

Abstract
Innate immune inflammatory responses are subject to complex layers of negative regulation at intestinal mucosal surfaces. While the type I interferon (IFN) system is critical for amplifying anti-viral immunity, it has been shown to play a homeostatic role in some models of autoimmune inflammation. Type I IFN is triggered in the gut by select bacterial pathogens, but whether and how the type I IFN might regulate innate immunity in the intestinal environment has not been investigated in the context of Salmonella enterica serovar Typhimurium (ST). ST infection of human or murine macrophages reveals that IFN-β selectively restricts the transcriptional responses mediated by both the Toll-Like Receptors (TLRs) and the NOD-Like Receptors (NLRs). Specifically, IFN-β potently represses ST-dependent innate induction of IL-1 family cytokines and neutrophil chemokines. This IFN-β-mediated transcriptional repression was independent of the effects of IFN-β on ST-induced macrophage cell death, but significantly dependent on IL-10 regulation. We further evaluated ST pathogenesis in vivo following oral inoculation of mice lacking IFN-β. We show that IFN-β−/− mice exhibit greater resistance to oral ST infection and a slower spread of ST to distal sterile sites. This work provides mechanistic insight into the relationship between ST and type I IFN, and demonstrates an additional mechanism by which IFN-β may promote spread of enteric pathogens.
Keywords: TLR4, Macrophage, Salmonella, Interferon Beta
Introduction
Salmonella enterica serovar Typhimurium (ST) is a Gram-negative enteric pathogen most often acquired from contaminated food or water that is associated clinically with severe gastroenteritis in humans and a systemic disease resembling typhoid fever in mice. A robust innate immune response in the gut is required to provide early control of ST infection, slow dissemination to distal sterile sites, and mitigate subsequent morbidity and mortality (1). Over 30 years ago, it was recognized that innate immunity is important in controlling ST (2). Specifically, the TLR system is critical, as animals deficient in the key bacterial recognition sensors TLR4 and TLR2 are significantly more susceptible to ST (3-6).
Despite the clear importance of innate immune responses in controlling infection, ST displays a remarkable capacity for subversion and co-opting of these same responses. Among the evasion mechanisms employed by ST are the type III secretion system (T3SS) effectors encoded by the Salmonella Pathogenicity Island 2 (SPI2) locus that blunt the anti-microbial oxidative burst driven by iNOS and NADPH oxidase in phagocytes (7). The ability of T3SS effectors to subvert endogenous host NLR chaperones to regulate NLR activity (8), as well as the ability to “reprogram” transcriptional responses in epithelial cells (9), have also been identified as mechanisms of ST subversion of the immune response.
However, it has recently become abundantly clear that evasion of innate responses is not the only strategy employed by ST, but that ST actively utilizes, and in some instances requires, host innate immune responses. Indeed, the process of gut inflammation itself can promote the pathogenesis of ST by enabling competition with microflora (10) and by generating novel energy sources (11). Additionally, ST takes advantage of TLR-governed phagosomal acidification to induce pH-dependent expression of SPI2 effectors in macrophages that facilitate intracellular ST survival (3, 12, 13).
Significantly, it has recently been reported that type I IFN receptor-null macrophages (IFNAR−/−) are refractory to ST-initiated necroptotic cell death (14). The type I IFNs are pleiotropic cytokines produced downstream of many innate receptors that function in both autocrine and paracrine fashions and possess remarkable antiviral properties (15, 16). However, the relationship between type I IFNs and bacterial pathogens is far more complex, and can be deleterious or protective, depending on the specific pathogen (17-19). In nearly all cases examined, however, the molecular mechanisms by which IFN might promote pathogenesis are poorly defined. We postulated that type I IFN may have a significant capacity to govern innate responses to ST. To investigate this hypothesis, we examined the response to infection by ST in IFN-β−/− macrophages, and found that the autocrine or paracrine action of IFN-β profoundly shapes the ST-induced TLR and NLR transcriptional responses in murine macrophages. This selective type I IFN “sculpting” of the TLR inflammatory response likely dampens cell-mediated innate immunity to ST by repressing neutrophil chemotaxis and IL-1 cytokine family activity, thereby enabling bacterial dissemination.
Materials and Methods
Ethics statement
Animal work performed for this study complied with all applicable provisions of the Animal Welfare Act, the U.S. Government Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training, the Public Health Services (PHS) Policy on the Humane Care and Use of Laboratory Animals and the Guide for the Care and Use of Laboratory Animals (8th Ed.). The protocol for this work was approved by the Institutional Animal Care and Use Committee of the University of Maryland, School of Medicine. Clinical ST strains isolated from pediatric patients were obtained from the Salmonella culture collection stored at the Center for Vaccine Development, University of Maryland Baltimore. These isolates are labeled with a specimen identifier only and lack information that can link them to individual patients. These isolates were collected as part of a previous study and IRB approval information is contained therein (20).
Cell Lines and Mice
Primary peritoneal macrophages were prepared as described previously (21). Briefly, 3 ml of 3% sterile thioglycollate was injected i.p into 6-8 week old, wild-type (WT) C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME). Four days later, macrophages were harvested by peritoneal lavage with sterile saline. IFN-β null mice (IFN-β−/−), MyD88-null mice (MyD88−/−) and TLR4-null mice (TLR4−/−) backcrossed onto a C57BL/6J background (N≥ 9), were bred in-house as described previously (22). Peritoneal macrophages from IL-10-null (IL-10−/−) mice were a kind gift of Dr. Jay Bream (Johns Hopkins School of Public Health). Peritoneal macrophages from Caspase-11 null (caspase-11−/−) mice were a kind gift of Dr. Joao Pedra (University of Maryland, Baltimore) that were obtained under a Materials Transfer Agreement with Genentech (South San Francisco, CA). The THP-1 cell line was maintained in RPMI supplemented with 10% FBS. Prior to infection, cells were plated and differentiated using PMA (50 ng/ml) fro 48 hrs. The MODEK cell line was a kind gift of Dr. Dana Philpott (University of Toronto, Toronto Canada) and these cells were maintained in DMEM supplemented with 10% FBS.
Antibodies and Reagents
Antibodies against total p38, phospho-p38, total p-38, phospho-ERK1/2, total ERK 1/2, total p65, phospho p65, and total IL-1β were purchased from Cell Signaling (Danvers, MA). Anti-IL-10 and anti-IL-27 neutralizing antibodies were purchased from e-Bioscience (San Diego, CA). All antibodies used in Flow Cytometry were purchased from Biolegend (San Diego, CA). Recombinant murine IL-10 and IL-27 was purchased from Biolegend. Protein-free phenol/water extracted Escherichia-coli K235 LPS was prepared as described elsewhere (23). S-[2,3-Bis(palmitoyloxy)-(2-RS)-propyl]-N-palmitoyl-(R)-Cys-Ser-Lys4-OH (Pam3Cys) was purchased from InVivogen (San Diego, CA). Purified mammalian-expressed recombinant IFN-β was purchased from PBL (Piscataway NJ). Recombinant murine IFN-γ was kindly provided by Genentech, Inc. (San Francisco, CA). Necrostatin was purchased from Santa Cruz (Santa Cruz, CA). L18-MDP and C12-iE-DAP were purchased from Invivogen (San Diego, CA).
Bacterial Strains
Salmonella Typhimurium strain SL1344 was the kind gift of Dr. Robert Ernst (University of Maryland, Baltimore). Strains I77 (ST19) and D65 (ST313) are clinical isolates of ST isolated from blood of febrile pediatric patients in Bamako, Mali and have been previously described (20).
Quantitative Real Time-PCR (qRT-PCR)
Total mRNA was isolated from peritoneal macrophages using TRIZOL (Invitrogen Carlsbad, CA) reagent according to manufacturer's instructions. A total of 1 μg of RNA was utilized in oligo (dT) cDNA synthesis (Bio-Rad RT system). qRT-PCR was carried out using a an ABI Prism 7900HTSequence Detection System (Applied Biosystems) utilizing SYBR Green reagent (Applied Biosystems) and transcript-specific primers. mRNA expression profiles were normalized to levels of housekeeping gene hypoxanthine-guanine phosphoribosyltransferase (HPRT) in each sample and the fold-change in expression was calculated by the 2−ΔΔCt method (24).
Western Blot Analysis
Whole cell lysates from primary murine macrophages were obtained by the addition of lysis buffer (20 mM HEPES, 1.0% TRITON X-100, 0.1% SDS, 150 mM NaCl, 10 mM NaF, 1 mM PMSF) and subsequent incubation at 4° C. Cell lysates were separated by electrophoresis in a denaturing SDS-PAGE gel and subsequent transfer to PVDF membrane. Blots were incubated overnight in relevant primary antibodies at 4° C and washed 3X with PBS, and then incubated with appropriate HRP-conjugated secondary antibody (Jackson Immunochemicals, west Grove, PA). Blots were developed following incubation in ECL PLUS Western Blotting Detection Reagent (Amersham Bioscience, Piscataway NJ).
In Vitro Bacterial Infections
A single colony of ST strain SL1344 was inoculated into 5 ml of HS media and grown overnight at 37° C with shaking. The following morning, an additional 10 ml of HS media was inoculated with 200 µl of overnight culture and incubated at 37° C with shaking until OD600 reached 1.5. One ml of the culture was pelleted by centrifugation and re-suspended into 1 ml of sterile PBS. Four hundred µl of bacterial suspension was mixed with 600 µl of sterile PBS to obtain a concentration of approximately 4×106 bacteria per 10 µl of culture. An appropriate volume of this re-suspension was mixed with 37° C antibiotic-free RPMI to obtain the desired multiplicity of infection. Bacteria were added to macrophages in culture plates and infections were synchronized by centrifugation for 5 min at 700 rpm. Infected cultures were incubated at 37° C for 30 min and the infection media was removed and replaced with RPMI containing 50 µg/ml gentamicin and incubated for an additional 45 min at 37° C. Following gentamicin incubation, media was removed and the cultures washed 2X with sterile PBS, and the media replaced with antibiotic-free RPMI. For infections involving IFN-β priming, cells were pre-treated for 1 hr (unless otherwise stated) with 100 IU/ml of murine or human IFN-β (in mouse or human cells, respectively) in antibiotic-free RPMI, and subsequently washed 2X in PBS prior to addition of infection medium.
In Vivo Salmonella infections
SL1344 cultures were grown overnight at 37°C without shaking. The following morning, 1 ml of culture was pelleted and re-suspended in 10 ml of ice cold PBS. For oral infection model, mice were inoculated by oral gavage utilizing 100 μl of PBS inoculum. For the sepsis infection model, animals were given a single IP injection of 1×108 ST Sl1344 in 100 μl PBS. Animals were sacrificed 8 hours later and livers were harvested for RNA expression analysis by qRT-PCR.
Stimulation of Macrophages
For TLR ligand experiments, macrophages were stimulated with 50 ng/ml purified E. coli LPS or 250 ng/ml P3C for the indicated times prior to harvest for RNA expression analysis. For IL-10 neutralizing experiments, 10 μg/ml of anti-IL-10 or an isotype-matched control antibody was added prior to IFN-β priming and maintained in media throughout stimulation with LPS. For NOD ligand experiments, primary peritoneal macrophages were treated overnight either with 20 μg/ml of NOD1 ligand (C12-iEDAP) or NOD2 ligand (L18-MDP) directly added to culture medium. Cell supernatants were harvested the following day for analysis of CXCL1 protein levels by ELISA.
Macrophage Cytotoxicity Assays
Primary peritoneal macrophages were infected with ST strain SL1344 at an MOI of 4 and samples of culture supernatant were harvested at the indicated time-points for analysis of Lactate Dehydrogenase (LDH) levels. LDH assay was carried out utilizing the CytoTox 96 (Promega, Madison WI) LDH release assay from according to manufacturer’s directions.
Flow Cytometry/ TLR4 internalization
Peritoneal macrophages were pretreated with or without IFN-β, washed 2x with PBS and stimulated with E. coli LPS (100ng/ml) for indicated times. Cells were subsequently washed with FACS buffer (PBS with 2% FBS), blocked using anti CD16/32 antibody (BioLegend San Diego, CA) for 20 min and specifically stained with anti-TLR4-PE (BioLegend San Diego, CA) or isotype control (BioLegend San Diego, CA) antibody for 40 min. Cells were washed and staining was assessed with BD FACS Canto. All antibody staining was carried out on ice. Isotype control staining was used to calculate TLR4 internalization based on mean fluorescence intensity (MFI) and expressed as a percentage of surface TLR4 staining on non-LPS-stimulated cells.
Myeloperoxidase Activity Assay
Excised tissues were weighed then homogenized using an electric homogenizer in 0.5% hexa-decyl-trimethyl-ammonium bromide in 50 mmol/L potassium phosphate buffer (pH6) and centrifuged at 12,000 rpm for 10 min. Aliquots of the supernatant were mixed with a chromogenic peroxidase substrate (BM blue, Roche) and allowed to react for 20 min. Absorbance was measured by spectrophotometry at 450 nm. Human purified myeloperoxidase enzyme was used to create standard curves. Myeloperoxidase activity was determined by calculating OD/mass of tissue sample.
Statistical Analysis
Statisical analysis when applied was done using the Prism software v5.0. All t tests were done using a two tailed analysis.
Results
Paracrine IFN-β selectively suppresses TLR-dependent inflammatory responses to ST
As previous work had described an increase in ST-induced cell death in IFNAR−/− macrophages, but did not describe altered innate receptor responses in this background (14), we sought to quantify the effect of IFN-β on the innate transcriptional response to ST in primary peritoneal murine macrophages. To confirm the primacy of TLRs in governing the early transcriptional responses of macrophages, WT or MyD88−/− peritoneal macrophages were infected with St SL1344 at an MOI of 4, followed by examination of classic pro-inflammatory gene transcripts by qRT-PCR (Figure 1A). We observed a complete dependence on MyD88 for ST-induced expression of inflammatory mediators (e.g., IL-1β, IL-6) within the first 6 hours p.i. (Figure 1A). As TLR4 has been specifically ascribed the largest role in mediating the early inflammation (3, 25), we similarly infected WT and TLR4−/− macrophages. Like MyD88−/− macrophages, TLR4−/− macrophages showed a nearly complete loss of induction of IL-1β and IL-6 mRNA, (Figure 1B) as well as other pro-inflammatory genes (data not shown). In vivo, type I IFN secreted by infected tissue acts on and establishes ant-microbial states in adjacent uninfected cells in advance of infection in a paracrine mode of action. We therefore sought to model the paracrine effects of IFN-β on macrophages. WT peritoneal macrophages were pre-treated with recombinant murine IFN-β (100 IU/ml) for 60 min prior to infection with ST SL1344 (MOI = 4). Remarkably, exposure of macrophages to IFN-β selectively suppressed inflammatory cytokine responses to ST (Figure 1C). Induction of IL-1β and IL-18 was significantly suppressed in IFN-β-primed macrophages over the first 6 h of infection (Figure 1C). Additionally, we found the neutrophil chemokines, i.e., CXCL1, CXCL2, and CXCL5, were suppressed by pretreatment of macrophages with IFN-β. IFN-β-mediated suppression of ST-induced cytokine/chemokine mRNA expression was not a global phenomenon, as no significant suppression of either IL-6 or TNF-α induction was observed (Figure 1D). Interestingly, prior exposure to IFN-β markedly enhanced subsequent ST-induced production of IFN-β mRNA, (Figure 1E), consistent with previous findings showing that type I IFN priming of macrophages increased induction of type I IFN in response to LPS (26). Each of these observations made at the mRNA level were independently confirmed at the protein level (Figure S1 A). As reduced pro-inflammatory gene induction might have resulted from differential infection of media- and IFN-β-pre-treated macrophages, the potential effects of IFN-β on the phagocytosis or early survival of ST in macrophages was ruled out by recovering equivalent numbers of intracellular bacteria from media- or IFN-β-pre-treated macrophages 6 hrs after infection (Figure 1F). To determine if the IFN-mediated cytokine suppression we observed was generalizable to a human system, the human macrophage-like cell line, THP-1, was also infected with at an MOI of 4 and human CXCL1 homologue, IL-8 mRNA, measured. A similar ST suppression of IL-8 mRNA was observed as seen with murine CXCL1 (Figure 1G). To address a second relevant tissue type in ST infection, we examined the suppressive effects of type I IFN on small intestinal epithelia. IFN-β-mediated suppression of ST induced CXCL2 was not observed in the ileal epithelial cell line, MODE-K, suggesting that this effect may be limited to innate myeloid cells (Figure S1 B).
Figure 1. Early Neutrophil Chemokine and IL-1 Family Cytokine Responses to ST Infection of Macrophages are TLR4 dependent and Suppressed by Paracrine IFN-β.

(A) MyD88−/− or MyD88+/+ peritoneal macrophages were infected with ST SL1344 at an moi of 4 for the indicated times and total RNA was used for mRNA analysis by qRT PCR. (B) TLR4 +/+ and TLR4−/− peritoneal macrophages were infected as in (A) and total RNA was used for cytokine mRNA analysis by qRT PCR. (C-E ) WT C57BL6/J macrophages were pretreated for 60 minutes with media alone or 100 u/ml murine IFN-β and infected with ST SL1344 for indicated times prior to mRNA analysis. Data are presented as mean ± SEM (F) WT C57BL6/J macrophages were infected with strain SL1344 at an moi of 4 for 6 h. The cells were lysed and recoverable bacteria counted after plating on LB agar. (G) Human THP-1 monocytic cells were differentiated for 48 hrs in PMA prior to infection with SL1344 at an moi of 4 for 6 h. RNA was harvested and expression of IL8 analyzed by qRT PCR. p = .003. Data in (A-G) is representative of at least three independent experiments.
IFN-β−/− macrophages are selectively hyper-responsive to ST infection
To confirm our findings, we also sought to establish a model for autocrine IFN action, in which IFN produced by infected cells acts back on these same cells to slow infection in the absence of exogenous IFN-β. To this end, primary peritoneal macrophages from IFN-β−/− mice were infected with ST SL1344 at (MOI = 4) for 4 hrs. Remarkably, in the IFN-β−/− macrophages, significantly enhanced induction of IL-1β and the neutrophil chemokines CXCL1, CXCL2, and CXCL5 mRNA was observed (Figure 2). The specificity of the IFN-β effect was supported by examining the induction of IL-6, TNF-α, and the monocyte chemokine, CCL5, none of which were enhanced in the absence of IFN-β (Figure 2), a pattern of gene induction that was also observed at multiple time points (data not shown). Each of the changes observed at the mRNA level were recapitulated by measuring protein levels (Figure S2).
Figure 2. Neutrophil Chemokine and IL-1 family cytokine expression are augmented following ST infection of IFN-β−/− macrophages.

(A) WT C57BL6/J or IFN-β−/− peritoneal macrophages were infected with SL1344 at an moi of 4 for 6 hours and RNA harvested for analysis by qRT PCR. Data is representative of three independent experiments and shown as mean ± SEM. ND = not detectable.
IFN-β suppression of ST-induced transcriptional responses is dissociable from macrophage cell death
The ST SL1344 strain has been used extensively for studies on the innate response to ST, however, this strain has been passaged for a prolonged period under laboratory conditions (27), and we considered the possibility that this laboratory passage had influenced the relationship of ST SL1344 with IFN-β. To rule out this possibility, we obtained clinical isolates from Mali North Africa in two sequence type variants (20, 28). Both the I77 and the D65 clinical strains exhibited an IFN-β-dependent suppression of IL-1β and neutrophil chemokines, confirming the potential clinical relevance of our findings (Figure 3A).
Figure 3. Clinical Isolates of ST Exhibit IFN-β Dependent Suppression of Inflammatory Responses, and Co-operativity with IFN-β is Independent of Cell Death.

(A) WT C57BL6/J peritoneal macrophages were pre-treated for 60 minutes with media alone or 100 u/ml IFN-β and subsequently infected with either ST strain I77 or strain D65 for 6 h. RNA was harvested and expression of CXCL1 and CXCL2 mRNA analyzed by qRT PCR. (B) WT C57BL6/J peritoneal macrophages were pre-treated for 60 minutes with media alone or 100u/ml IFN-β. Additionally half of the cells were pretreated with the cell death inhibitor Necrostatin-1. Macrophages were infected with SL1344 at an moi of 4 for 6 or 24 hours. At each time point samples of cell supernatants were taken and the levels of LDH in the supernatant quantified by colorimetric assay. (C) WT C57BL6/J or caspase-11−/− peritoneal macrophages were pre-treated for 60 minutes with media alone or 100u/ml IFN-β and subsequently infected with either ST strain SL1344 for 6 hours. RNA was harvested and expression of CXCL1 mRNA analyzed by qRT PCR.. Data is representative of three independent experiments. Data are shown as mean ± SEM.
While we observed striking alterations of innate inflammatory transcriptional responses following ST infection of IFN-β-primed macrophages, a previous study found that type I IFN enhanced ST killing of macrophages in a manner consistent with necroptosis (14). As it remained formally possible that the IFN-dependent altered transcriptional responses we observed were related to the onset of a cell death program, we examined ST-induced cytotoxicity in media- or IFN-β-treated macrophages by LDH release assay. Macrophages were treated with media alone or IFN-β for 4 h prior to infection for 6 or 24 h (MOI = 4) in the absence or presence of the necroptosis inhibitor necrostatin-1 (Nec-1; 33 μM). Samples of cell supernatant were analyzed for the release of LDH. Direct lysis of the cell monolayer by detergent was utilized as a positive control. We did not observe significant cytotoxicity at 6 h p.i. (the time point at which we made our mRNA observations) (Figure 3B, left panel). However, there was significant ST-induced cytotoxicity observable by 24 hrs that could be substantially inhibited by Nec-1 (Figure 3B, right panel). This result strongly argues that early IFN-β-mediated transcriptional suppression is dissociable from its effects on macrophage viability. Recently, it has been reported that caspase-11 is an IFN-β-inducible executioner caspase capable of triggering macrophage necroptosis in response to ST (29). As an additional control for IFN-regulated cell death pathways, we infected WT C57BL/6J or caspase-11−/− macrophages with ST following treatment with IFN-β. The absence of caspase-11 did not diminish the ability of IFN-β to suppress TLR4 responses (Figure 3C).
Multiple innate sensing systems are suppressed by IFN-β
As ST expresses a number of Microbe-Associated Molecular Patterns (MAMPs) and is capable of simultaneously ligating multiple PRRs, we sought to determine if the IFN-β-mediated cytokine and chemokine suppression seen with live bacteria could be recapitulated using a single purified TLR ligand. As the early inflammatory cytokine responses to ST in peritoneal macrophages were highly TLR4-dependent (Figure 1, A and B), peritoneal macrophages were stimulated with E. coli LPS (50 ng/ml) after pretreatment with medium- or IFN-β, and cytokine mRNA assayed over a 6 hr time course. The previously observed pattern of selective gene suppression seen when macrophages were infected with live ST (Figure 1) was reproduced by LPS stimulation (Figure 4A). Similar trends were observed when cytosolic pro-IL-1β (Figure 4B) and secreted levels of CXCL1 and CXCL2 protein (Figure 4C) were measured by Western blot and ELISA, respectively. To define further the kinetics of IFN-β action, peritoneal macrophages were pre-treated with IFN-β for time periods ranging from 30 min to 24 h and then stimulated with E. coli LPS for 4 h, at which time, IL-1β mRNA was examined by qRT-PCR. As little as 30 min exposure to recombinant IFN-β was sufficient to achieve selective inhibition of LPS-induced genes, while inhibition could be observed as late as 24 hrs post-IFN treatment (Figure 4D). As our experiments had heretofore been concentrated on TLR4 responses, we sought to determine if the suppressive effects of IFN-β were generalizable to other TLRs, as well as other potentially relevant innate sensing systems. We stimulated media- or IFN-β-primed macrophages with the synthetic TLR2 ligand Pam3Cys (250 ng/ml) for 4 hrs and assayed pro-IL-1β gene induction by qRT-PCR. IFN-β priming effectively suppressed IL-1β mRNA induced via TLR2 (Figure 4E). While TLRs are the most significant innate sensing system contributing to cytokine induction within the initial hours of ST infection of macrophages, there is significant literature documenting a role for the NOD-like receptors (NLRs) in contributing to responses to ST, and in particular, in contributing to the production of neutrophil chemokines (30, 31). We therefore assayed for the ability of IFN-β to govern NLR chemokines by stimulating medium- or IFN-β-primed macrophages with the synthetic NOD1 ligand (C12-iE-DAP; 20 μg/ml) or a synthetic NOD2 ligand (L-18-MDP; 20 μg/ml). CXCL1 secretion derived from either NOD1 or NOD2 ligand was also diminished by IFN-β priming (Figure 4F).
Figure 4. IFN-β Suppresses Transcriptional Responses to Purified Ligands Across Multiple Innate Receptor Systems.

(A) WT C57BL6/J macrophages were pretreated for 60 minutes either with media alone or 100 U/ml IFN-β. Macrophages were stimulated with 50 ng/ml purified E.coli LPS for the indicated times. RNA was harvested and transcript levels assayed by qRT PCR. (B) Macrophages were pre-treated as in (A) and stimulated with 50 ng/ml LPS for 6 hours. Whole cell lysates were probed by western blot for pro-IL1β protein. (C) Samples of cell supernatants from macrophages treated as in (A) were collected at a 24 hour time-point and assayed for chemokine content by ELISA. (D) WT C57BL6/J peritoneal macrophages were pretreated with 100 U/ml mammalian IFN-β for the indicated times and subsequently re-stimulated with 50 ng/ml E. coli LPS for 6 hours. RNA was harvested and IL-1β transcript expression quantified by qRT PCR. (E) WT C57BL6/J peritoneal macrophages were pretreated with 100 U/ml mammalian IFN-β for 60 minutes prior to stimulation for the indicated times with the TLR2 ligand Pam3Cys (250ng/ml). (F) Peritoneal macrophages pre-treated with media alone or IFN-β were subsequently treated overnight with the NOD-1 ligand C12-iE-DAP (20μg/ml) or the NOD-2 ligand L18-MDP (20μg/ml). Samples of cell supernatant were harvested and CXCL1 levels assayed by ELISA. Data in A-F is representative of three independent experiments. Data is shown as mean ± SEM.
Having established an important role for IFN-β in shaping the TLR- and NOD-dependent response to ST, we wanted to address the question of underlying molecular mechanism. While the regulation of IFN-β induction by upstream TLR ligation has been studied in great detail, the effect of prior exposure to type I IFN on the biology of TLRs themselves has not been systematically investigated. We therefore characterized TLR receptor expression, trafficking, and signaling following LPS stimulation of media or IFN-β treated macrophages. We did not observe a reduction in steady-state mRNA levels of TLRs 2 or 4 or the adaptors MyD88 and TRIF, but did observe IFN-dependent enhanced transcription of TLR3 and TLR9. TLR4 protein surface expression was slightly enhanced by IFN-β. LPS induced signal transduction, as judged by MAPK activation (Figure S3), as well as NF-κB and IRF3 activation (data not shown), was not diminished by IFN-β. TLR4 receptor internalization and endosomal trafficking were similarly unaffected (Figure S3).
IFN-β governs ST production of anti-inflammatory cytokines
Having found no IFN-β-mediated suppression of TLR4 expression, trafficking, or signaling, we speculated that IFN-β may act on innate immune receptor stimulation by augmenting production of secreted anti-inflammatory mediators that act as a negative feedback loop. The best-characterized secreted anti-inflammatory mediator induced by TLRs is IL-10. Therefore, we examined IL-10 production in medium- or IFN-β-treated macrophages during ST infection. Strikingly, we found that IL-10 mRNA and protein production was dramatically enhanced in macrophages primed with IFN-β (Figure 5A). We complemented this result by examining IL-10 production, as well as production of another reportedly immunosuppressive cytokine, IL-27, in IFN-β−/− macrophages. In the absence of IFN-β, ST-infected macrophages produced extremely low levels of IL-10 mRNA and protein (Figure 5B) and IL-27 (Figure 5C). Conversely, we tested the ability of recombinant IL-10 and IL-27 to suppress cytokines downstream of TLR4 with the kinetics observed in our system. Recombinant IL-10 and IL-27 potently suppressed IL-1β and KC mRNA expression even when added 24 hrs prior to LPS treatment (Figure 5D and E). Finally, to test the hypothesis that IL-10 underlies the observed IFN-β-mediated suppression, WT macrophages were stimulated with IFN-β in the presence of anti-IL-10 or anti-IL-27 neutralizing antibody, followed by 4 hrs of LPS treatment. Neutralization of IL-10 significantly, but incompletely, reversed the suppression of IL-1β mRNA caused by IFN-β (Figure 5F), while neutralization of IL-27 had no effect (Figure 5G). Reversal of IL-1β suppression by anti-IL-10 was also observed at the protein level (Figure 5H). As the incomplete reversal of IFN-β suppression of IL-1β may have been due to the incomplete neutralization of IL-10 by antibody, we tested the capacity of IFN-β to inhibit TLR4-mediated gene expression in IL-10−/− peritoneal macrophages. WT and IL-10−/− macrophages were pre-treated with medium or IFN-β for 60 min prior to infection with ST SL1344 for 4 hrs. Substantial rescue of CXCL1 mRNA expression was seen in the IL-10−/− background (Figure 6A). In agreement with our antibody neutralization data, lack of IL-10 significantly, but incompletely, reversed the suppression of IL-1β by IFN-β (Figure 6A) at this time point. These results argue for an important role for autocrine IL-10 in mediating the transcriptional repression due to IFN-β.
Figure 5. IFN-β Governs S. typhimurium Production of Anti-Inflammatory IL10 and IL10 Contributes to IFN-β Mediated Suppression.

(A) WT C57BL6/J peritoneal macrophages were pre-treated with 100 U/ml IFN-β and subsequently infected with ST at an moi of 4 for 6 hours. mRNA and protein levels of IL10 were assayed (B) WT and IFN-β−/− macrophages were infected with SL1344 for 6 hours, and mRNA and IL10 protein levels assayed. (C) IL27 mRNA expression assayed following 6 hours of SL1344 infection as in (B). (D) WT peritoneal macrophages were pre-treated with recombinant Il-10 for 60 minutes prior to stimulation with 50 ng/ml E. coli LPS for 4 hours. (E) WT peritoneal macrophages were pre-treated with recombinant Il-27 for 60 minutes prior to stimulation with 50 ng/ml E. coli LPS for 4 hours (F and G) Macrophages were pre-treated with IFN-β in the presence or absence of neutralizing IL-10 (10 μg/ml)(F) or IL-27 (10 μg/ml)(G) antibody followed by subsequent stimulation with 50 ng/ml E. coli LPS for 4 hours. Levels of IL-1β mRNA were assayed by qRT PCR (H) Treatment as in (F) with analysis of pro-IL-1β levels in whole cell lysate by western blot. Data in A-G is representative of three independent experiments.
Figure 6. Loss IFN-β Mediated Transcriptional Suppression in IL-10−/− Macrophages.
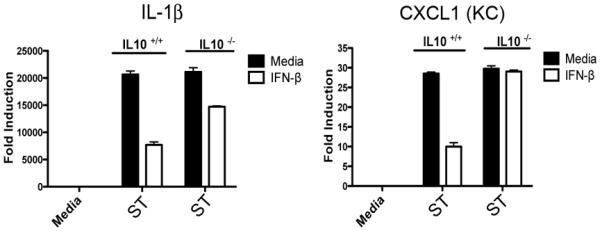
(A) WT and IL-10−/− macrophages were pretreated with media alone or 100 U/ml IFN-β for 60 minutes prior to infection with SL1344 for 4 hours. IL-1β and CXCL1 mRNA was assayed by qRT-PCR.
IFN-β promotes susceptibility to oral Salmonella infection
While our investigations have established a clear capacity for IFN-β to regulate ST innate transcriptional responses in peritoneal macrophages, the inflammatory responses of intestinal macrophage populations during in vivo ST infection may not be identical. To interrogate further a role for IFN-β in the gut during ST infection, wild-type (WT) C57BL/6 or IFN-β−/− mice were inoculated with 1 × 108 CFU of ST strain SL1344 by oral gavage and were monitored for lethality over time (Figure 7A). We observed a significant increase in mean time to death in the IFN-β−/− mice (p < 0.05). Additional experiments with lower doses of ST (~ 5×106) revealed even greater differences (Figure 7B). When we assessed organ burden from the livers of WT and IFN-β−/− mice 48 hrs post-infection, in all cases, livers from ST-infected IFN-β−/− mice displayed a markedly reduced bacterial burden (Figure 7D), with similar results at 24 hrs post-infection (data not shown). A similar difference in bacterial burden was also observed in the spleen (data not shown). We sought to determine whether the presence of IFN-β in vivo governed early innate inflammatory responses. Serial sections of small intestine proximal to the cecum were excised from PBS-treated or ST-infected WT and IFN-β−/− mice 48 hrs p.i. and mRNA extracted for analysis of inflammation-related transcripts. At early time points (24 and 48 hrs), we did not detect an appreciable up-regulation of classic pro-inflammatory genes (e.g., TNF-α, IL-6) in either strain (data not shown). The failure to detect significant inflammatory cytokine mRNA in whole gut tissue is not unexpected, as our infection model does not involve pretreatment of the animals with antibiotics to reduce levels of competing micro-flora, and therefore, to enhance ST colonization and attendant inflammation (32). We did, however, consistently observe a marked increase in the expression of the neutrophil chemo-attractant, CXCL2 (MIP2), in the small intestine of IFN-β−/− compared to WT mice at 48 hrs p.i. (Figure 7C). This difference in chemokine expression led us to speculate that there may be enhanced early neutrophil responses in the IFN-β−/− animals that may, in part, account for the early increased resistance to oral ST infection. To quantify the presence of infiltrating neutrophils in the small intestine, myeloperoxidase activity (MPO), a hallmark of neutrophil activity, was measured in homogenates from sections of WT and IFN-β−/− mice 48 hrs p.i. We observed significantly greater MPO expression in the small intestine of IFN-β−/− mice at 48 hrs compared to WT animals (Figure 7E), consistent with enhanced CXCL2 activity.
Figure 7. Oral ST infection Elicits Differential Inflammatory Responses in WT and IFN-β−/− Mice.

(A and B) WT C57BL6/J or IFNβ−/− were inoculated with approx. 2×107 or 5×106 CFU St strain SL1344 in PBS by oral gavage. Animals were monitored twice daily for morbidity and mortality. Data are representative of three experiments. Significance determined by Mantel-Cox test p = .0012 (C) WT C57BL6/J or IFNβ−/− animals were infected as in (A) and sections of small bowel harvested at 48 hrs. RNA was prepared from sections and mRNA levels of CXCL2 was analyzed by qRT PCR. p =.0211 (D) WT C57BL6/J or IFNβ−/− animals were infected with 2x107 CFU of Sl1344 by oral gavage. The livers were removed 48 hrs later. Livers were weighed and homogenates plated on LB agar. p = .002 ((E) WT C57BL6/J or IFNβ−/− animals were infected as in (A) and sections of small bowel harvested at 48 hrs. Tissue homogenates were analyzed for peroxidase activity by colorimetric assay (p = .0255. Data from B-D representative of three independent experiments and shown as mean ± range. Significance determined by students t test. (F-J) WT C57BL6/J or IFNβ−/− animals were given a single IP injection of 1×108 CFU SL1344 or PBS. 8 hours later the liver was harvested and RNA was isolated for analysis of indicated genes by qRT PCR. (K) Animals were infected as in (F-J) but harvested livers were homogenized and plated on LB agar for colony counts.
As we were unable to quantify levels of induced IFN-β or IL-10 in our model of oral infection, we utilized an additional model of systemic “septic” infection. WT or IFN-β−/− animals were infected intra-peritoneally and cytokine and chemokine levels as well as bacterial colony counts in the liver were determined after 8 hours. We observed an induction of IFN-β mRNA in the liver of infected WT but not the IFN-β−/− using this model (Figure 7F) Importantly, we also observed a marked reduction in IL-10 levels in the livers of infected IFN-β−/− animals when compared to WT, in agreement with our in vitro studies (Figure 7G). The levels of IL-6 were comparable in both infected genotypes, while CXCL1 and IL-1β were consistently elevated in infected IFN-β−/− mice (Figure 7H-J). We did observed lower levels of recoverable bacteria in the livers of IFN-β−/− animals at 8 hours post-infection (Figure 7K).
Discussion
Among the mammalian IFNs, a common schema assigns antibacterial functions to the Type II IFN (IFN-γ), while the type I IFN system is widely regarded as an inducer of antiviral responses. However, within the last decade, there has been a growing interest in the relationship in vivo between the type I IFN systems and bacterial infections. Salmonella enterica serovar Typhimurium is a significant human pathogen that induces an in vivo type I IFN response through its interactions with TLRs (25). Our current work argues that type I IFN plays a significant role in shaping the TLR-induced transcriptional response to ST (and likely other enteric pathogens), and in particular, in governing the cellular aspects of the innate response through regulation of IL-1 family and chemokine gene expression. A recent manuscript studying IFN-α/β and ST identified a role for type I IFN in regulating ST-induced macrophage cell death, an effect that may well be a result of IFN-induced caspase-11 (29, 33). However, it should be noted that a RIP3K−/− animal model that is defective in the induction of macrophage programmed cell death failed to recapitulate the dramatic resistance to ST seen in the IFNAR−/− mice (14), arguing for the existence of additional IFN-regulated pathways in addition to necroptosis.
The role we have identified for IFN-β in suppressing aspects of the TLR/NLR-driven innate inflammatory response is congruent with a significant literature detailing a homeostatic as well as anti-inflammatory role for type I IFNs in a variety of inflammatory and autoimmune conditions (34-36), particularly in the intestine (37-39). Of distinct relevance for the present study is work showing that lactic acid bacteria, a common commensal bacterium, constitutively stimulate low level production of IFN-β, but not IFN-α, in the small intestine via double-stranded RNA ligation of TLR3. This ‘homeostatic’ production of IFN-β has significant protective effects in an induced colitis model, including reduction of infiltrating neutrophils (25). Importantly, however, the previous work does not address critical questions of molecular mechanism to explain the action of type I IFN in the gut. The ability of IFN-β to “sculpt” the TLR/NLR-induced transcriptional response in the intestinal environment may be critical to its palliative capacities.
While low levels of type I IFN production are required to maintain homeostasis in the intestine, the present work reveals that induced production of type I IFN, and specifically, IFN-β, can be utilized by a pathogenic bacteria to suppress an innate inflammatory response. We have identified the immunosuppressive cytokine IL-10 as a significant mediator of IFN-β action. IL-10 and TLR action have been described as uniquely significant in the gut (40, 41), and elevated IL-10 has been shown to impair clearance of non-typhoidal ST in multiple infection models (42, 43). How the selective suppression of TLR4-induced genes is achieved by IFN-β requires further study. This question is made all the more interesting when considering that recombinant IL-10 as a single agent is generally suppressive of TLR responses in macrophages (44). Thus, there appears to be a combinatorial effect of type I IFN and IL-10. This selective, rather than general, suppression of TLR/NLR-mediated responses may be important for the biology of ST infection as ST has been shown to benefit in competition with intestinal microbiota from some aspects of the intestinal inflammation it induces (10).
In assessing the likely biological consequences of IFN-β-mediated innate immune suppression, it is important to consider the roles of the individual genes that we have identified as being IFN-sensitive. Both IL-1β and IL-18 have been shown in vivo to be important for the survival of mice following oral salmonella infection, with IL-18 expression having a greater effect (45). The contributions of IL-1β and IL-18 to resistance are likely manifold; however, it has been shown that IL-18, in particular, is a critical driver of IFN-γ expression in T cells and possibly neutrophils during salmonella infection (46-48). The contributions of CXCL1, CXCL2, and CXCL5 to neutrophil chemotaxis have been extensively documented (49), as have the importance of neutrophil responses in controlling ST infection in the gut (12, 50, 51) where, neutrophil influx during salmonella infection is driven by production of CXC chemokines that are largely governed at the mucosal surface by the IL-17/IL-23 axis (52, 53). Another point of note is that this present work may have implications for our understanding of the biology of ST and viral co-morbidities. In particular, a substantial correlation between underlying HIV infection and invasive cases of non-typhoidal salmonella has been demonstrated (54, 55). This may be notable as HIV infection induces abundant systemic type I IFN production (56). The promotion of ST dissemination from the gut in SIV-infected macaques has also been shown experimentally (53).
While we show clearly that the production of IFN-β in vivo rapidly accelerates the spread of ST infection to secondary sites (Figure 1B), this is clearly not the case for all pathogenic bacteria (19, 38) nor even all Gram negative bacteria. Indeed, Y. entercolitica, which shares a common oral route of infection with ST, is suppressed by TRIF-dependent IFN production (57). It has long been known that the type I IFNs mediate the up-regulation of hundreds of genes responsible for cell-intrinsic defense against viral infection, and a select number of genes with direct antibacterial function, such as iNOS. To take advantage of the suppressive effects of IFN, however, bacteria must be able to evade the cell-intrinsic defenses. Such behavior has been demonstrated for ST in that it is particularly good at evading the iNOS-driven reactive nitrogen species prevalent in macrophages, while being significantly more susceptible to the neutrophil-derived reactive oxygen species (58, 59). We have shown that the suppressive effects of type I IFN on ST-induced cytokine transcription can be dissociated from the capacity of type I IFN to potentiate ST-induced macrophage pyroptotic cell death through caspase-11 (Figure 3 B&C). As pyroptotic cell death mediated by IFN induced caspase-11 results in some mature IL-1β release, a legitimate question arises as to whether type I IFN has both a pro- and anti-inflammatory role during ST infection (33, 60). We believe that as caspase-11 does not directly process IL-1, but indirectly leads to auxiliary caspase-1 activation and IL-1β processing, the contribution of this caspase-11 pathway to total IL-1β release in vivo during ST infection is likely to be much smaller than that achieved by conventional inflammasome and caspase-1 activation induced by ST-encoded PAMPs such as flagellin. Instead, the chief effect of caspase-11 regulation by IFN may be to kill ST-infected macrophages that, while depriving ST of an intracellular niche for replication, may also limit the duration of non-IL-1 inflammatory cytokine production by the infected cell.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health R01 AI018797
References
- 1.Broz P, Ohlson MB, Monack DM. Innate immune response to Salmonella typhimurium, a model enteric pathogen. Gut Microbes. 2012;3:62–70. doi: 10.4161/gmic.19141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Brien AD, Metcalf ES, Rosenstreich DL. Defect in macrophage effector function confers Salmonella typhimurium susceptibility on C3H/HeJ mice. Cell Immunol. 1982;67:325–333. doi: 10.1016/0008-8749(82)90224-6. [DOI] [PubMed] [Google Scholar]
- 3.Arpaia N, Godec J, Lau L, Sivick KE, McLaughlin LM, Jones MB, Dracheva T, Peterson SN, Monack DM, Barton GM. TLR signaling is required for Salmonella typhimurium virulence. Cell. 2011;144:675–688. doi: 10.1016/j.cell.2011.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vazquez-Torres A, Vallance BA, Bergman MA, Finlay BB, Cookson BT, Jones-Carson J, Fang FC. Toll-like receptor 4 dependence of innate and adaptive immunity to Salmonella: importance of the Kupffer cell network. J Immunol. 2004;172:6202–6208. doi: 10.4049/jimmunol.172.10.6202. [DOI] [PubMed] [Google Scholar]
- 5.Weiss DS, Raupach B, Takeda K, Akira S, Zychlinsky A. Toll-like receptors are temporally involved in host defense. J Immunol. 2004;172:4463–4469. doi: 10.4049/jimmunol.172.7.4463. [DOI] [PubMed] [Google Scholar]
- 6.Talbot S, Totemeyer S, Yamamoto M, Akira S, Hughes K, Gray D, Barr T, Mastroeni P, Maskell DJ, Bryant CE. Toll-like receptor 4 signalling through MyD88 is essential to control Salmonella enterica serovar typhimurium infection, but not for the initiation of bacterial clearance. Immunology. 2009;128:472–483. doi: 10.1111/j.1365-2567.2009.03146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vazquez-Torres A, Xu Y, Jones-Carson J, Holden DW, Lucia SM, Dinauer MC, Mastroeni P, Fang FC. Salmonella pathogenicity island 2-dependent evasion of the phagocyte NADPH oxidase. Science. 2000;287:1655–1658. doi: 10.1126/science.287.5458.1655. [DOI] [PubMed] [Google Scholar]
- 8.Bhavsar AP, Brown NF, Stoepel J, Wiermer M, Martin DD, Hsu KJ, Imami K, Ross CJ, Hayden MR, Foster LJ, Li X, Hieter P, Finlay BB. The Salmonella type III effector SspH2 specifically exploits the NLR co-chaperone activity of SGT1 to subvert immunity. PLoS Pathog. 2013;9:e1003518. doi: 10.1371/journal.ppat.1003518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hannemann S, Gao B, Galan JE. Salmonella modulation of host cell gene expression promotes its intracellular growth. PLoS Pathog. 2013;9:e1003668. doi: 10.1371/journal.ppat.1003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, Chaffron S, Macpherson AJ, Buer J, Parkhill J, Dougan G, von Mering C, Hardt WD. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007;5:2177–2189. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, Roth JR, Baumler AJ. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. 2010;467:426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sivick KE, Arpaia N, Reiner GL, Lee BL, Russell BR, Barton GM. Toll-like receptor-deficient mice reveal how innate immune signaling influences Salmonella virulence strategies. Cell Host Microbe. 2014;15:203–213. doi: 10.1016/j.chom.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong CE, Sad S, Coombes BK. Salmonella enterica serovar typhimurium exploits Toll-like receptor signaling during the host-pathogen interaction. Infection and immunity. 2009;77:4750–4760. doi: 10.1128/IAI.00545-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13:954–962. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol. 2012;12:367–382. doi: 10.1038/nri3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5:675–687. doi: 10.1038/nri1684. [DOI] [PubMed] [Google Scholar]
- 19.Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, Tomasello F, Gambuzza M, Macri G, Ruggeri A, Leanderson T, Teti G. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol. 2007;178:3126–3133. doi: 10.4049/jimmunol.178.5.3126. [DOI] [PubMed] [Google Scholar]
- 20.Tennant SM, Diallo S, Levy H, Livio S, Sow SO, Tapia M, Fields PI, Mikoleit M, Tamboura B, Kotloff KL, Nataro JP, Galen JE, Levine MM. Identification by PCR of non-typhoidal Salmonella enterica serovars associated with invasive infections among febrile patients in Mali. PLoS Negl Trop Dis. 2010;4:e621. doi: 10.1371/journal.pntd.0000621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dobrovolskaia MA, Medvedev AE, Thomas KE, Cuesta N, Toshchakov V, Ren T, Cody MJ, Michalek SM, Rice NR, Vogel SN. Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR "homotolerance" versus "heterotolerance" on NF-kappa B signaling pathway components. J Immunol. 2003;170:508–519. doi: 10.4049/jimmunol.170.1.508. [DOI] [PubMed] [Google Scholar]
- 22.Shirey KA, Nhu QM, Yim KC, Roberts ZJ, Teijaro JR, Farber DL, Blanco JC, Vogel SN. The anti-tumor agent, 5,6-dimethylxanthenone-4-acetic acid (DMXAA), induces IFN-beta-mediated antiviral activity in vitro and in vivo. J Leukoc Biol. 89:351–357. doi: 10.1189/jlb.0410216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McIntire FC, Sievert HW, Barlow GH, Finley RA, Lee AY. Chemical, physical, biological properties of a lipopolysaccharide from Escherichia coli K-235. Biochemistry. 1967;6:2363–2372. doi: 10.1021/bi00860a011. [DOI] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Kawashima T, Kosaka A, Yan H, Guo Z, Uchiyama R, Fukui R, Kaneko D, Kumagai Y, You DJ, Carreras J, Uematsu S, Jang MH, Takeuchi O, Kaisho T, Akira S, Miyake K, Tsutsui H, Saito T, Nishimura I, Tsuji NM. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-beta. Immunity. 2013;38:1187–1197. doi: 10.1016/j.immuni.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 26.Thomas KE, Galligan CL, Newman RD, Fish EN, Vogel SN. Contribution of interferon-beta to the murine macrophage response to the toll-like receptor 4 agonist, lipopolysaccharide. J Biol Chem. 2006;281:31119–31130. doi: 10.1074/jbc.M604958200. [DOI] [PubMed] [Google Scholar]
- 27.Wray C, Sojka WJ. Experimental Salmonella typhimurium infection in calves. Research in veterinary science. 1978;25:139–143. [PubMed] [Google Scholar]
- 28.Okoro CK, Kingsley RA, Connor TR, Harris SR, Parry CM, Al-Mashhadani MN, Kariuki S, Msefula CL, Gordon MA, de Pinna E, Wain J, Heyderman RS, Obaro S, Alonso PL, Mandomando I, MacLennan CA, Tapia MD, Levine MM, Tennant SM, Parkhill J, Dougan G. Intracontinental spread of human invasive Salmonella Typhimurium pathovariants in sub-Saharan Africa. Nat Genet. 2012;44:1215–1221. doi: 10.1038/ng.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, Monack DM. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. 2012;490:288–291. doi: 10.1038/nature11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol. 2014;14:9–23. doi: 10.1038/nri3565. [DOI] [PubMed] [Google Scholar]
- 31.Geddes K, Rubino S, Streutker C, Cho JH, Magalhaes JG, Le Bourhis L, Selvanantham T, Girardin SE, Philpott DJ. Nod1 and Nod2 regulation of inflammation in the Salmonella colitis model. Infect Immun. 2010;78:5107–5115. doi: 10.1128/IAI.00759-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barthel M, Hapfelmeier S, Quintanilla-Martinez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Russmann H, Hardt WD. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun. 2003;71:2839–2858. doi: 10.1128/IAI.71.5.2839-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, Miao EA. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339:975–978. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Shaked I, Stanford SM, Zhou W, Curtsinger JM, Mikulski Z, Shaheen ZR, Cheng G, Sawatzke K, Campbell AM, Auger JL, Bilgic H, Shoyama FM, Schmeling DO, Balfour HH, Jr., Hasegawa K, Chan AC, Corbett JA, Binstadt BA, Mescher MF, Ley K, Bottini N, Peterson EJ. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity. 2013;39:111–122. doi: 10.1016/j.immuni.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. 2008;118:1680–1690. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–223. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 37.Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, Raz E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest. 2005;115:695–702. doi: 10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Long TM, Nisa S, Donnenberg MS, Hassel BA. Enteropathogenic Escherichia coli Inhibits Type I Interferon- and RNase L-Mediated Host Defense To Disrupt Intestinal Epithelial Cell Barrier Function. Infect Immun. 2014;82:2802–2814. doi: 10.1128/IAI.00105-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kole A, He J, Rivollier A, Silveira DD, Kitamura K, Maloy KJ, Kelsall BL. Type I IFNs regulate effector and regulatory T cell accumulation and anti-inflammatory cytokine production during T cell-mediated colitis. Journal of immunology. 2013;191:2771–2779. doi: 10.4049/jimmunol.1301093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rakoff-Nahoum S, Hao L, Medzhitov R. Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity. 2006;25:319–329. doi: 10.1016/j.immuni.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 41.Hoshi N, Schenten D, Nish SA, Walther Z, Gagliani N, Flavell RA, Reizis B, Shen Z, Fox JG, Iwasaki A, Medzhitov R. MyD88 signalling in colonic mononuclear phagocytes drives colitis in IL-10-deficient mice. Nat Commun. 2012;3:1120. doi: 10.1038/ncomms2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mooney JP, Butler BP, Lokken KL, Xavier MN, Chau JY, Schaltenberg N, Dandekar S, George MD, Santos RL, Luckhart S, Tsolis RM. The mucosal inflammatory response to non-typhoidal Salmonella in the intestine is blunted by IL-10 during concurrent malaria parasite infection. Mucosal Immunol. 2014 doi: 10.1038/mi.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neves P, Lampropoulou V, Calderon-Gomez E, Roch T, Stervbo U, Shen P, Kuhl AA, Loddenkemper C, Haury M, Nedospasov SA, Kaufmann SH, Steinhoff U, Calado DP, Fillatreau S. Signaling via the MyD88 adaptor protein in B cells suppresses protective immunity during Salmonella typhimurium infection. Immunity. 2010;33:777–790. doi: 10.1016/j.immuni.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 44.Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci U S A. 2005;102:8686–8691. doi: 10.1073/pnas.0500419102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raupach B, Peuschel SK, Monack DM, Zychlinsky A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect Immun. 2006;74:4922–4926. doi: 10.1128/IAI.00417-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Donnell H, Pham OH, Li LX, Atif SM, Lee SJ, Ravesloot MM, Stolfi JL, Nuccio SP, Broz P, Monack DM, Baumler AJ, McSorley SJ. Toll-like receptor and inflammasome signals converge to amplify the innate bactericidal capacity of T helper 1 cells. Immunity. 2014;40:213–224. doi: 10.1016/j.immuni.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Srinivasan A, Salazar-Gonzalez RM, Jarcho M, Sandau MM, Lefrancois L, McSorley SJ. Innate immune activation of CD4 T cells in salmonella-infected mice is dependent on IL-18. Journal of immunology. 2007;178:6342–6349. doi: 10.4049/jimmunol.178.10.6342. [DOI] [PubMed] [Google Scholar]
- 48.Spees AM, Kingsbury DD, Wangdi T, Xavier MN, Tsolis RM, Baumler AJ. Neutrophils are a source of gamma interferon during acute Salmonella enterica serovar Typhimurium colitis. Infection and immunity. 2014;82:1692–1697. doi: 10.1128/IAI.01508-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends in immunology. 2011;32:452–460. doi: 10.1016/j.it.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheminay C, Chakravortty D, Hensel M. Role of neutrophils in murine salmonellosis. Infect Immun. 2004;72:468–477. doi: 10.1128/IAI.72.1.468-477.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Conlan JW. Neutrophils prevent extracellular colonization of the liver microvasculature by Salmonella typhimurium. Infect Immun. 1996;64:1043–1047. doi: 10.1128/iai.64.3.1043-1047.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Godinez I, Raffatellu M, Chu H, Paixao TA, Haneda T, Santos RL, Bevins CL, Tsolis RM, Baumler AJ. Interleukin-23 orchestrates mucosal responses to Salmonella enterica serotype Typhimurium in the intestine. Infect Immun. 2009;77:387–398. doi: 10.1128/IAI.00933-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raffatellu M, Santos RL, Verhoeven DE, George MD, Wilson RP, Winter SE, Godinez I, Sankaran S, Paixao TA, Gordon MA, Kolls JK, Dandekar S, Baumler AJ. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med. 2008;14:421–428. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feasey NA, Dougan G, Kingsley RA, Heyderman RS, Gordon MA. Invasive non-typhoidal salmonella disease: an emerging and neglected tropical disease in Africa. Lancet. 2012;379:2489–2499. doi: 10.1016/S0140-6736(11)61752-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schreiber F, Lynn DJ, Houston A, Peters J, Mwafulirwa G, Finlay BB, Brinkman FS, Hancock RE, Heyderman RS, Dougan G, Gordon MA. The human transcriptome during nontyphoid Salmonella and HIV coinfection reveals attenuated NFkappaB-mediated inflammation and persistent cell cycle disruption. The Journal of infectious diseases. 2011;204:1237–1245. doi: 10.1093/infdis/jir512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bosinger SE, Utay NS. Type I interferon: understanding its role in HIV pathogenesis and therapy. Current HIV/AIDS reports. 2015;12:41–53. doi: 10.1007/s11904-014-0244-6. [DOI] [PubMed] [Google Scholar]
- 57.Sotolongo J, Espana C, Echeverry A, Siefker D, Altman N, Zaias J, Santaolalla R, Ruiz J, Schesser K, Adkins B, Fukata M. Host innate recognition of an intestinal bacterial pathogen induces TRIF-dependent protective immunity. J Exp Med. 2011;208:2705–2716. doi: 10.1084/jem.20110547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burton NA, Schurmann N, Casse O, Steeb AK, Claudi B, Zankl J, Schmidt A, Bumann D. Disparate impact of oxidative host defenses determines the fate of Salmonella during systemic infection in mice. Cell Host Microbe. 2014;15:72–83. doi: 10.1016/j.chom.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 59.Jacobs AT, Ignarro LJ. Lipopolysaccharide-induced expression of interferon-beta mediates the timing of inducible nitric-oxide synthase induction in RAW 264.7 macrophages. The Journal of biological chemistry. 2001;276:47950–47957. doi: 10.1074/jbc.M106639200. [DOI] [PubMed] [Google Scholar]
- 60.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.