35 (red), Ki67 (white), and CD169 (pink). The top images are tiled images of the spleens. The two images in the middle panels are 4X electronic zooms of the spleens from a non-immunized and an immunized mouse. The two images in the bottom panels are from the inguinal lymph node of a non-immunized and an immunized mouse. GC- Germinal center; B- B cell zone; and TCZ- T cell zone are indicated.
B cells from ric8fl/flmb1-cre mice enter the splenic white pulp and lymph nodes poorly, tend to reside near lymph node HEVs or in the red pulp of the spleen, and have reduced in vivo motility
Because the Ric-8A deficient B cells can enter into lymph nodes, we could assess their localization and motility by intravital 2-photon microscopy. B cells from ric8fl/flmb1-cre and WT mice were differentially fluorescently labeled and adoptively transferred to WT mice at a 1:1 or 3:1 ratio, respectively. The following day, the inguinal lymph node of the recipient mouse was prepared for intravital microscopy. The blood vessels were outlined by intravenous injection of Evans blue. As expected, fewer Ric-8A deficient B cells resided in the lymph node 24 hours after transfer. Those cells that had entered tended to reside near the high endothelial venules, many failing to enter into the lymph node follicle (Fig. 5A, Video 1). Following the imaging procedure the animals were sacrificed and the localization of the transferred B cells in the spleen was assessed. Fewer Ric-8A deficient B cells than wild type entered the splenic white pulp as most of the transferred mutant B cells resided in the red pulp (Fig. 5B). Analysis of the imaging data from the inguinal lymph node allowed us to track the movement of the transferred B cells over time and to derive motility parameters. We focused only on the B cells that had entered into the follicle. The B cells from the ric8fl/flmb1-cre mice moved slower and more erratically than did the WT B cells. There were also less polarized (Fig. 5C). These results are consistent with an impaired input from chemoattractant receptors, which help support much of the spontaneous motility of B cells in lymph nodes.
Figure 5.
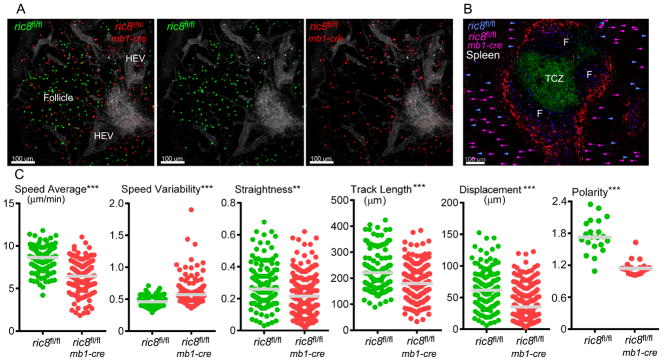
Intravital microscopy of the inguinal lymph node and imaging of spleen sections reveal that loss of Ric-8A leads to B cells with decreased motility that access B cell niches poorly. (A) Images acquired during intravital microscopy of the inguinal lymph node of a recipient mouse that had received an intravenous transfer WT (green) and ric8Afl/flmb1-cre (red) B cells (1:1 ratio) 18h prior to imaging. Evans blue was infused intravenously immediately before the imaging to outline blood vessels (grey). (B) Standard confocal microscopy of a thick spleen section from the same mouse used for the intravital microscopy focused on a splenic primary follicle. The T cell zone and B cell zones are indicated. The section was immunostained for CD4 (green) and CD169 (red). ric8fl/flmb1-cre B cells (pink) and ric8fl/fl (blue) B cells are shown. The location of the transferred B cells in the surrounding red pulp is indicated with arrowheads of the corresponding color. (C) Motility parameters of the ric8fl/flmb1-cre B cells and ric8fl/fl (control) B cells in the lymph node follicle of the recipient mice. Also shown are polarity measurements (long axis/short axis) of the control and ric-8fl/flmb1-cre B cells. Results are based on the analysis of two separate imaging experiments. ** p < 0.005; *** p < 0.0005.
Loss of Ric-8A reduced anti-IgM induced increases in intracellular calcium, did not impact B cell proliferation, but enhanced B cell survival
As B cells that lack Gαi2 or both Gαi2/3 have a reduced anti-IgM induced intracellular cellular response (27), we checked the response of the B cells from the ric8fl/flmb1-cre mice. We found that similar to Gαi deficient B cells, the Ric-8A deficient B cells also demonstrated a sub-optimal peak intracellular calcium response to anti-Ig stimulation. We verified these results using B cells purified by the ric8fl/flvav1-cre mice. We noted a decrease following stimulation with either a low or a high concentration of anti-IgM (Fig. 6A). Using a CFSE dye dilution assay we checked the proliferative response of B cells from the ric8fl/flmb1-cre and WT mice. While we found little difference in the proliferation index (the average number of cell divisions that a cell in the original population undergoes) or the division index (the average number of cell divisions of the responding cells), we did note a modest reduction in the % of Ric-8A deficient B cells that divided when Il-4 was present in the culture. We also found that at the end of the culture period that we recovered more Ric-8A deficient B cells following CD40 or CD40 and IgM stimulation suggesting that the loss of Ric-8A had led to an improved B cell survival rate (Fig. 6B). B cells from mice lacking Gαq are known to have an intrinsic survival advantage over normal B cells (31) suggesting that the loss of Gαq in the Ric-8A deficient B cells may have accounted for their enhanced survival.
Figure 6.

While Ric-8A deficient B cells respond relatively normally to in vitro B cell proliferative signals, Ric8fl/flmb1-cre mice have low serum immunoglobulins levels and generate poor antibody responses following immunization. (A) Control (mb1-cre), ric8fl/flmb1-cre, and ric8fl/flvav1-cre purified B cells were stimulated with anti-IgM and the induced changes in intracellular calcium were monitored over 3 minutes. The top graph shows results from ric8fl/fl versus ric8fl/flmb1-cre B cells. The data shown below is shown as a percentage of the maximal response of control cells. The experimental value is the mean of three determinations. Experiments were repeated 2 times with similar results. (B) Analysis of flow cytometry results from CFSE dye dilution assays using B cells purified from 3 WT (ric8fl/fl) and 3 ric8fl/flmb1-cre mice and stimulated for 96 hours as indicated. The division index, % cell divided, and the proliferation index were calculated using FlowJo. The numbers of viable B cells recovered at the end of the experiment are shown. Each assay was done in duplicate. Similar results in two other experiments. (C) ELISA assay to measure immunoglobulin isotypes in the sera of control (ric8Afl/fl, vav1-cre, and mb1-cre) and ric8Afl/flmb1-cre mice. Sera analyzed were from 6 mice for each genotype. (D) ELISA assay measuring specific antibody in the serum of NP-Ficoll immunized control and ric8fl/flmb1-cre mice. Four mice of each genotype were immunized and sera collected at the indicated time points. ELISA results from individual time points were compared. (E) ELISA assay measuring specific antibody in the serum of NP-KLH immunized control and ric8fl/flmb1-cre mice. Four mice of each genotype were immunized and serum collected at the indicated time points. Mice were boosted at day 60. Results from individual time points were compared. * p < 0.05; ** p < 0.005; ***.
The lack of Ric-8A in B cells reduces serum immunoglobulin levels and significantly impairs the humoral immune response to a thymus dependent and an independent antigen
The reduction in marginal zone B cells and the reduced number of peripheral B cells portended lower levels of serum immunoglobulins (Ig) in the ric8fl/flmb1-cre mice. As anticipated, we found decreased serum levels of IgA, IgM, and IgG isotypes. In contrast, the mb1-cre mice had a similar serum immunoglobulin profile as did the vav1-cre mice arguing that a loss of single mb-1 allele does not significantly compromised humoral immunity. Next, we check specific antibody responses following immunization with a thymus dependent or a thymus independent antigen. The ric8fl/flmb1-cre mice had little or no increase in serum Ig specific for NP following immunization with NP-Ficoll, which will elicit a thymus independent B cell response (Fig. 6D). This is consistent with the marked loss of marginal zone B cells in these mice. Immunization with NP-KLH, which triggers a thymus dependent response, led to a relatively normal primary and secondary NP specific IgM response. In contrast, both the primary and secondary NP IgG responses were very poor in the immunized ric8fl/flmb1-cre mice (Fig. 6E). This result is consistent with the poor GC response and the reduction in IgG1 positive B cells noted following immunization of the ric8fl/flmb1-cre mice with sRBCs. These results show a marked impairment in humoral immunity particularly in eliciting responses to new antigens.
The ric8fl/flmb1-cre B cells exhibit an occasional delay in cytokinesis abscission, decreased F-actin levels, and a reduced incidence of asymmetric cell divisions
We had previously noted that in human cell lines that Gαi proteins, RGS14, LGN, and Ric-8A all accumulated at the midbody during cytokinesis (10, 11). We had also shown that lowering Ric-8A expression delayed the abscission time of dividing human cells, which correlated with increased intercellular bridge length and multinucleation (12). To visualize dividing ric8fl/flmb1-cre, B cells we generated ric8fl/flmb1-cre EGFP-Lifeact mice. Lifeact accurately labels F-actin and is useful for visualizing dynamic changes in F-actin networks (32). B cells from these mice and control mice were cultured on ICAM1 coated plates and imaged following LPS stimulation. The stimulated B cells from ric8fl/flmb1-cre mice underwent symmetrical mitosis similar to B cells from wild type EGFP-Lifeact mice. However, as we had observed with the human cells lines we noted that some of the ric8fl/flmb1-cre EGFP-Lifeact B cells had a delayed abscission time following mitosis (Fig. 7A & B, video 2 and 3). The B cells from the ric8fl/flmb1-cre mice also exhibited a reduced motility, had difficulty in maintaining a polarized phenotype, and had lower overall levels of F-actin (Fig. 7C, data not shown).
Figure 7.

Occasional cytokinesis failure and reduced asymmetric cell division in B cells from ric8fl/flmb1-cre mice. (A) Confocal microscopy of dividing of a ric8wt/wt Lifeact B cell and ric8Afl/flmb1-cre Lifeact B cells. Splenic B cells purified from ric8wt/wt Lifeact and ric8Afl/flmb1-cre Lifeact mice were placed on ICAM-1 coated plates, stimulated with LPS, and imaged using time lapse live cell confocal microscopy. Image stacks (3–5 slices) were acquired every 15–20 seconds for 1 hour and dividing B cells identified by morphology changes and varying Lifeact expression. Single slice confocal images of wild type (top panel) and ric8fl/flmb1-cre B cell (bottom 3 panels) are shown. Scale bar 5 is μm. (B) Delayed cytokinesis in the ric8fl/flmb1-cre B cells. LPS activated B cells plated as in (A) were imaged and the number of minutes required from initial cleavage furrow formation to completion of cytokinesis was determined. Wild type Lifeact and ric8fl/flmb1-cre Lifeact B cells were compared. The results are from three separate imaging experiments. (C) Confocal microscopy images and F-actin levels in LPS stimulated wild type (Lifeact) and ric8fl/flmb1-cre B cells plated on ICAM-1 coated plates. The level of Lifeact immunofluorescence in single cells was quantitated using Imaris and results shown in the graph on the right. (D) Imagestream analysis to measure the number of asymmetrically dividing ric8Afl/fl and ric8Afl/flmb1-cre B cells. Prior to the analysis purified B cells were stimulated, or not, with CD40 + IgM or LPS for 72h. The B cells were immunostained for B220-PE-cy7, fixed, immunostained for PKCζ, and counterstained with DRAQ5 to outline the nuclei. A representative graph showing the number of cells harboring an asymmetric PKCζ distribution is shown (left panel) and below are images from a symmetrical, above, and asymmetrical, below, dividing cell. The numbers of cells with asymmetric PKCζ are shown (middle panel). The populations proliferative status are shown (right panel). (E) Imagestream analysis to determine the percentage of germinal center B cells showing an asymmetric PKCζ distribution among the germinal center B cells analyzed from the spleens from WT or ric8fl/flmb1-cre mice immunized 10 days previously with sRBCs. The B cells were immunostained for B220-PE-cy7, fixed, immunostained for aPKC, and counterstained with DRAQ5 to outline the nuclei. * p < 0.05; ** p < 0.005.
As heterotrimeric G protein signaling proteins can shape and regulate the orientation of the spindle body with the axis of polarity during asymmetric cell division (ACD) (33), we assessed the role of the B-cell specific ric8 deletion in ACD. Polarity is regulated by complexes of evolutionarily conserved polarity proteins known as the Scribble (including Scribble, lethal giant larvae, and discs large [Dlg]) and Par (including Par3, Par6, and atypical protein kinase C [PKCζ]) complexes, which antagonize each other to define molecularly distinct regions of the cell (34). We used a fast and unbiased methodology to quantify the frequency of dividing B cells with an asymmetric PKCζ distribution (Fig. 7D, left panel). This approach showed that the loss of Ric-8A expression decrease the propensity of dividing B cells to undergo asymmetric polarization of PCKCζ following in vitro stimulation with LPS or CD40 plus IgM (Fig. 7D, middle panel). The decrease in asymmetric localization of PKCζ cannot be accounted for by a reduction in the dividing cells as we found no significant decrease in the % of dividing cells in ric8fl/flmb1-cre B cells at 72 hours post-stimulation using this assay (Fig. 7D, right panel). Although there is a severe reduction in germinal center B cells in the immunized ric8fl/flmb1-cre mice we could use the same high-throughput methodology to analyze splenic B cells from immunized mice to determine the % of germinal center cells with an asymmetric localization of PKCζ. We found that 10 days following sRBC immunization approximately 6% of the phenotypic germinal center B cells (B220+GL7+Fas+) from the WT mice had an asymmetric distribution of PKCζ while only 2% of the phenotypically similar cells from the ric8fl/flmb1-cre B cells had an asymmetric distribution (Fig. 7E). Together these results point to the importance of heterotrimeric G protein signaling proteins in the regulation of polarity components during B cell activation and differentiation.
Discussion
Ric-8A is a guanine nucleotide exchange factor for a subset of Gα subunits and is needed for the normal levels of Gαi and Gαq and likely Gα13 proteins in lymphocytes and macrophages. Mice with hematopoietic cell specific loss of Ric-8A had lymphocytosis, anemia, and a shortened lifespan. Yet blood neutrophil, eosinophil, basophil, and platelet numbers in the blood were relatively normal. This indicates that hematopoietic stem cell function and bone marrow egress were largely intact. Hematopoietic cell differentiation proceeded despite the lack of Ric-8A and decreased levels of Gαi, Gαq, and Gα13 proteins in hematopoietic progenitors. Nevertheless, functional defects in the mature hematopoietic cells can be expected. The causes of the anemia and decreased longevity in these mice are unknown, but one possibility is bleeding due to impaired platelet function. The absence of Gαq, Gαi, or Gα13 is known to impair platelet function and can cause a bleeding diathesis (35–37). The lymphocytosis was accompanied by a reduction in B cells in secondary lymphoid organs, which suggested a B lymphocyte trafficking defect. Surprisingly T cell trafficking was less affected and thymocyte development proceeded with only evidence of a mild egress defect. Nevertheless further studies using a T cell specific Cre are warranted. Here, we focused on the role of Ric-8A in B cells and B cell function by generating mice with a B lymphocyte specific loss of Ric-8A. Study of these mice confirmed the importance of Ric-8A for B cell trafficking; chemokine receptor and BCR signaling; germinal center and memory B cell responses; asymmetric cell division; and humoral responses to novel antigens.
As indicated in the introduction the steady state levels of Gαi, Gαq, and Gα13 were reduced by 85% in the ric8−/− embryonic stem cells compared to similar cells from WT mice. Our study shows that absence of Ric-8A in lymphocytes and bone marrow derived macrophages causes a similar reduction in Gαi2/3, Gαq, and Gα13 although Gα12 levels were unaffected. Presumably other hematopoietic cells in the ric8fl/flvav1-cre mice are similarly affected although we have not directly examined them. All hematopoietic cells have ric8 mRNA transcripts although neutrophils conspicuously have the highest levels (18). Consistent with a major loss of Gαi proteins in neutrophils, the ric8fl/flvav1-cre neutrophils were very poorly recruited to inflammatory sites in vivo (O.K., unpublished observation).
By a variety of in vitro and in vivo assays the responses of the ric8fl/flmb1-cre B cells to chemoattractants is severely degraded. Essentially none of the ric8fl/flmb1-cre B cells migrated to near optimal concentrations of CXCL12 and CCL19 and only at the highest concentration of CXCL13 did we observe some migration. This is likely accounted for residual expression of Gαi2 and Gαi3 in the ric8fl/flmb1-cre B cells as the Gαi2/3 deficient B cells are refractory (27). The loss of marginal zone B cell in the ric8fl/flmb1-cre mice also argues for a physiologically significant loss in Gαi signaling. A reduction in marginal zone B cells was noted in Gαi2 deficient mice and the sphingosine 1-phosphate receptor 1 and cannabinoid receptor 2 are needed for the proper positioning of marginal zone B cells (38–40).
Engagement of chemoattractant receptors also elicits a rise in intracellular Ca2+, which is loss in the absence of Gαi signaling. As previously mentioned it is possible that the marked reduction in intracellular Ca2+ following exposure to chemokines may have arisen from the reduction Gαq in the setting of a reduced, but not absent Gαi in these mice. The disruption of the lymphoid organ architecture is also consistent with a major defect in B cell trafficking. In the spleen Gαi nucleotide exchange is needed to enter into the white pulp (41) and loss of CXCR5 leads to a failure to populate B cell follicles (42). In the ric8fl/flmb1-cre mice the splenic white pulp is poorly populated with B cells, the B/T borders are indistinct, marginal zone B cells are rare, and many B cells resided in the red pulp. Lymph nodes also had small B cell zones with indistinct B/T borders. Although not shown Peyer’s Patches were small and poorly developed in the ric8fl/flmb1-cre mice. This was also observed in the mice with Gαi2/Gαi3 deficient B cells and as previously discussed may be secondary to a reduction in B cell recruitment into the developing Peyer’s patches (27). The adoptively transfer studies supported the lymphoid architecture changes as many ric8fl/flmb1-cre B cells resided near HEVs failing to enter into the lymph node follicle and the splenic white pulp. Those B cells that did enter the follicle exhibited a motility pattern consistent with diminished Gαi signaling (43). Thus, the loss of Ric-8A in B cells reduces Gαi2 and Gαi3 levels sufficiently to severely impair chemoattractant receptor signaling. This loss was not accompanied by any significant change in chemoattractant receptor expression. This contrast to the Gαi2/3 deficient B cells where a modest decrease in CXCR4, CCR7, and CXCR5 expression was noted (27).
The reduction of BCR mediated increases in intracellular calcium found with the Ric-8A deficient B cells was also seen with the Gαi2 and Gαi2/3 deficient B cells (27). In contrast, Gαq−/− B cells are hyper-responsive to BCR crosslinking exhibiting higher amounts of phosphorylated AKT, ERK, and PLCγ2 following stimulation. They also had an augmented proliferative response to LPS and enhanced B cell viability (31). While the ric8fl/flmb1-cre B cells had major reductions in both Gαq and Gαi, the ric8fl/flmb1-cre B cell phenotype more reflected the loss of Gαi than Gαq. Similar to the Gnai3−/−Gnai2fl/flmb1-cre mice, the ric8fl/flmb1-cre mice had a reduced number of splenic B cells and a severe reduction in marginal zone B cell, while the Gnaq−/− mice had an expansion of splenic B cells including marginal zone B cells. We did note that with some inductive signals in in vitro cultures that we recovered more ric8fl/flmb1-cre B cells than control B cells, a result consistent with the improved B cell survival noted with the Gαq−/− B cells. We saw no evidence of autoimmunity in the ric8fl/flmb1-cre mice as had been noted in the Gnaq−/− mice.
Interestingly the serum immunoglobulin profiles and antibody responses to immunization in the ric8fl/flmb1-cre mice differed from both the Gnai3−/−Gnai2fl/flmb1-cre and the Gnai2fl/flmb1-cre mice. The Gnai3−/−Gnai2fl/flmb1-cre mice had a more severe phenotype exhibiting a hyper IgM-like syndrome with elevated serum IgM and severely depressed other Ig isotypes (27). The Gnai2fl/flmb1-cre mice had a more modest phenotype with reductions only in serum IgG1 and IgG2b. When immunized with a thymus independent antigen these mice had a slightly decreased IgM and IgG3 response and paradoxically an elevated IgG2C response. For unclear reasons the ric8fl/flmb1-cre mice had a more severe phenotype with essentially no specific response to the same thymus independent antigen. Similarly, a thymus dependent antigen elicited a much poorer response in the ric8fl/flmb1-cre mice than had been observed with the Gnai2fl/flmb1-cre mice. The ric8fl/flmb1-cre mice had a poor primary and essentially no secondary response. This is likely a consequence of the much poorer induction of germinal center cells in the ric8fl/flmb1-cre mice (approximately 10 fold decrease) than had been observed in either the Gnai2fl/flmb1-cre or the Gnai3−/− mice (approximately 2 fold reduction in each strain). The germinal center response in the ric8fl/flmb1-cre mice much more resembled that previously observed with the Gnai3−/−Gnai2fl/flmb1-cre mice. While B cell chemotaxis is highly dependent upon Gαi2, and less so on Gαi3, evidently the germinal center response depends upon the presence of both Gαi2 and Gαi3 (27). CXCR4, CXCR5, and Ebi2 signaling help orchestrate germinal center B cell dynamics (44–46) and there combined loss would likely recapitulate the severe germinal center phenotype noted in the ric8fl/flmb1-cre and Gnai3−/−Gnai2fl/flmb1-cre mice.
In the past years, a conserved general mechanism for asymmetric cell division has been discovered: asymmetric localization of Par-proteins polarizes the cell cortex, orients the mitotic spindle through heterotrimeric G-proteins and directs the segregation of determinants into only one of the two daughter cells. The polarity axis can be established by either cell intrinsic or extrinsic environmental signal (47). These observations have been extended to hematopoiesis and lymphocyte differentiation although not without controversies (48). In dividing T cells an unequal partitioning of membrane and cytoplasmic proteins can lead to the generation of two phenotypically distinct populations of daughter cells (24, 49). In B cells, the polarized secretion of lysosomes at the B cell synapse couples antigen extraction to presentation, and the asymmetric segregation of antigen during cell division shape the subsequent capacity of B cells to present antigen (50, 51). Furthermore, germinal center B cells asymmetrically segregate the transcriptional regulator Bcl6, the IL-21 receptor, and an atypical protein kinase C (aPKC) to one side of the plane of division by a mechanism that depends upon extrinsic signals from the microenvironment. However, another study indicated that Scribble-mediated asymmetric cell division is not required for humoral immunity (52). In the present study we addressed whether the loss of Ric-8A affected asymmetric partitioning of PKCζ. Our results showed that loss of Ric-8A expression and function switched off the molecular mechanisms in activated and germinal center B cells that had led to the asymmetric distribution of PKCζ. This is likely secondary to the loss of the activities of Ric-8A as an exchange factor and a chaperone, both of which have been implicated in mitotic spindle orientation. Despite this observation and those from others, many questions remain unanswered. For example, it remains unclear whether asymmetric cell divisions affects the decisions of germinal B cell to re-enter the germinal center, to adopt one of two alternative fates, a plasma cell or a memory B cell. More detailed studies combining fate mapping techniques (53) with sorting of cells harboring an asymmetric distribution (54) should help reveal the role that asymmetric cell division plays in lymphocyte fate decisions. Another interesting question is how do G-proteins act on microtubules and how is this process regulated? A growing body of evidences links a polarized Cdc42 activation to the initiation of asymmetric cell division (51, 55). One model that could explain an asymmetric activation of Cdc42 is a spatial organization of GoLoco proteins that could free Gα protein. Their subsequent activation could be Ric-8A dependent to mediate Cdc42 activation (56).
This study shows that hematopoiesis, bone marrow B cell development, and thymopoiesis occur in the absence of Ric-8A. The loss of Ric-8A from splenocytes, B cells, and likely other hematopoietic cell types leads to major decreases in the cellular content of Gαi, Gαq, and likely Gα13 proteins. The B lymphocyte specific loss of Ric-8A impairs B cell development in the spleen and severely impairs B lymphocyte trafficking and differentiation. B cells respond poorly to chemoattractants and fail to appropriately populate B cell niches. Humoral immunity is adversely affected with a major loss in germinal center formation, a marked reduction in antibody responses to novel antigens, and a nearly absent IgG memory response. The loss of Ric-8A in B cells interferes with asymmetric cell divisions although their importance in B cell differentiation remains unclear. The ric8fl/fl mice will be a valuable resource for studying the roles of Ric-8A, Gαi, Gαq, and Gα13 proteins in immune and hematopoietic cells.
Supplementary Material
Acknowledgments
This research was supported by the intramural program of the National Institutes of Allergy and Infectious Diseases.
The authors would like to thank Dr. Anthony Fauci for his continued support.
Abbreviations used in this article
- ACD
asymmetric cell division
- AGS3
Activator of G-protein signaling 3
- BCR
B cell antigen receptor
- GDI
guanine nucleotide dissociation inhibitor
- GEF
guanine nucleotide exchange factor
- GoLoco
Gαi/o-Loco interaction
- GPCR
G-protein coupled receptor
- GPR
G-protein regulatory
- LGN
Leu-Gly-Asn-enriched protein
- RGS
Regulator of G-protein Signaling
- Ric-8A
Resistance to inhibitors of cholinesterase 8A
Footnotes
Disclosures
The authors declare no competing financial interests.
Contributions
C.B., I-Y. H, and J.H.K. planned the experiments; C.B. backcrossed the mice, performed the initial phenotyping, western blotting, ELISA assays, and Image Stream analysis, I-Y. H. did the majority of the flow cytometry, chemotaxis assays, and intracellular calcium assays; C.P. did the intravital microscopy and the immunohistochemistry; O.K. did the live cell imaging of the purified B cells; K.H. helped with western blotting; Z.H. provided the ric8fl/fl mice; and C.B. and J.H.K. wrote the manuscript.
References
- 1.Cho H, Kehrl JH. Regulation of immune function by G protein-coupled receptors, trimeric G proteins, and RGS proteins. Prog Mol Biol Transl Sci. 2009;86:249–298. doi: 10.1016/S1877-1173(09)86009-2. [DOI] [PubMed] [Google Scholar]
- 2.Tall GG. Ric-8 regulation of heterotrimeric G proteins. J Recept Signal Transduct Res. 2013;33:139–143. doi: 10.3109/10799893.2013.763828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Afshar K, Willard FS, Colombo K, Johnston CA, McCudden CR, Siderovski DP, Gonczy P. RIC-8 is required for GPR-1/2-dependent Galpha function during asymmetric division of C. elegans embryos. Cell. 2004;119:219–230. doi: 10.1016/j.cell.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 4.Afshar K, Willard FS, Colombo K, Siderovski DP, Gonczy P. Cortical localization of the Galpha protein GPA-16 requires RIC-8 function during C. elegans asymmetric cell division. Development. 2005;132:4449–4459. doi: 10.1242/dev.02039. [DOI] [PubMed] [Google Scholar]
- 5.Couwenbergs C, Spilker AC, Gotta M. Control of embryonic spindle positioning and Galpha activity by C. elegans RIC-8. Curr Biol. 2004;14:1871–1876. doi: 10.1016/j.cub.2004.09.059. [DOI] [PubMed] [Google Scholar]
- 6.Woodard GE, Huang NN, Cho H, Miki T, Tall GG, Kehrl JH. Ric-8A and Gi alpha recruit LGN, NuMA, and dynein to the cell cortex to help orient the mitotic spindle. Mol Cell Biol. 2010;30:3519–3530. doi: 10.1128/MCB.00394-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siderovski DP, Willard FS. The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci. 2005;1:51–66. doi: 10.7150/ijbs.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parmentier ML, Woods D, Greig S, Phan PG, Radovic A, Bryant P, O’Kane CJ. Rapsynoid/partner of inscuteable controls asymmetric division of larval neuroblasts in Drosophila. J Neurosci. 2000;20:RC84. doi: 10.1523/JNEUROSCI.20-14-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doherty D, Chudley AE, Coghlan G, Ishak GE, Innes AM, Lemire EG, Rogers RC, Mhanni AA, Phelps IG, Jones SJ, Zhan SH, Fejes AP, Shahin H, Kanaan M, Akay H, Tekin M, Consortium FC, Triggs-Raine B, Zelinski T. GPSM2 mutations cause the brain malformations and hearing loss in Chudley-McCullough syndrome. Am J Hum Genet. 2012;90:1088–1093. doi: 10.1016/j.ajhg.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blumer JB, Chandler LJ, Lanier SM. Expression analysis and subcellular distribution of the two G-protein regulators AGS3 and LGN indicate distinct functionality. Localization of LGN to the midbody during cytokinesis. J Biol Chem. 2002;277:15897–15903. doi: 10.1074/jbc.M112185200. [DOI] [PubMed] [Google Scholar]
- 11.Cho H, Kehrl JH. Localization of Gi alpha proteins in the centrosomes and at the midbody: implication for their role in cell division. J Cell Biol. 2007;178:245–255. doi: 10.1083/jcb.200604114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boularan C, Kamenyeva O, Cho H, Kehrl JH. Resistance to inhibitors of cholinesterase (Ric)-8A and Galphai contribute to cytokinesis abscission by controlling vacuolar protein-sorting (Vps)34 activity. PLoS One. 2014;9:e86680. doi: 10.1371/journal.pone.0086680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blumer JB, Lord K, Saunders TL, Pacchioni A, Black C, Lazartigues E, Varner KJ, Gettys TW, Lanier SM. Activator of G protein signaling 3 null mice: I. Unexpected alterations in metabolic and cardiovascular function. Endocrinology. 2008;149:3842–3849. doi: 10.1210/en.2008-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Regner KR, Nozu K, Lanier SM, Blumer JB, Avner ED, Sweeney WE, Jr, Park F. Loss of activator of G-protein signaling 3 impairs renal tubular regeneration following acute kidney injury in rodents. FASEB J. 2011;25:1844–1855. doi: 10.1096/fj.10-169797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vural A, McQuiston TJ, Blumer JB, Park C, Hwang IY, Williams-Bey Y, Shi CS, Ma DZ, Kehrl JH. Normal autophagic activity in macrophages from mice lacking Galphai3, AGS3, or RGS19. PLoS One. 2013;8:e81886. doi: 10.1371/journal.pone.0081886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Branham-O’Connor M, Robichaux WG, 3rd, Zhang XK, Cho H, Kehrl JH, Lanier SM, Blumer JB. Defective chemokine signal integration in leukocytes lacking activator of G protein signaling 3 (AGS3) J Biol Chem. 2014;289:10738–10747. doi: 10.1074/jbc.M113.515031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamakura S, Nomura M, Hayase J, Iwakiri Y, Nishikimi A, Takayanagi R, Fukui Y, Sumimoto H. The cell polarity protein mInsc regulates neutrophil chemotaxis via a noncanonical G protein signaling pathway. Dev Cell. 2013;26:292–302. doi: 10.1016/j.devcel.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Boularan C, Kehrl JH. Implications of non-canonical G-protein signaling for the immune system. Cell Signal. 2014;26:1269–1282. doi: 10.1016/j.cellsig.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tonissoo T, Lulla S, Meier R, Saare M, Ruisu K, Pooga M, Karis A. Nucleotide exchange factor RIC-8 is indispensable in mammalian early development. Dev Dyn. 2010;239:3404–3415. doi: 10.1002/dvdy.22480. [DOI] [PubMed] [Google Scholar]
- 20.Chan P, Thomas CJ, Sprang SR, Tall GG. Molecular chaperoning function of Ric-8 is to fold nascent heterotrimeric G protein alpha subunits. Proc Natl Acad Sci U S A. 2013;110:3794–3799. doi: 10.1073/pnas.1220943110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gabay M, Pinter ME, Wright FA, Chan P, Murphy AJ, Valenzuela DM, Yancopoulos GD, Tall GG. Ric-8 proteins are molecular chaperones that direct nascent G protein alpha subunit membrane association. Sci Signal. 2011;4:ra79. doi: 10.1126/scisignal.2002223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma S, Kwon HJ, Huang Z. Ric-8a, a guanine nucleotide exchange factor for heterotrimeric G proteins, regulates bergmann glia-basement membrane adhesion during cerebellar foliation. J Neurosci. 2012;32:14979–14993. doi: 10.1523/JNEUROSCI.1282-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruisu K, Kask K, Meier R, Saare M, Raid R, Veraksits A, Karis A, Tonissoo T, Pooga M. Ablation of RIC8A function in mouse neurons leads to a severe neuromuscular phenotype and postnatal death. PLoS One. 2013;8:e74031. doi: 10.1371/journal.pone.0074031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oliaro J, Van Ham V, Sacirbegovic F, Pasam A, Bomzon Z, Pham K, Ludford-Menting MJ, Waterhouse NJ, Bots M, Hawkins ED, Watt SV, Cluse LA, Clarke CJ, Izon DJ, Chang JT, Thompson N, Gu M, Johnstone RW, Smyth MJ, Humbert PO, Reiner SL, Russell SM. Asymmetric cell division of T cells upon antigen presentation uses multiple conserved mechanisms. J Immunol. 2010;185:367–375. doi: 10.4049/jimmunol.0903627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, Reth M. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci U S A. 2006;103:13789–13794. doi: 10.1073/pnas.0605944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, Kioussis D. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol. 2003;33:314–325. doi: 10.1002/immu.200310005. [DOI] [PubMed] [Google Scholar]
- 27.Hwang IY, Park C, Luong T, Harrison KA, Birnbaumer L, Kehrl JH. The loss of Gnai2 and Gnai3 in B cells eliminates B lymphocyte compartments and leads to a hyper-IgM like syndrome. PLoS One. 2013;8:e72596. doi: 10.1371/journal.pone.0072596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chai Q, Onder L, Scandella E, Gil-Cruz C, Perez-Shibayama C, Cupovic J, Danuser R, Sparwasser T, Luther SA, Thiel V, Rulicke T, Stein JV, Hehlgans T, Ludewig B. Maturation of lymph node fibroblastic reticular cells from myofibroblastic precursors is critical for antiviral immunity. Immunity. 2013;38:1013–1024. doi: 10.1016/j.immuni.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park C, I, Hwang Y, Sinha RK, Kamenyeva O, Davis MD, Kehrl JH. Lymph node B lymphocyte trafficking is constrained by anatomy and highly dependent upon chemoattractant desensitization. Blood. 2012;119:978–989. doi: 10.1182/blood-2011-06-364273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stein SJ, Baldwin AS. Deletion of the NF-kappaB subunit p65/RelA in the hematopoietic compartment leads to defects in hematopoietic stem cell function. Blood. 2013;121:5015–5024. doi: 10.1182/blood-2013-02-486142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Misra RS, Shi G, Moreno-Garcia ME, Thankappan A, Tighe M, Mousseau B, Kusser K, Becker-Herman S, Hudkins KL, Dunn R, Kehry MR, Migone TS, Marshak-Rothstein A, Simon M, Randall TD, Alpers CE, Liggitt D, Rawlings DJ, Lund FE. G alpha q-containing G proteins regulate B cell selection and survival and are required to prevent B cell-dependent autoimmunity. J Exp Med. 2010;207:1775–1789. doi: 10.1084/jem.20092735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riedl J, Flynn KC, Raducanu A, Gartner F, Beck G, Bosl M, Bradke F, Massberg S, Aszodi A, Sixt M, Wedlich-Soldner R. Lifeact mice for studying F-actin dynamics. Nat Methods. 2010;7:168–169. doi: 10.1038/nmeth0310-168. [DOI] [PubMed] [Google Scholar]
- 33.Bellaiche Y, Gotta M. Heterotrimeric G proteins and regulation of size asymmetry during cell division. Curr Opin Cell Biol. 2005;17:658–663. doi: 10.1016/j.ceb.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 34.Tajbakhsh S, Rocheteau P, Le Roux I. Asymmetric cell divisions and asymmetric cell fates. Annu Rev Cell Dev Biol. 2009;25:671–699. doi: 10.1146/annurev.cellbio.24.110707.175415. [DOI] [PubMed] [Google Scholar]
- 35.Jantzen HM, Milstone DS, Gousset L, Conley PB, Mortensen RM. Impaired activation of murine platelets lacking G alpha(i2) J Clin Invest. 2001;108:477–483. doi: 10.1172/JCI12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weig HJ, Bott-Flugel L, Stadele C, Winter K, Schmidt R, Gawaz M, Laugwitz KL, Seyfarth M. Impaired platelet function reduces myocardial infarct size in Galphaq knock-out mice in vivo. J Mol Cell Cardiol. 2008;44:143–150. doi: 10.1016/j.yjmcc.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 37.Moers A, Nieswandt B, Massberg S, Wettschureck N, Gruner S, Konrad I, Schulte V, Aktas B, Gratacap MP, Simon MI, Gawaz M, Offermanns S. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat Med. 2003;9:1418–1422. doi: 10.1038/nm943. [DOI] [PubMed] [Google Scholar]
- 38.Dalwadi H, Wei B, Schrage M, Spicher K, Su TT, Birnbaumer L, Rawlings DJ, Braun J. B cell developmental requirement for the G alpha i2 gene. J Immunol. 2003;170:1707–1715. doi: 10.4049/jimmunol.170.4.1707. [DOI] [PubMed] [Google Scholar]
- 39.Muppidi JR, Arnon TI, Bronevetsky Y, Veerapen N, Tanaka M, Besra GS, Cyster JG. Cannabinoid receptor 2 positions and retains marginal zone B cells within the splenic marginal zone. J Exp Med. 2011;208:1941–1948. doi: 10.1084/jem.20111083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cinamon G, Matloubian M, Lesneski MJ, Xu Y, Low C, Lu T, Proia RL, Cyster JG. Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat Immunol. 2004;5:713–720. doi: 10.1038/ni1083. [DOI] [PubMed] [Google Scholar]
- 41.Cyster JG, Goodnow CC. Pertussis toxin inhibits migration of B and T lymphocytes into splenic white pulp cords. J Exp Med. 1995;182:581–586. doi: 10.1084/jem.182.2.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 43.Han SB, Moratz C, Huang NN, Kelsall B, Cho H, Shi CS, Schwartz O, Kehrl JH. Rgs1 and Gnai2 regulate the entrance of B lymphocytes into lymph nodes and B cell motility within lymph node follicles. Immunity. 2005;22:343–354. doi: 10.1016/j.immuni.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 44.Gatto D, Wood K, Brink R. EBI2 operates independently of but in cooperation with CXCR5 and CCR7 to direct B cell migration and organization in follicles and the germinal center. J Immunol. 2011;187:4621–4628. doi: 10.4049/jimmunol.1101542. [DOI] [PubMed] [Google Scholar]
- 45.Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, Cyster JG. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5:943–952. doi: 10.1038/ni1100. [DOI] [PubMed] [Google Scholar]
- 46.Yi T, Wang X, Kelly LM, An J, Xu Y, Sailer AW, Gustafsson JA, Russell DW, Cyster JG. Oxysterol gradient generation by lymphoid stromal cells guides activated B cell movement during humoral responses. Immunity. 2012;37:535–548. doi: 10.1016/j.immuni.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams SE, Fuchs E. Oriented divisions, fate decisions. Curr Opin Cell Biol. 2013;25:749–758. doi: 10.1016/j.ceb.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pham K, Sacirbegovic F, Russell SM. Polarized cells, polarized views: asymmetric cell division in hematopoietic cells. Front Immunol. 2014;5:26. doi: 10.3389/fimmu.2014.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang JT, V, Palanivel R, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth SA, Vinup KE, Mrass P, Oliaro J, Killeen N, Orange JS, Russell SM, Weninger W, Reiner SL. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315:1687–1691. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 50.Thaunat O, Granja AG, Barral P, Filby A, Montaner B, Collinson L, Martinez-Martin N, Harwood NE, Bruckbauer A, Batista FD. Asymmetric segregation of polarized antigen on B cell division shapes presentation capacity. Science. 2012;335:475–479. doi: 10.1126/science.1214100. [DOI] [PubMed] [Google Scholar]
- 51.Yuseff MI, Reversat A, Lankar D, Diaz J, Fanget I, Pierobon P, Randrian V, Larochette N, Vascotto F, Desdouets C, Jauffred B, Bellaiche Y, Gasman S, Darchen F, Desnos C, Lennon-Dumenil AM. Polarized secretion of lysosomes at the B cell synapse couples antigen extraction to processing and presentation. Immunity. 2011;35:361–374. doi: 10.1016/j.immuni.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 52.Hawkins ED, Oliaro J, Kallies A, Belz GT, Filby A, Hogan T, Haynes N, Ramsbottom KM, Van Ham V, Kinwell T, Seddon B, Davies D, Tarlinton D, Lew AM, Humbert PO, Russell SM. Regulation of asymmetric cell division and polarity by Scribble is not required for humoral immunity. Nat Commun. 2013;4:1801. doi: 10.1038/ncomms2796. [DOI] [PubMed] [Google Scholar]
- 53.Gerlach C, van Heijst JW, Swart E, Sie D, Armstrong N, Kerkhoven RM, Zehn D, Bevan MJ, Schepers K, Schumacher TN. One naive T cell, multiple fates in CD8+ T cell differentiation. J Exp Med. 2010;207:1235–1246. doi: 10.1084/jem.20091175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bocharov G, Luzyanina T, Cupovic J, Ludewig B. Asymmetry of Cell Division in CFSE-Based Lymphocyte Proliferation Analysis. Front Immunol. 2013;4:264. doi: 10.3389/fimmu.2013.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dehapiot B, Carriere V, Carroll J, Halet G. Polarized Cdc42 activation promotes polar body protrusion and asymmetric division in mouse oocytes. Dev Biol. 2013;377:202–212. doi: 10.1016/j.ydbio.2013.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ueda H, Morishita R, Yamauchi J, Itoh H, Kato K, Asano T. Regulation of Rac and Cdc42 pathways by G(i) during lysophosphatidic acid-induced cell spreading. J Biol Chem. 2001;276:6846–6852. doi: 10.1074/jbc.M007541200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.