

Abstract
Genome instability, primarily caused by faulty DNA repair mechanisms, drives tumorigenesis. Therapeutic interventions that exploit deregulated DNA repair in cancer have made considerable progress by targeting tumor-specific alterations of DNA repair factors, which either induces synthetic lethality or augments the efficacy of conventional chemotherapy and radiotherapy. The study of Fanconi anemia (FA), a rare inherited blood disorder and cancer predisposition syndrome, has been instrumental in understanding the extent to which DNA repair defects contribute to tumorigenesis. The FA pathway functions to resolve blocked replication forks in response to DNA interstrand cross-links (ICLs), and accumulating knowledge of its activation by the ubiquitin-mediated signaling pathway has provided promising therapeutic opportunities for cancer treatment. Here, we discuss recent advances in our understanding of FA pathway regulation and its potential application for designing tailored therapeutics that take advantage of deregulated DNA ICL repair in cancer.
Keywords: cancer therapeutics, DNA interstrand cross-link, DNA repair, Fanconi anemia, ubiquitin signaling
MECHANISM OF DNA ICL REPAIR IN THE FA PATHWAY
Fanconi anemia (FA) is a chromosomal instability syndrome characterized by developmental abnormalities, progressive bone marrow failure, and increased cancer susceptibility (D’Andrea, 2010). Bone marrow failure is the main cause of childhood fatality in FA patients, and unfortunately, most children with FA develop myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) (Garaycoechea and Patel, 2014; Kee et al., 2012). Moreover, patients who live into adulthood are predisposed to additional malignancies, including head and neck, gynecological, and gastrointestinal cancers. Thus, the study of FA has provided important insights into the pathogenesis of cancer and could lead to the development of anti-cancer therapeutics for the general population.
FA is caused by germ-line mutations in one of the 17 currently known FA genes whose gene products participate in a common DNA repair pathway called the FA pathway (Kim and D’Andrea, 2012). The pathway functions to resolve DNA interstrand cross-links (ICLs), lethal DNA lesions that block DNA replication and transcription (Clauson et al., 2013; Deans and West, 2011). It is also required to buffer against the genotoxic effects of reactive aldehydes naturally produced inside the cells (Langevin et al., 2011). The FA pathway coordinates various steps in ICL repair: recognition and unhooking of ICLs, translesion DNA synthesis (TLS), and homologous recombination (HR) (Kottemann and Smogorzewska, 2013). Central to the FA pathway is the conjugation of monoubiquitin to FANCD2 at Lys561 and, to a lesser extent, its binding partner FANCI at Lys523 in human (Garcia-Higuera et al., 2001; Smogorzewska et al., 2007). The ubiquitinated FANCD2-FANCI complex promotes downstream nucleolytic incisions near the ICL and TLS past the lesion (Knipscheer et al., 2009). This step is activated by the multi-subunit ubiquitin E3 ligase complex, also known as the FA core complex, which is composed of eight FANC proteins (FANCA/B/C/E/F/G/L/M) along with the accessory proteins, FAAP100, FAAP20, and FAAP24 (Walden and Deans, 2014). The FA pathway senses an ICL-mediated stalled replication fork, and the FANCM-FAAP24 heterodimer in the complex recognizes DNA damage caused by the DNA ICL and induces Ataxia-telangiectasia and Rad3-related (ATR)-dependent activation of the FA core complex (Collis et al., 2008). The auxiliary proteins, histone-fold containing kinetochore protein MHF1 and MHF2, promote constitutive association of FANCM to chromatin (Singh et al., 2010; Yan et al., 2010). FANCM contains a DEAH translocase domain at its N-terminus (Meetei et al., 2005). Although not required for FANCD2 monoubiquitination, ATPase activity of FANCM is essential for remodeling and stabilization of replication forks, and activation of the cell cycle checkpoint (Blackford et al., 2012; Gari et al., 2008; Schwab et al., 2010).
Although the mechanisms of ICL repair are not yet fully understood, the most detailed model derived from studies in Xenopus egg extracts suggests that two DNA replication forks converge on an ICL to initiate repair (Fig. 1A) (Zhang et al., 2015). The ubiquitinated FANCD2-FANCI heterodimer is relocalized to a converged fork near the ICL and controls nucleolytic incision to release the ICL from one of the two strands (Knipscheer et al., 2009). FANCD2-monoubiquitin (FANCD2-Ub) has been implicated in the recruitment of several structure-specific nucleases, which are responsible for unhooking the DNA ICL (Fig. 1B). FANCP/SLX4 functions as a scaffold that targets structure-specific nucleases such as the ERCC1-FANCQ/XPF heterodimer and SLX1 to a lesion, utilizing its UBZ4 (ubiquitin-binding zinc finger 4) motif that recognizes FANCD2-Ub (Cybulski and Howlett, 2011; Yamamoto et al., 2011). Recent biochemical and genetic studies indicated that nuclease activity of the ERCC1-XPF complex plays a prominent role in carrying out unhooking incisions, which is controlled by the presence of SLX4 (Hodskinson et al., 2014; Klein Douwel et al., 2014). Interestingly, mutations in FA-Q patients disrupt the function of XPF in ICL repair, without altering its function in nucleotide excision repair (NER), highlighting the multifunctional activity of the XPF endonuclease (Bogliolo et al., 2013). The MUS81-EME1 nuclease complex also associates with SLX4 and functions in ICL repair, but is dispensable for initial ICL processing in Xenopus egg extracts (Klein Douwel et al., 2014; Räschle et al., 2015), suggesting that the complex may process different intermediates in ICL repair such as Holliday junction (HJ) resolution during double-strand break (DSB) repair (Chen et al., 2001). FAN1, another structure-specific nuclease implicated in ICL repair, contains a UBZ4 motif required for interaction with FANCD2-Ub (Smogorzewska et al., 2010). However, FAN1 deficiency leads not to FA, but rather to a rare kidney disease called karyomegalic interstitial nephritis (KIN), suggesting that FAN1 may play a redundant role with other nucleases in the FA pathway or deal with lesions independently of the FA pathway (Zhou et al., 2012). SLX4 also participates in diverse genome maintenance pathways including telomere maintenance and regulation of DNA damage checkpoints (Kim, 2014). Interestingly, the SLX4 complex was recently shown to function as a SUMO E3 ligase that sumoylates XPF and counteracts replication stress (Guervilly et al., 2015).
Fig. 1.
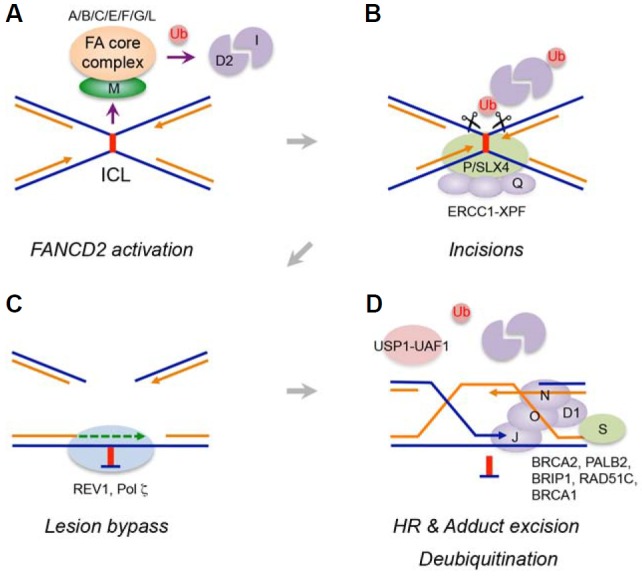
Overview of the FA pathway. (A) A stalled replication fork at a DNA ICL activates the FA core complex to monoubiquitinate the FANCD2-FANCI heterodimer. (B) Ubiquitinated FANCD2 recruits a nuclease complex to initiate nucleolytic incisions flanking the ICL. (C) The unhooked lesion is processed and bypassed by TLS polymerases to restore a nascent leading strand. (D) Downstream FA proteins promote HR to repair double-stranded breaks, and the unhooked ICL is removed by NER. The USP1-UAF1 complex removes ubiquitin from FANCD2 to complete the repair.
The incision step converts the lagging strand into a DSB at the stalled fork. TLS, a DNA damage tolerance mechanism mediated by specialized TLS polymerases (Chang and Cimprich, 2009), allows a nascent leading strand to bypass the unhooked adduct and extend from primer-template termini to resume replication (Fig. 1C). The 5′ exonuclease SNM1A may promote this process by digesting the unhooked cross-link intermediate, generating a preferred substrate for TLS polymerases (Wang et al., 2011). The active site for TLS polymerases is less restricted, and TLS polymerases can thus accommodate base mismatches and distorted base-parings. The precise mechanism for the recruitment of TLS polymerases to DNA ICL lesions remains elusive, but the FA core complex has been shown to facilitate this process (Budzowska et al., 2015; Kim et al., 2012). Among TLS polymerases, Pol ξ (a REV3-REV7 heterodimer) and REV1 play a key role in replication-dependent ICL repair. Previous genetic studies in chicken DT40 cells indicated that Rev1 and Rev3 are epistatic with FANCC in cisplatin sensitivity (Niedzwiedz et al., 2004). Pol ξ has been shown to execute the lesion bypass step of ICL repair that occurs in Xenopus egg extracts (Raschle et al., 2008). Although REV1 has deoxycytidyl transferase activity to insert dCMP opposite an ICL, it plays a more structural role to facilitate polymerase switching between different TLS polymerases, and coordinates insertion and extension steps (Lehmann et al., 2007). Indeed, recent structural analysis revealed the formation of a quaternary TLS polymerase complex consisting of the C-terminal domain (CTD) of REV1, heterodimeric Pol ξ and Pol κ, thereby highlighting the role of the REV1 CTD in a scaffold that simultaneously binds these polymerases (Wojtaszek et al., 2012). Given the diverse structures formed by distinct ICL-inducing agents, each ICL lesion may be processed by a combination of specific TLS polymerases with unique substrate preferences (Guainazzi and Schärer, 2010).
Another important step following nucleolytic incision is repairing replication-associated DSBs, which is mediated by HR. A sister chromatid restored by TLS is used as a template for strand invasion by the 3′ overhang of a lagging strand to restore information lost during the incision process (Fig. 1D). FANCD2 physically interacts with CtIP, a protein required for end resection, to channel repair to the HR process (Unno et al., 2014). Downstream FA gene products directly regulate HR. RAD51 coats a single-stranded DNA to initiate strand invasion, and FANCD1/BRCA2 is required for its loading onto stalled forks (Moynahan et al., 2001). FANCN/PALB2 interacts with BRCA1 to promote this process (Xia et al., 2006). The RAD51 paralog FANCO/RAD51C also contributes to replication-associated DSB repair by participating in strand invasion and HJ resolution (Liu et al., 2007; Vaz et al., 2010). Biallelic mutations in BRCA1 were recently found in a breast cancer patient with a FA-like disorder, and thus BRCA1 has been designated as a new FA gene, FANCS (Sawyer et al., 2015). BRCA1 plays unique roles in the FA pathway. It associates with BRCA2 and promotes resection of the double-stranded DNA ends for RAD51 loading (Zhang et al., 2009). It is also required for unloading of the Cdc45-MCM-GINS (CMG) helicase complex from stalled forks and loading of FANCD2-Ub onto DNA lesions, which functions independently of HR (Bunting et al., 2012; Long et al., 2014). Copying information from a sister chromatid through HR restores a replication fork, and the unhooked adduct is believed to be removed by NER.
The deubiquitinating enzyme USP1 regulates the level of FANCD2-Ub (Nijman et al., 2005). USP1 associates with its activating factor UAF1, and the USP1-UAF1 complex removes monoubiquitin from FANCD2 to complete the repair (Cohn et al., 2007) (Fig. 1D). In addition to its stimulatory role, UAF1 is also necessary for recruiting the FANCD2-FANCI complex to USP1 (Yang et al., 2011). The Usp1 knockout mouse exhibits FA phenotypes, and Usp1−/− cells are hypersensitive to DNA cross-linking agents, indicating that timely deubiquitination process is essential for the integrity of the FA pathway (Kim et al., 2009). However, the precise timing and location of deubiquitination of FANCD2 remain to be resolved.
REGULATION OF FANCD2 ACTIVATION BY THE FA CORE COMPLEX
Functional modules of the FA core complex and its regulation
FANCD2 monoubiquitination by the FA core complex is a key regulatory step in the FA pathway as it connects an upstream DNA damage response (DDR) to downstream enzymatic DNA repair processes. Therefore, the activity of the FA core complex needs to be tightly controlled. A variety of posttranslational modifications and protein-protein interactions maintain the integrity of the complex and regulate its enzymatic activity (Fig. 2). Among the components of the complex, only the FANCL subunit has known catalytic activity mediated by a RING domain (Meetei et al., 2003). The other subunits appear to function as structural and regulatory elements. Notably, disruption of the interaction between FANCL and its ubiquitin E2 conjugating enzyme UBE2T caused by UBE2T mutations in FA patients leads to compromised FANCD2 monoubiquitination, suggesting that UBE2T (FANCT) mutations define a new FA subtype (Hira et al., 2015). Recent biochemical and genetic experiments classified the FA core complex into three distinct modules, namely, the FANCB-FANCL-FAAP100 core catalytic unit, and the FANCA-FANCG-FAAP20 and FANCC-FANCE-FANCF auxiliary units (Huang et al., 2014; Rajendra et al., 2014). A minimal FANCB-FANCL-FAAP100 subcomplex is sufficient to monoubiquitinate FANCD2 in vitro (Rajendra et al., 2014). The other modules are necessary for stabilizing the FA core complex and achieving its maximal activity. For instance, the N-terminus of FANCF connects three modules to the FANCM anchor complex (Deans and West, 2009). The C-terminus of FANCE is required for recruiting the FANCD2-FANCI heterodimer to the FA core complex to facilitate FANCD2 monoubiquitination (Polito et al., 2014). The N-terminus of FAAP20 interacts with FANCA and prevents it from undergoing uncontrolled degradation (Kim et al., 2012; Leung et al., 2012). Deficiency in FANCA also results in destabilization of its binding partners FANCG and FAAP20, and hypersensitivity to ICL-inducing agents, highlighting its role as a scaffold to preserve the integrity of the complex. However, it remains unclear why the FA core complex consists of at least ten subunits that do not appear to have any homology or evolutionary connections, and what precise roles each module exerts.
Fig. 2.
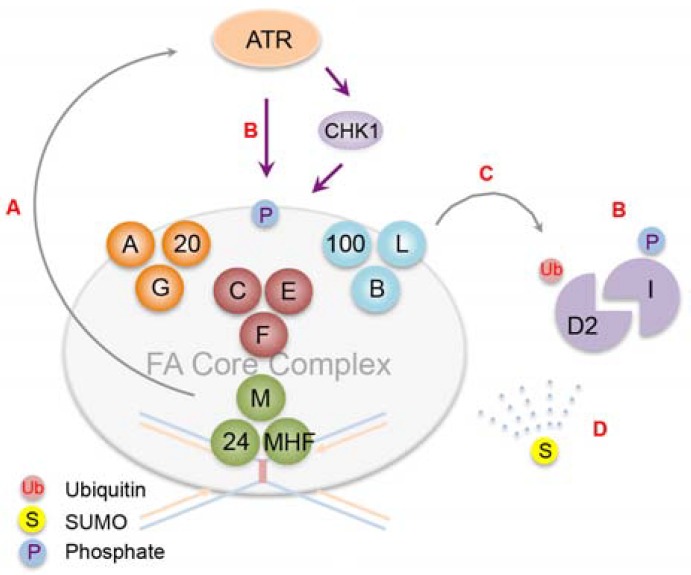
Posttranslational modifications involved in the activation of FANCD2. (A) The FA core complex consists of three modules plus a lesion recognition unit, FANCM. Recognition of a DNA ICL by the FANCM-FAAP24-MHF complex activates ATR checkpoint signaling. (B) ATR and its effector CHK1 phosphorylate components of the FA core complex and the FANCD2-FANCI complex to control their activities. (C) The FANCB-FANCL-FAAP100 module constitutes a minimal catalytic core to monoubiquitinate FANCD2. FANCI phosphorylation potentiates FANCD2 activation. (D) ‘SUMO spray’ of FANC proteins may ensure balanced protein dosage required for the functional integrity of the FA pathway during DNA repair. For instance, FANCA and FANCD2 undergo controlled degradation by the integrated SUMO-ubiquitin signaling.
DNA damage response and FA pathway activation
ATR-dependent phosphorylation is essential for activation of the FA pathway. Recognition of ICL-stalled replication forks by the FANCM-FAAP24 heterodimer, which recruits replication protein A (RPA) and the human homolog of the Caenorhabditis elegans biological clock protein CLK-2 (HCLK2), initiates the activation of the ATR-mediated checkpoint and the FA core complex (Collins et al., 2009; Huang et al., 2010). Multiple components of the FA core complex are activated by phosphorylation during the stress response. For instance, disruption of ATR-dependent FANCM phosphorylation at Ser1045 compromises CHK1 activation and FANCD2 monoubiquitination (Singh et al., 2013). CHK1-mediated phosphorylation of FANCE at Thr346 and Ser374, and ATR-dependent FANCA phosphorylation at Ser1449 are required for the functional integrity of the FA pathway (Collins et al., 2009; Wang et al., 2007). The FANCD2-FANCI heterodimer itself is phosphorylated in a DNA damage-dependent manner. FANCD2 phosphorylation at Thr691 and Ser717 promotes FANCD2 monoubiquitination and the intra-S-phase checkpoint (Ho et al., 2006). FANCI contains a cluster of SQ/TQ phosphorylation sites near its ubiquitination site, and mutations of these residues abrogate FANCD2 monoubiquitination (Ishiai et al., 2008). Conversely, FANCI phosphomimetic mutations enhance FANCD2 activation, indicating that the level of FANCI phosphorylation dictates FANCD2 activation. Interestingly, the three-dimensional structure of the FANCD2-FANCI heterodimer revealed a unique FANCD2 interacting domain in FANCI, which undergoes a conformational change that stabilizes its interaction with FANCD2 (Joo et al., 2011). Structural reorganization of the heterodimer mediated by ATR-dependent FANCI phosphorylation may render FANCD2 suitable for monoubiquitination, although how FANCD2 monoubiquitination is coordinated with FANCI phosphorylation is unclear.
Fine-tuning FA pathway activation
Recent studies demonstrated roles of SUMOylation that regulate FANCD2 activation. The FANCD2-FANCI heterodimer is SUMOylated following replication stress in an ATR-dependent manner, and SUMOylated FANCD2 is a target for the SUMO-targeted ubiquitin E3 ligase (STUbL) RNF4, which results in polyubiquitination and removal of FANCD2 from damage sites via the DVC1-p97 ubiquitin segregase complex (Gibbs-Seymour et al., 2015). Improper clearance of FANCD2 due to disruption of SUMO signaling compromises cellular survival against replication stress, suggesting that timely inactivation of FANCD2 is required for the functional integrity of the FA pathway.
Integrated SUMO-ubiquitin signaling also regulates FANCA degradation. Characterization of a breast cancer patient with a unique FANCA mutation that disrupts the interaction with FAAP20 revealed that in the absence of FAAP20 interaction, a FANCA SUMOylation site becomes exposed, which initiates proteasome-dependent FANCA degradation mediated by RNF4 (Xie et al., 2015). Aberrant accumulation of FANCA at sites of repair by the loss of RNF4 may prevent replication fork restarting and thus inhibit completion of DNA repair. Notably, SUMO modification has been shown to occur simultaneously at multiple sites of several proteins during DSB repair and stabilize physical interactions between the proteins (Psakhye and Jentsch, 2012). This protein group SUMOylation may be relevant to the activation of the FA core complex as well; pervasive SUMOylation may stabilize the FA core complex assembly and promote its activation, which is followed by selective degradation of its components by a STUbL such as RNF4. Several other components of the FA core complex in addition to FANCA are expected to be SUMOylated in a similar manner (Xie et al., 2015), and thus the role of SUMOylation in regulating the FA pathway awaits further characterization.
Several factors have been implicated in the regulation of FANCD2 activation in addition to the FA core complex. Deficiency of RAD18, an ubiquitin E3 ligase involved in postreplication repair of UV-damaged DNA, delays the kinetics of FANCD2 monoubiquitination (Williams et al., 2011a). The MutS complex, a damage sensor required for mismatch repair, has been shown to facilitate the recruitment of the FA core complex to chromatin, thereby playing a redundant role with the FANCM complex (Huang et al., 2011; Williams et al., 2011c). Ubiquitin-like with PHD and ring finger domains 1 (UHRF1), a key epigenetic regulator of chromatin modification at replication forks, was recently identified as a DNA ICL recognition factor that initiates ICL repair (Liang et al., 2015; Tian et al., 2015). UHRF1 is rapidly targeted to a DNA ICL, which promotes recruitment of FANCD2 and structure-specific nucleases required for ICL processing. It will be interesting to determine if these factors crosstalk with the FA core complex to fine-tune the steps of FANCD2 activation.
DNA REPAIR FACTORS AS THERAPEUTIC TARGETS IN CANCER
The DNA repair system functions as a critical tumor suppressor network to preserve the integrity of the genome and prevent malignancy. Accordingly, genome instability is one of the most pervasive characteristics of cancer cells. Germ-line mutations or promoter hyper-methylation of DNA repair genes confer increased risk for multiple cancers (Negrini et al., 2010). In addition, replication stress resulting from high levels of DNA damage that interfere with DNA replication and progression is augmented by faulty DNA repair mechanisms caused by selection of somatic mutations that disrupt DNA repair process (Gaillard et al., 2015). For instance, DNA hyper-replication induced by oncogene activation triggers DDR as a natural barrier to prevent malignancy; thus, inactivation of DDR promotes cellular transformation, and mutations in DDR factors are frequently found in various human cancers (Bartkova et al., 2006; Di Micco et al., 2006; Kandoth et al., 2013). However, although defects in DNA repair confer survival advantages on cancer cells by increasing their adaptability, these defects may reveal a weakness that can be therapeutically exploited. This is possible because cancer cells either become more susceptible to conventional chemotherapy that causes DNA damage due to a decreased capacity to address genotoxicity, or become hyperdependent on another compensatory DNA repair pathway, which provides a therapeutic window for specific killing of cancer cells. Hence, ICL-inducing agents such as nitrogen mustards and platinum compounds are some of the most widely used chemotherapeutic regimens to treat leukemia as well as a variety of solid tumors.
In-depth analysis of the FA signaling pathway has helped to understand the molecular mechanism of the chemotherapeutic effects of DNA ICL-inducing agents and allowed for targeted anti-cancer therapy. This effect could be achieved either by inhibiting the intact or upregulated FA pathway to chemosensitize cancer cells, or by exploiting the synthetic lethality of cancer cells that are defective in the FA pathway (Fig. 3). Resistance to ICL-inducing chemotherapy constitutes a significant barrier to improving patient outcomes. As ICL-inducing chemotherapy directly causes DNA damage, the DNA repair capacity of cancer cells plays a major role in determining the effectiveness of DNA-damaging drugs. For instance, a role for TLS has been implicated in acquired drug resistance following chemotherapy. Suppression of REV1 or Pol ξ not only sensitized cancer cells to platinum agents but also limited mutagenesis and acquired chemoresistance in mouse B-cell lymphoma and lung adenocarcinoma models (Doles et al., 2010; Xie et al., 2010). Upregulation of REV3L mRNA levels was observed in human glioma, and stable overexpression of REV3L attenuated cisplatin-induced toxicity, while downregulation enhanced its cytotoxicity (Wang et al., 2009). These results indicate that upregulation of TLS represents one of the critical mechanisms that confer acquired chemoresistance on tumor cells, and targeting the enhanced TLS pathway using specific TLS polymerase inhibitors could be utilized as an adjuvant therapy for treating chemoresistant malignancy. Selective inhibition of the active site of one of the many TLS polymerases may be difficult to achieve, but recent structural data on protein-protein interactions that regulate TLS polymerase activities may be helpful for identifying small molecules that specifically interfere with such interactions (Kikuchi et al., 2012; Wojtaszek et al., 2012).
Fig. 3.
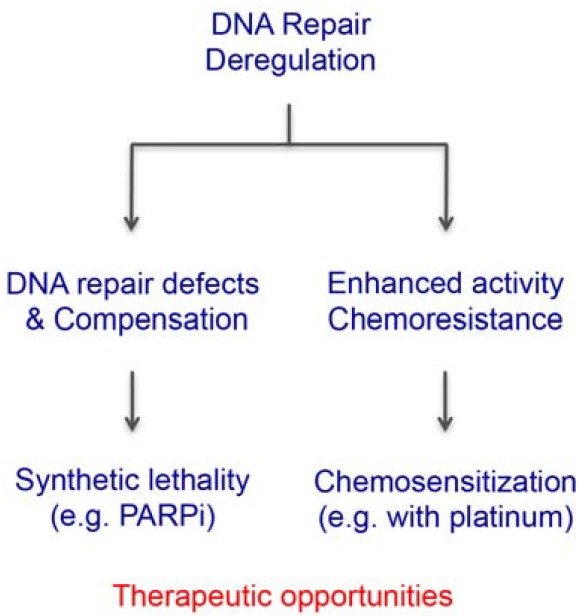
Strategies to exploit deregulated DNA repair for anticancer therapy. Deregulation of DNA repair leads to hyperdependency on a compensatory repair pathway or acquired platinum resistance due to an enhanced DNA repair capacity of cancer cells, which can be exploited by synthetic lethal interactions or resensitization to cross-linking agents.
Hyperdependency on compensatory DNA repair due to defects in the FA pathway can be exploited for achieving synthetic lethality. The best example is development of poly-ADP ribose polymerase (PARP) inhibitors to treat breast and ovarian cancers harboring BRCA1/2 mutations (Bryant et al., 2005; Farmer et al., 2005). Because DNA lesions that cannot be repaired by base excision repair (BER) due to the inhibition of PARP enzymes should be repaired by HR during the S-phase, cancer cells that are defective in HR are selectively sensitive to PARP inhibition owing to the concomitant loss of two DNA repair pathways. Theoretically, tumors that share common HR defects (i.e., BRCAness) could be managed with PARP inhibitors. Indeed, impaired BRCA1 phosphorylation and subsequent defects in HR caused by CDK1 inhibition was shown to sensitize BRCA1-proficient tumors to PARP inhibition, suggesting that functional disruption of HR signaling could expand the utility of PARP inhibitors (Johnson et al., 2011). PARP enzymes are also involved in the error-prone microhomology-mediated end joining (MMEJ) pathway. DNA polymerase θ promotes this process, and a recent genetic study showed that, unlike single knockouts, Fancd2−/−Polq−/− double knockout mice die in utero (Ceccaldi et al., 2015; Mateos-Gomez et al., 2015). Given that Pol θ was found to be highly upregulated in epithelial ovarian cancers, this result indicates that the synthetic lethal relationship between the HR pathway and the Pol θ-dependent MMEJ could be employed as a new therapeutic target for cancers with defective HR, or the FA pathway in general.
THERAPEUTIC POTENTIAL OF FA PATHWAY INHIBITORS
Although inhibition of the FA pathway can occur at multiple levels, FANCD2 monoubiquitination has been a primary target for pharmacological interventions. A previous study demonstrated that proteasome inhibition by bortezomib or depletion of proteasome subunits leads to suppression of FANCD2 monoubiquitination and foci formation (Jacquemont and Taniguchi, 2007). Bortezomib also downregulates FANCD2 gene expression by inhibiting NF-κB signaling and enhances cytotoxicity of melphalan-resistant multiple myeloma cells (Yarde et al., 2009). Bortezomib has been used as standard care for relapsed/refractory multiple myeloma and mantle cell lymphoma, and eventually, its use could be expanded to include a combination therapy, for instance with PARP inhibition in HR-proficient tumors. The natural compound curcumin and its analogs such as EF24 and 4H-TTD have been shown to inhibit FANCD2 activation and sensitize a variety of cancer cells to DNA damage (Chirnomas et al., 2006; Landais et al., 2009). In addition, MLN4924, an inhibitor of a Nedd8 activating enzyme, was shown to impair FANCD2 activation, rendering cancer cells hypersensitive to ICL-inducing agents (Kee et al., 2012).
The USP1-UAF1 deubiquitinating enzyme complex plays an essential role in the FA pathway by antagonizing the level of FANCD2-Ub. Thus, disrupting the ubiquitin-deubiquitination cycle of FANCD2 could lead to the inhibition of the FA pathway. Indeed, several inhibitors of the USP1-UAF1 complex have been developed to target the FA pathway. Small molecule inhibitors such as pimozide and GW7647, as well as a more selective inhibitor ML323, inhibit FANCD2 deubiquitination and potentiate cisplatin cytotoxicity of chemoresistant cancer cells (Chen et al., 2011; Liang et al., 2014). Inhibiting the USP1-UAF1 complex also compromises TLS by increasing levels of monoubiquitinated PCNA, another substrate of USP1, suggesting that inhibition of the USP1-UAF1 complex can simultaneously target two major steps in the FA pathway (Liang et al., 2014). USP1 also prevents the Inhibitor of DNA-binding-1 (ID1) transcription factor from undergoing destruction and thus maintains the stemness of malignant cells (Williams et al., 2011b). Another USP1 inhibitor C527 was shown to promote ID1 degradation and induce differentiation of leukemic cells (Mistry et al., 2013). Therefore, USP1 inhibitors can play versatile roles in targeting multiple cancers.
An siRNA-based synthetic lethal screening identified several genes including ATM, PARP1, CDK1, NBS1, and PLK1 that are required for the survival of cells deficient in FANCG, indicating that these genes could be targeted in conjunction with an FA pathway inhibitor (Kennedy et al., 2007). As deficiency of ATM signaling is found in several types of leukemia and triple-negative breast cancer, the FA pathway inhibitor could be used as a single agent in these cancers as well. In addition, CHK1 inhibition was shown to be synthetically lethal with FANCA deficiency following cisplatin treatment (Chen et al., 2009). These results suggest that targeting the FA pathway can become more effective by inhibiting DNA damage signaling simultaneously, and appropriate combinations of DDR and DNA repair inhibitors could be customized for increasing the efficacy of chemotherapy.
CONCLUSION
Since the discovery of FANCD2 monoubiquitination in the early 2000s, the study of FA has integrated important topics in biology, including ubiquitin signaling, DNA repair, and the pathogenesis of cancer. FANCD2 activation acts as a surrogate marker for the DNA ICL repair process, and knowledge of how FANCD2 is activated to regulate downstream repair steps has opened new therapeutic opportunities for cancer treatment. However, many of the details are still missing. We must increase our understanding of the regulatory mechanisms underlying FANCD2 activation by the FA core complex. More specifically, a comprehensive understanding of the complex network of posttranslational modifications that regulate the FA core complex may lead to the identification of additional targets for therapeutic interventions. Moreover, finding ways to inhibit the enzymatic steps of the FA pathway, including incisions and TLS, may be useful. Basic information on DNA repair regulation could also have a broad translational impact. Profiling DNA repair in individuals with nonfunctional or upregulated DNA repair capacity could provide targeted and personalized therapeutic options. Optimizing the selection of a chemotherapy approach requires the identification of reliable DNA repair biomarkers. Thus, development of fast and affordable ways to monitor individual DNA repair activity is highly desirable. Furthermore, synthetic lethality could be extended beyond DNA repair pathways, to include aberrant signaling of activated oncogenes and growth factors, or enhanced anti-apoptotic signaling. Overall, further deciphering of the complex regulatory network underlying the FA pathway will enable development of new strategies to exploit aberrant regulation of DNA repair in cancer.
Acknowledgments
We thank Dr. Orlando Schärer for critically reading the manuscript. This work was supported by startup funds from the Office of the Vice President for Research and the Cancer Center at Stony Brook University.
REFERENCES
- Bartkova J., Rezaei N., Liontos M., Karakaidos P., Kletsas D., Issaeva N., Vassiliou L.V., Kolettas E., Niforou K., Zoumpourlis V.C., et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Blackford A.N., Schwab R.A., Nieminuszczy J., Deans A.J., West S.C., Niedzwiedz W. The DNA translocase activity of FANCM protects stalled replication forks. Human molecular genetics. 2012;21:2005–2016. doi: 10.1093/hmg/dds013. [DOI] [PubMed] [Google Scholar]
- Bogliolo M., Schuster B., Stoepker C., Derkunt B., Su Y., Raams A., Trujillo J.P., Minguillon J., Ramirez M.J., Pujol R., et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. American journal of human genetics. 2013;92:800–806. doi: 10.1016/j.ajhg.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N.J., Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Budzowska M., Graham T.G., Sobeck A., Waga S., Walter J.C. Regulation of the Rev1-pol zeta complex during bypass of a DNA interstrand cross-link. EMBO J. 2015;pii:e201490878. doi: 10.15252/embj.201490878. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting S.F., Callen E., Kozak M.L., Kim J.M., Wong N., Lopez-Contreras A.J., Ludwig T., Baer R., Faryabi R.B., Malhowski A., et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell. 2012;46:125–135. doi: 10.1016/j.molcel.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R., Liu J.C., Amunugama R., Hajdu I., Primack B., Petalcorin M.I., O’Connor K.W., Konstantinopoulos P.A., Elledge S.J., Boulton S.J., et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature. 2015;518:258–262. doi: 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D.J., Cimprich K.A. DNA damage tolerance: when it’s OK to make mistakes. Nat. Chem. Biol. 2009;5:82–90. doi: 10.1038/nchembio.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.C., Kennedy R.D., Sidi S., Look A.T., D’Andrea A. CHK1 inhibition as a strategy for targeting Fanconi Anemia (FA) DNA repair pathway deficient tumors. Mol. Cancer. 2009;8:24. doi: 10.1186/1476-4598-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.B., Melchionna R., Denis C.M., Gaillard P.H., Blasina A., Van de Weyer I., Boddy M.N., Russell P., Vialard J., McGowan C.H. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol. Cell. 2001;8:1117–1127. doi: 10.1016/s1097-2765(01)00375-6. [DOI] [PubMed] [Google Scholar]
- Chen J., Dexheimer T.S., Ai Y., Liang Q., Villamil M.A., Inglese J., Maloney D.J., Jadhav A., Simeonov A., Zhuang Z. Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem. Biol. 2011;18:1390–1400. doi: 10.1016/j.chembiol.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirnomas D., Taniguchi T., de la Vega M., Vaidya A.P., Vasserman M., Hartman A.R., Kennedy R., Foster R., Mahoney J., Seiden M.V., et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol. Cancer Ther. 2006;5:952–961. doi: 10.1158/1535-7163.MCT-05-0493. [DOI] [PubMed] [Google Scholar]
- Clauson C., Scharer O.D., Niedernhofer L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harbor Perspect. Biol. 2013;5:a012732. doi: 10.1101/cshperspect.a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn M.A., Kowal P., Yang K., Haas W., Huang T.T., Gygi S.P., D’Andrea A.D. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol. Cell. 2007;28:786–797. doi: 10.1016/j.molcel.2007.09.031. [DOI] [PubMed] [Google Scholar]
- Collins N.B., Wilson J.B., Bush T., Thomashevski A., Roberts K.J., Jones N.J., Kupfer G.M. ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood. 2009;113:2181–2190. doi: 10.1182/blood-2008-05-154294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collis S.J., Ciccia A., Deans A.J., Horejsi Z., Martin J.S., Maslen S.L., Skehel J.M., Elledge S.J., West S.C., Boulton S.J. FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol. Cell. 2008;32:313–324. doi: 10.1016/j.molcel.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Cybulski K.E., Howlett N.G. FANCP/SLX4: a Swiss army knife of DNA interstrand crosslink repair. Cell Cycle. 2011;10:1757–1763. doi: 10.4161/cc.10.11.15818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Andrea A.D. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl. J. Med. 2010;362:1909–1919. doi: 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans A.J., West S.C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell. 2009;36:943–953. doi: 10.1016/j.molcel.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Deans A.J., West S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer. 2011;11:467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R., Fumagalli M., Cicalese A., Piccinin S., Gasparini P., Luise C., Schurra C., Garre M., Nuciforo P.G., Bensimon A., et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- Doles J., Oliver T.G., Cameron E.R., Hsu G., Jacks T., Walker G.C., Hemann M.T. Suppression of Rev3, the catalytic subunit of Pol{zeta}, sensitizes drug-resistant lung tumors to chemotherapy. Proc. Natl. Acad. Sci. USA. 2010;107:20786–20791. doi: 10.1073/pnas.1011409107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H., McCabe N., Lord C.J., Tutt A.N., Johnson D.A., Richardson T.B., Santarosa M., Dillon K.J., Hickson I., Knights C., et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Gaillard H., Garcia-Muse T., Aguilera A. Replication stress and cancer. Nat. Rev. Cancer. 2015;15:276–289. doi: 10.1038/nrc3916. [DOI] [PubMed] [Google Scholar]
- Garaycoechea J.I., Patel K.J. Why does the bone marrow fail in Fanconi anemia? Blood. 2014;123:26–34. doi: 10.1182/blood-2013-09-427740. [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I., Taniguchi T., Ganesan S., Meyn M.S., Timmers C., Hejna J., Grompe M., D’Andrea A.D. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Gari K., Decaillet C., Delannoy M., Wu L., Constantinou A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc. Natl. Acad. Sci. USA. 2008;105:16107–16112. doi: 10.1073/pnas.0804777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs-Seymour I., Oka Y., Rajendra E., Weinert B.T., Passmore L.A., Patel K.J., Olsen J.V., Choudhary C., Bekker-Jensen S., Mailand N. Ubiquitin-SUMO circuitry controls activated fanconi anemia ID complex dosage in response to DNA damage. Mol. Cell. 2015;57:150–164. doi: 10.1016/j.molcel.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guainazzi A., Schärer O.D. Using synthetic DNA interstrand crosslinks to elucidate repair pathways and identify new therapeutic targets for cancer chemotherapy. Cell Mol Life Sci. 2010;67:3683–3697. doi: 10.1007/s00018-010-0492-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guervilly J.H., Takedachi A., Naim V., Scaglione S., Chawhan C., Lovera Y., Despras E., Kuraoka I., Kannouche P., Rosselli F., et al. The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Mol. Cell. 2015;57:123–137. doi: 10.1016/j.molcel.2014.11.014. [DOI] [PubMed] [Google Scholar]
- Hira A., Yoshida K., Sato K., Okuno Y., Shiraishi Y., Chiba K., Tanaka H., Miyano S., Shimamoto A., Tahara H., et al. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi Anemia. Am. J. Hum. Genet. 2015;96:1001–1007. doi: 10.1016/j.ajhg.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho G.P, Margossian S., Taniguchi T., D’Andrea A.D. Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Mol. Cell. Biol. 2006;26:7005–7015. doi: 10.1128/MCB.02018-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodskinson M.R., Silhan J., Crossan G.P., Garaycoechea J.I., Mukherjee S., Johnson C.M., Scharer O.D., Patel K.J. Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF-ERCC1 in DNA crosslink repair. Mol. Cell. 2014;54:472–484. doi: 10.1016/j.molcel.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M., Kim J.M., Shiotani B., Yang K., Zou L., D’Andrea A.D. The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Mol. Cell. 2010;39:259–268. doi: 10.1016/j.molcel.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M., Kennedy R., Ali A.M., Moreau L.A., Meetei A.R., D’Andrea A.D., Chen C.C. Human MutS and FANCM complexes function as redundant DNA damage sensors in the Fanconi Anemia pathway. DNA Repair. 2011;10:1203–1212. doi: 10.1016/j.dnarep.2011.09.006. [DOI] [PubMed] [Google Scholar]
- Huang Y., Leung J.W., Lowery M., Matsushita N., Wang Y., Shen X., Huong D., Takata M., Chen J., Li L. Modularized functions of the Fanconi anemia core complex. Cell Rep. 2014;7:1849–1857. doi: 10.1016/j.celrep.2014.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiai M., Kitao H., Smogorzewska A., Tomida J., Kinomura A., Uchida E., Saberi A., Kinoshita E., Kinoshita-Kikuta E., Koike T., et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat. Struct. Mol. Biol. 2008;15:1138–1146. doi: 10.1038/nsmb.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont C., Taniguchi T. Proteasome function is required for DNA damage response and fanconi anemia pathway activation. Cancer Res. 2007;67:7395–7405. doi: 10.1158/0008-5472.CAN-07-1015. [DOI] [PubMed] [Google Scholar]
- Johnson N., Li Y.C., Walton Z.E., Cheng K.A., Li D., Rodig S.J., Moreau L.A., Unitt C., Bronson R.T., Thomas H.D., et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat. Med. 2011;17:875–882. doi: 10.1038/nm.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo W., Xu G., Persky N.S., Smogorzewska A., Rudge D.G., Buzovetsky O., Elledge S.J., Pavletich N.P. Structure of the FANCI-FANCD2 complex: insights into the Fanconi anemia DNA repair pathway. Science. 2011;333:312–316. doi: 10.1126/science.1205805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A., et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee Y., Huang M., Chang S., Moreau L.A., Park E., Smith P.G., D’Andrea A.D. Inhibition of the Nedd8 system sensitizes cells to DNA interstrand cross-linking agents. Mol. Cancer Res. 2012;10:369–377. doi: 10.1158/1541-7786.MCR-11-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy R.D, Chen C.C., Stuckert P., Archila E.M., De la Vega M.A., Moreau L.A., Shimamura A., D’Andrea A.D. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J. Clin. Invest. 2007;117:1440–1449. doi: 10.1172/JCI31245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi S., Hara K., Shimizu T., Sato M., Hashimoto H. Structural basis of recruitment of DNA polymerase zeta by interaction between REV1 and REV7 proteins. J. Biol. Chem. 2012;287:33847–33852. doi: 10.1074/jbc.M112.396838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. Nuclease delivery: versatile functions of SLX4/FANCP in genome maintenance. Mol. Cells. 2014;37:569–574. doi: 10.14348/molcells.2014.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., D’Andrea A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–1408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.M., Parmar K., Huang M., Weinstock D.M., Ruit C.A., Kutok J.L., D’Andrea A.D. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev. Cell. 2009;16:314–320. doi: 10.1016/j.devcel.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Yang K., Dejsuphong D., D’Andrea A.D. Regulation of Rev1 by the Fanconi anemia core complex. Nat. Struct. Mol. Biol. 2012;19:164–170. doi: 10.1038/nsmb.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Douwel D., Boonen R.A., Long D.T., Szypowska A.A., Raschle M., Walter J.C., Knipscheer P. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell. 2014;54:460–471. doi: 10.1016/j.molcel.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P., Raschle M., Smogorzewska A., Enoiu M., Ho T.V., Scharer O.D., Elledge S.J., Walter J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann M.C., Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landais I., Sobeck A., Stone S., LaChapelle A., Hoatlin M.E. A novel cell-free screen identifies a potent inhibitor of the Fanconi anemia pathway. Int. J. Cancer. 2009;124:783–792. doi: 10.1002/ijc.24039. [DOI] [PubMed] [Google Scholar]
- Langevin F., Crossan G.P., Rosado I.V., Arends M.J., Patel K.J. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475:53–58. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- Lehmann A.R., Niimi A., Ogi T., Brown S., Sabbioneda S., Wing J.F., Kannouche P.L., Green C.M. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair. 2007;6:891–899. doi: 10.1016/j.dnarep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Leung J.W., Wang Y., Fong K.W., Huen M.S., Li L., Chen J. Fanconi anemia (FA) binding protein FAAP20 stabilizes FA complementation group A (FANCA) and participates in interstrand cross-link repair. Proc. Natl. Acad. Sci. USA. 2012;109:4491–4496. doi: 10.1073/pnas.1118720109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q., Dexheimer T.S., Zhang P., Rosenthal A.S., Villamil M.A., You C., Zhang Q., Chen J., Ott C.A., Sun H., et al. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat. Chem. Biol. 2014;10:298–304. doi: 10.1038/nchembio.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C.C, Zhan B., Yoshikawa Y., Haas W., Gygi S.P., Cohn M.A. UHRF1 is a sensor for DNA interstrand crosslinks and recruits FANCD2 to initiate the Fanconi anemia pathway. Cell Rep. 2015;10:1947–1956. doi: 10.1016/j.celrep.2015.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Tarsounas M., O’Regan P., West S.C. Role of RAD51C and XRCC3 in genetic recombination and DNA repair. J. Biol. Chem. 2007;282:1973–1979. doi: 10.1074/jbc.M609066200. [DOI] [PubMed] [Google Scholar]
- Long D.T., Joukov V., Budzowska M., Walter J.C. BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol. Cell. 2014;56:174–185. doi: 10.1016/j.molcel.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos-Gomez P.A., Gong F., Nair N., Miller K.M., Lazzerini-Denchi E., Sfeir A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature. 2015;518:254–257. doi: 10.1038/nature14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meetei A.R, de Winter J.P., Medhurst A.L., Wallisch M., Waisfisz Q., van de Vrugt H.J., Oostra A.B., Yan Z., Ling C., Bishop C.E., et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet. 2003;35:165–170. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- Meetei A.R, Medhurst A.L., Ling C., Xue Y., Singh T.R., Bier P., Steltenpool J., Stone S., Dokal I., Mathew C.G., et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 2005;37:958–963. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry H., Hsieh G., Buhrlage S.J., Huang M., Park E., Cuny G.D., Galinsky I., Stone R.M., Gray N.S., D’Andrea A.D., et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol. Cancer Ther. 2013;12:2651–2662. doi: 10.1158/1535-7163.MCT-13-0103-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan M.E., Pierce A.J., Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell. 2001;7:263–272. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- Negrini S., Gorgoulis V.G., Halazonetis T.D. Genomic instability--an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- Niedzwiedz W., Mosedale G., Johnson M., Ong C.Y., Pace P., Patel K.J. The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol. Cell. 2004;15:607–620. doi: 10.1016/j.molcel.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Nijman S.M., Huang T.T., Dirac A.M., Brummelkamp T.R., Kerkhoven R.M., D’Andrea A.D., Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol. Cell. 2005;17:331–339. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Polito D., Cukras S., Wang X., Spence P., Moreau L., D’Andrea A.D., Kee Y. The carboxyl terminus of FANCE recruits FANCD2 to the Fanconi Anemia (FA) E3 ligase complex to promote the FA DNA repair pathway. J. Biol. Chem. 2014;289:7003–7010. doi: 10.1074/jbc.M113.533976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psakhye I., Jentsch S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell. 2012;151:807–820. doi: 10.1016/j.cell.2012.10.021. [DOI] [PubMed] [Google Scholar]
- Rajendra E., Oestergaard V.H., Langevin F., Wang M., Dornan G.L., Patel K.J., Passmore L.A. The genetic and biochemical basis of FANCD2 monoubiquitination. Mol. Cell. 2014;54:858–869. doi: 10.1016/j.molcel.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle M., Knipscheer P., Enoiu M., Angelov T., Sun J., Griffith J.D., Ellenberger T.E., Scharer O.D., Walter J.C. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Räschle M., Smeenk G., Hansen R.K., Temu T., Oka Y., Hein M.Y., Nagaraj N., Long D.T., Walter J.C., Hofmann K., et al. DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. Science. 2015;348:1253671. doi: 10.1126/science.1253671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer S.L., Tian L., Kahkonen M., Schwartzentruber J., Kircher M., University of Washington Centre for Mendelian, G. Consortium F.C., Majewski J., Dyment D.A., Innes A.M., et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015;5:135–142. doi: 10.1158/2159-8290.CD-14-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab R.A, Blackford A.N., Niedzwiedz W. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. 2010;29:806–818. doi: 10.1038/emboj.2009.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T.R., Saro D., Ali A.M., Zheng X.F., Du C.H., Killen M.W., Sachpatzidis A., Wahengbam K., Pierce A.J., Xiong Y., et al. MHF1-MHF2, a histone-fold-containing protein complex, participates in the Fanconi anemia pathway via FANCM. Mol. Cell. 2010;37:879–886. doi: 10.1016/j.molcel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T.R, Ali A.M., Paramasivam M., Pradhan A., Wahengbam K., Seidman M.M., Meetei A.R. ATR-dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions. Cancer Res. 2013;73:4300–4310. doi: 10.1158/0008-5472.CAN-12-3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A., Matsuoka S., Vinciguerra P., McDonald E.R., Hurov K.E., 3rd, Luo J., Ballif B.A., Gygi S.P., Hofmann K., D’Andrea A.D., et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A., Desetty R., Saito T.T., Schlabach M., Lach F.P., Sowa M.E., Clark A.B., Kunkel T.A., Harper J.W., Colaiacovo M.P., et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell. 2010;39:36–47. doi: 10.1016/j.molcel.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y., Paramasivam M., Ghosal G., Chen D., Shen X., Huang Y., Akhter S., Legerski R., Chen J., Seidman M.M., et al. UHRF1 Contributes to DNA Damage Repair as a Lesion Recognition Factor and Nuclease Scaffold. Cell Rep. 2015;10:1957–1966. doi: 10.1016/j.celrep.2015.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unno J., Itaya A., Taoka M., Sato K., Tomida J., Sakai W., Sugasawa K., Ishiai M., Ikura T., Isobe T., et al. FANCD2 binds CtIP and regulates DNA-end resection during DNA interstrand crosslink repair. Cell Rep. 2014;7:1039–1047. doi: 10.1016/j.celrep.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Vaz F., Hanenberg H., Schuster B., Barker K., Wiek C., Erven V., Neveling K., Endt D., Kesterton I., Autore F., et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat. Genet. 2010;42:406–409. doi: 10.1038/ng.570. [DOI] [PubMed] [Google Scholar]
- Walden H., Deans A.J. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Ann. Rev. Biophys. 2014;43:257–278. doi: 10.1146/annurev-biophys-051013-022737. [DOI] [PubMed] [Google Scholar]
- Wang X., Kennedy R.D., Ray K., Stuckert P., Ellenberger T., D’Andrea A.D. Chk1-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Mol. Cell. Biol. 2007;27:3098–3108. doi: 10.1128/MCB.02357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Zhang S.Y., Wang S., Lu J., Wu W., Weng L., Chen D., Zhang Y., Lu Z., Yang J., et al. REV3L confers chemoresistance to cisplatin in human gliomas: the potential of its RNAi for synergistic therapy. Neuro Oncol. 2009;11:790–802. doi: 10.1215/15228517-2009-015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A.T., Sengerova B., Cattell E., Inagawa T., Hartley J.M., Kiakos K., Burgess-Brown N.A., Swift L.P., Enzlin J.H., Schofield C.J., et al. Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 2011;25:1859–1870. doi: 10.1101/gad.15699211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S.A., Longerich S., Sung P., Vaziri C., Kupfer G.M. The E3 ubiquitin ligase RAD18 regulates ubiquitylation and chromatin loading of FANCD2 and FANCI. Blood. 2011a;117:5078–5087. doi: 10.1182/blood-2010-10-311761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S.A., Maecker H.L., French D.M., Liu J., Gregg A., Silverstein L.B., Cao T.C., Carano R.A., Dixit V.M. USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell. 2011b;146:918–930. doi: 10.1016/j.cell.2011.07.040. [DOI] [PubMed] [Google Scholar]
- Williams S.A., Wilson J.B., Clark A.P., Mitson-Salazar A., Tomashevski A., Ananth S., Glazer P.M., Semmes O.J., Bale A.E., Jones N.J., et al. Functional and physical interaction between the mismatch repair and FA-BRCA pathways. Hum. Mol. Genet. 2011c;20:4395–4410. doi: 10.1093/hmg/ddr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszek J., Lee C.J., D’Souza S., Minesinger B., Kim H., D’Andrea A.D., Walker G.C., Zhou P. Structural basis of Rev1-mediated assembly of a quaternary vertebrate translesion polymerase complex consisting of Rev1, heterodimeric polymerase (Pol) zeta, and Pol kappa. J. Biol. Chem. 2012;287:33836–33846. doi: 10.1074/jbc.M112.394841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia B., Sheng Q., Nakanishi K., Ohashi A., Wu J., Christ N., Liu X., Jasin M., Couch F.J., Livingston D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Xie K., Doles J., Hemann M.T., Walker G.C. Error-prone translesion synthesis mediates acquired chemoresistance. Proc. Natl. Acad. Sci. USA. 2010;107:20792–20797. doi: 10.1073/pnas.1011412107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J., Kim H., Moreau L.A., Puhalla S., Garber J., Al Abo M., Takeda S., D’Andrea A.D. RNF4-mediated polyubiquitination regulates the Fanconi anemia/BRCA pathway. J. Clin. Invest. 2015;125:1523–1532. doi: 10.1172/JCI79325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K.N., Kobayashi S., Tsuda M., Kurumizaka H., Takata M., Kono K., Jiricny J., Takeda S., Hirota K. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc. Natl. Acad. Sci. USA. 2011;108:6492–6496. doi: 10.1073/pnas.1018487108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z., Delannoy M., Ling C., Daee D., Osman F., Muniandy P.A., Shen X., Oostra A.B., Du H., Steltenpool J., et al. A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol. Cell. 2010;37:865–878. doi: 10.1016/j.molcel.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K., Moldovan G.L., Vinciguerra P., Murai J., Takeda S., D’Andrea A.D. Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev. 2011;25:1847–1858. doi: 10.1101/gad.17020911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarde D.N., Oliveira V., Mathews L., Wang X., Villagra A., Boulware D., Shain K.H., Hazlehurst L.A., Alsina M., Chen D.T., et al. Targeting the Fanconi anemia/BRCA pathway circumvents drug resistance in multiple myeloma. Cancer Res. 2009;69:9367–9375. doi: 10.1158/0008-5472.CAN-09-2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Ma J., Wu J., Ye L., Cai H., Xia B., Yu X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 2009;19:524–529. doi: 10.1016/j.cub.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Dewar J.M., Budzowska M., Motnenko A., Cohn M.A., Walter J.C. DNA interstrand cross-link repair requires replication-fork convergence. Nat. Struct. Mol. Biol. 2015;22:242–247. doi: 10.1038/nsmb.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Otto E.A., Cluckey A., Airik R., Hurd T.W., Chaki M., Diaz K., Lach F.P., Bennett G.R., Gee H.Y., et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet. 2012;44:910–915. doi: 10.1038/ng.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]