

Abstract
We investigated the role of HDAC3 in anti-cancer drug-resistance. The expression of HDAC3 was decreased in cancer cell lines resistant to anti-cancer drugs such as celastrol and taxol. HDAC3 conferred sensitivity to these anti-cancer drugs. HDAC3 activity was necessary for conferring sensitivity to these anti-cancer drugs. The down-regulation of HDAC3 increased the expression of MDR1 and conferred resistance to anti-cancer drugs. The expression of tubulin β3 was increased in drug-resistant cancer cell lines. ChIP assays showed the binding of HDAC3 to the promoter sequences of tubulin β3 and HDAC6. HDAC6 showed an interaction with tubulin β3. HDAC3 had a negative regulatory role in the expression of tubulin β3 and HDAC6. The down-regulation of HDAC6 decreased the expression of MDR1 and tubulin β3, but did not affect HDAC3 expression. The down-regulation of HDAC6 conferred sensitivity to taxol. The down-regulation of tubulin β3 did not affect the expression of HDAC6 or MDR1. The down-regulation of tubulin β3 conferred sensitivity to anti-cancer drugs. Our results showed that tubulin β3 serves as a downstream target of HDAC3 and mediates resistance to microtubule-targeting drugs. Thus, the HDAC3-HDAC6-Tubulin β axis can be employed for the development of anti-cancer drugs.
Keywords: anti-cancer drug-resistance, expression regulation, HDAC3, tubulin beta 3
INTRODUCTION
Among the numerous HDACs, histone deactylase-3 (HDAC3) is ubiquitously expressed and conserved in a wide range of species (Mahlknecht et al., 1999). HDAC3 forms large corepressor complexes containing N-CoR/SMRT and additional proteins (Li et al., 2000). HDAC3 regulates the JNK pathway (Zhang et al., 2002), NF-κB activity (Chen et al., 2001), and MAPK activation (Mahlknecht et al., 2004). HDAC3 represses CREB3-mediated transcription and migration of metastatic breast cancer cells (Kim et al., 2010a). It localizes to the mitotic spindle and is required for kinetochore-microtubule attachment (Ishii et al., 2008). Aurora kinase B plays a critical role in mitosis. Aurora kinase B activity is required for mitotic processes, including kinetochore-microtubule attachment and chromosome congression (Fadri-Moskwik et al., 2012). Aurora kinase B activity is regulated by histone acetylation/deacetylation (Fadri-Moskwik et al., 2012). HDAC3 transiently interacts with Aurora kinase B and leads to reduction of acetylation of Aurora kinase (Fadri-Moskwik et al., 2012). Aurora kinase B is active in its deacetylated state (Fadri-Moskwik et al., 2012). Over-expression of Aurora kinase has been reported in multiple tumors and the selective inhibition of Aurora kinase B results in apoptosis induction (Xie et al., 2013). Aurora kinase B is regulated by the MAPK/ERK pathways and is a potential target for overcoming resistance to vemurafenib in metastatic melanomas (Bonet et al., 2012)
Phase I trial revealed that albumin-bound paclitaxel shows encouraging activity against advanced metastatic melanomas (Ott et al., 2013). Resistance to taxol, a microtubule-targeting drug, in hepatoma cells is related to JNK activation and prohibition into mitosis (Chae et al., 2012). Taxol-resistance results from MAPK activation (Xu et al., 2011). The inhibition of MAPK enhances taxol-induced apoptosis (Xu et al., 2009). As HDAC3 suppresses JNK (Zhang et al., 2002) and MAPK activation (Mahlknecht et al., 2004), it is likely that HDAC3 may regulate taxol-resistance.
The increased expression level of tubulin β3 is closely related with resistance to taxol (Kavallaris et al., 1997). High tubulin β3 expression is closely related with non-responsiveness to chemotherapy and is regulated by multiple signaling pathways, including PI3 kinase/Akt, Ras and MAP-ERK kinase (Levallet et al., 2012). HDAC6 deacetylates alpha-tubulin and regulates microtubule-dependent cell motility (Hubbert et al., 2002). This suggests that histone acetylation/deacetylation may also regulate activity and/or expression of tubulins. These reports suggested potential role of HDAC3 in resistance to microtubule-targeting drugs, including taxol. However, the role of HDAC3 in resistance to microtubule-targeting drugs in cancer cell lines in relation with tubulin β3 remains unknown. In this study, we investigated the molecular relationship between HDAC3 and tubulin β3. We showed that the low expression level of HDAC3 is related with the resistance to microtubule-targeting drugs. We showed that HDAC3 confers sensitivity to microtubule-targeting drugs. HDAC3 directly regulates the expression of MDR1 by binding to its promoter sequence. The expression of tubulin β3 was increased in cancer cell lines resistant to microtubule-targeting drugs and HDAC3 decreased the expression of tubulin β3. HDAC3 showed direct binding to the promoter sequences of tubulin β3. HDAC6 showed an interaction with tubulin β3 that was disrupted by HDAC3. HADC6 acted upstream of tubulin β3, and the down-regulation of HDAC6 enhanced sensitivity to microtubule-targeting drugs. Tubulin β3 did not affect expression MDR1. The down-regulation of MDR1 did not affect the expression of tubulin β3, either. Our results indicated that tubulin β3 is an independent target of HDAC3.
MATERIALS AND METHODS
Materials
Anti mouse and anti rabbit IgG-horse radish peroxidase conjugate antibodies were purchased from Pierce Company. An ECL (enhanced chemiluminiscence) kit was purchased from Amersham. Lipofectamin and PlusTM reagent were purchased from Invitrogen (USA). Bioneer (Korea) synthesized all primers and oligonucleotides oused in this study. All antibodies used in this study were purchased from Santa Cruz Company.
Cell lines and cell culture
Cancer cell lines used in this study were cultured in Dulbecco’s modified minimal essential medium (DMEM; Gibco, USA) supplemented with heat-inactivated 10% fetal bovine serum (FBS, Gibco) and antibiotics at 37°C in a humidified incubator with a mixture of 95% air and 5% CO2. Cancer cell lines (SNU387R and Malme3MR) made resistant to microtubule-targeting drugs were established by stepwise addition of the respective drug. Cells surviving drug treatment (attached fraction) were obtained and used throughout this study. SNU387/SNU387R or Malme3M/Malme3MR cells that stably express anti-sense HDAC3 cDNA, HDAC3-Flag or mutant HDAC3 were selected by G418 (400 μg/ml).
Western blot analysis
Western blot analysis, immunoprecipitation and cellular fractionation were performed according to the standard procedures (Kim et al., 2010b).
Cell viability determination
The cells were assayed for their growth activity using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT; Sigma). Viable cell number counting was carried out by trypan blue exclusion assays.
HDAC3 constructs
HDAC3S424A-Myc/His(6) expression plasmid (catalytically inactive HDAC3 mutant) was derived from pHDAC3-Myc/His(6) with the Quick-change site-directed mutagenesis kit (Stratagene).
Transfection
All transfections were performed according to the manufacturer’s instructions. Lipofectamine and Plus reagents (Invitrogen) were used.
Histone deacetylase activity assays
Histone deacetylase activity was measured according to the manufacturer’s instructions (Cayman Chemical, USA). The activity was measured according to the manufacturer’s instructions. For immunoprecipitation, cells were lysed with ice-cold buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 15 mM MgCl2, 250 mMsucrose, 0.12 mM EDTA, 0.5% Nonidet P-40, and a mixture of protease inhibitors). The lysates were suspended with nuclear extraction buffer (50 mM HEPES, pH 7.5, 420 mM NaCl, 0.5 mMn EDTA, 0.1 mM EGTA, and 10% glycerol), sonicated for 30 s, and centrifuged at 10,000 g for 10 min at 4°C. The supernatant containing the nuclear extract was immunoprecipitated with anti-HDAC3 (2 μg/ml), anti-HDAC2 (2 μg/ml), or anti-IgG antibody (2 μg/ml). The immunoprecipitants were incubated with 200 μM acetylated fluorometric substrate for 30 min at 37°C, and 40 μl of developer was added. After 15 min, the fluorescencewas measured using an excitation wavelength of 340–360 nm and an emission wavelength of 440–460 nm.
Caspase-3 activity assays
Caspase-3 activity was measuredaccording to the manufacturer’s instructions (BioVision, USA). Cells were lysed in 0.1 M HEPES buffer, pH 7.4, containing 2 mM dithiothreitol, 0.1% CHAPS, and 1% sucrose. Cell lysates were incubated with a colorimetric substrate, 200 μM Ac-DEVD-p-nitroanilide, for 30 min at 30°C. The fluorescence was measured at 405 nm using a microtiter plate reader.
ChIP assays
Assays were performed according to manufacturer’s instruction (Upstate). The immunoprecipitates were reverse cross-linked. PCR was done on the phenol-chloroform-extracted DNA. PCR was done on the phenol-chloroform-extracted DNA with specific primers of tubulin β3 promoter-1 [5′-GCAGCAGTCGCCCAAGCAGA-3′ (sense)] and [5′-CAGCCCACCTGCACTGAGCC-3′ (antisense)], tubulin β3 promoter-2 [5′-GCTCAGTGCAGGTGGGCTGG-3′ (sense)] and [5′-CCTGCCCCACAGTGTGCTCG-3′ (antisense)], HDAC6 promoter-1[5′-TACAGAAACACCTGTGACCC-3′ (sense)] and [5′-ATCTGTGCTGTAGTGTCAGG-3′ (antisense)], HDAC6 promoter-2 [5′-TGCTTATCTCTCCGGTCCCA-3′ (sense)] and [5′-CTGCGGTGCAAGCTTTTTCT-3′ (antisense)], HDAC6 promoter-3 [5′-AGAAAAAGCTTGCACCGCAG-3′ (sense)] and [5′-CCCCATTCCCAGACCCTCTA-3′ (antisense)] sequences were used.
Preparation of SiRNA duplexes
The SiRNA duplexes were constructed with the following target sequences. Tubulin β3, sense (5′-AAGCCTCTTCCTCACAAGTACGCCTGTCTC-3′); antisense (5′-AACGGAGAAGAGTGTTCATGCCCTGTCTC-3′); HDAC6, sense (5′-AAGGTGTCACCTGAGGGTTATCCTGTCTC-3′); antisense (5′-AAATAACCCTCAGGTGACACCCCTGTCTC-3′); MDR1-1, sense (5′-AATCCAAGGCATCAATTTCACCCTGTCTC-3′); antisense (5′-AAGTGAAATTGATGCCTTGGACCTGTCTC-3′); MDR1-2, sense (5′-AATTGCATACGCTAAGAGTTCCCTGTCTC-3′); antisense (5′-AAGAACTCTTAGCGTATGCAACCTGTCTC-3′); Control, sense (5′-AATTCTCCGAACGTGTCACGTCCTGTCTC-3′); antisense (5′-AAACGTGACACGTTCGGAGAACCTGTCTC-3′). The construction of SiRNA was carried out according to the instruction manual provided by the manufacturer (Ambion, USA).
Statistical analysis
Statistical differences in this were determined by using the Student’s t test.
RESULTS
Taxol-resistant cancer cell lines show lower expression level of HDAC3 than taxol-sensitive cancer cell lines
We established cancer cell lines selected for resistance to celastrol, a microtubule-targeting drug. We examined the role of HDAC(s) in resistance to microtubule-targeting drugs such as celastrol and taxol. Hepatoma cell lines i.e., HepG2, Hep3B and SUN387R cells showed resistance to taxol (Fig. 1A). Melanoma cell line WM266-4 and Malme3MR showed resistance to taxol (Fig. 1B). SNU387R and Malme3MR cells are cancer cells selected for resistance to celastrol. HepG2, Hep3B and SUN387R showed lower expression of HDAC3 than taxol-sensitive SNU387 cells (Fig. 1C). Furthermore, WM266-4 and Malme3MR showed lower expression of HDAC3 than taxol-sensitive Malme3M cells (Fig. 1C). The expression of HDAC3 in these cancer cell lines showed correlation with the expression of p53 (Fig. 1C). HDAC3 expression was inversely related to HDAC2 expression (Fig. 1C). SNU387 and Malme3M cells showed higher caspase-3 activity than the respective controls (Fig. 1D). This finding suggested that sensitivity of SNU387 and Malme3M cells to taxol results from caspase-3 activation. HDAC3 expression level is correlated with HDAC3 activity, similar to HDAC2 (Fig. 1E). The expression level of HDAC3 and HDAC2 showed correlation with their activity in melanoma cells employed in this study (data not shown). These results suggested that the expression level of HDAC3 might determine the response to microtubule-targeting drugs.
Fig. 1.
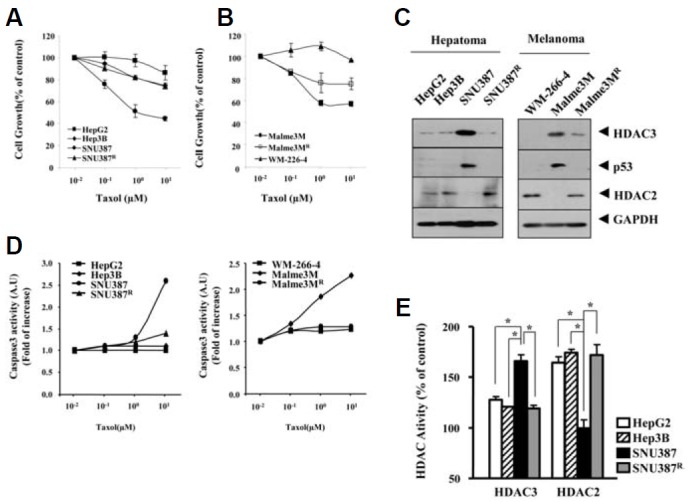
Low expression of HDAC3 is correlated with resistance to taxol. (A) The indicated hepatoma cell line was treated with various concentrations of taxol for 48 h, followed by MTT assays. Hep3B, SNU387 and SNU387R are the hepatocellular carcinoma cell lines, and HepG2 is a hepatoma cell line. The numbers are average of three independent experiments. Each experiment consists of triplicate measurement. (B) The indicated hepatoma cell line was treated with various concentrations of taxol for 48 h, followed by MTT assays. Each value represents an average obtained from 3 independent experiments. Data is expressed as a mean ± SD. (C) Cell lysates from the indicated cancer cell line were subjected to Western blot analysis. The representative figures are provided from three independent experiments. (D) The indicated hepatoma cell line was treated with various concentrations of taxol for 48 h, followed by MTT assays. (E) Cell lysates isolated from the indicated cancer cell line were subjected to HDAC activity assays as described. *p < 0.05.
HDAC3 confers sensitivity to microtubule-targeting drugs
Because the down-regulation of HDAC 3was correlated with resistance to taxol, we hypothesized that over-expression of HDAC3 confers sensitivity to microtubule-targeting drugs. In this study, we employed catalytically inactive mutant HDAC3S424A to determine whether HDAC3 activity is necessary to confer sensitivity to microtubule-targeting drugs. Ser424 is a non-conserved residue among class I HDACs. Ser 424 is the the protein kinase CK2 phosphoacceptor site in HDAC3 (Zhang et al., 2005). HDAC3Ser424 lacks histone deacetylase activity (Zhang et al., 2005). Wild type HDAC3, but not HDAC3S424A, enhanced the sensitivity of HepG2, Hep3B and WM-266-4 cells (Figs. 2A, 2C, and 2E) to microtubule-targeting drugs such as celsatrol, taxol and vinblastine. The enhanced sensitivity was accompanied by enhanced cleavages of PARP and caspase-3 in these cancer cells (Figs. 2B, 2D, and 2F). These results suggested that HDAC3 activity is necessary for conferring sensitivity to microtubule-targeting drugs. We established cancer cell lines that stably express anti-sense HDAC3 (SNU387-As-HDAC3, Mame3MAs-HDAC3) to further confirm the role of HDAC3. SNU387-As-HDAC3 cells and Malme3M-As-HDAC3 cells showed higher resistance to microtubule-targeting drugs than the respective controls (Table 1). They also showed increased expression of MDR1 (Figs. 3A and 3B). Wild type HDAC3, but not HDAC3Ser424A, enhanced cleavages of PARP and caspase-3 in SNU387-As-HDAC3 and Malme3M-As-HDAC3 in response to microtubule-targeting drugs (Figs. 3C and 3D). Wild type HDAC3, but not HDAC3S424A, enhanced the sensitivity of SNU387-As-HDAC3 cells and Malme3M-As-HDAC3 cells to microtubule-targeting drugs (Table 1). This result confirmed that resistance to microtubule-targeting drugs results from the down-regulation of HDAC3. Taken together, these results suggested that HDAC3 regulates response to microtubule-targeting drugs.
Fig. 2.
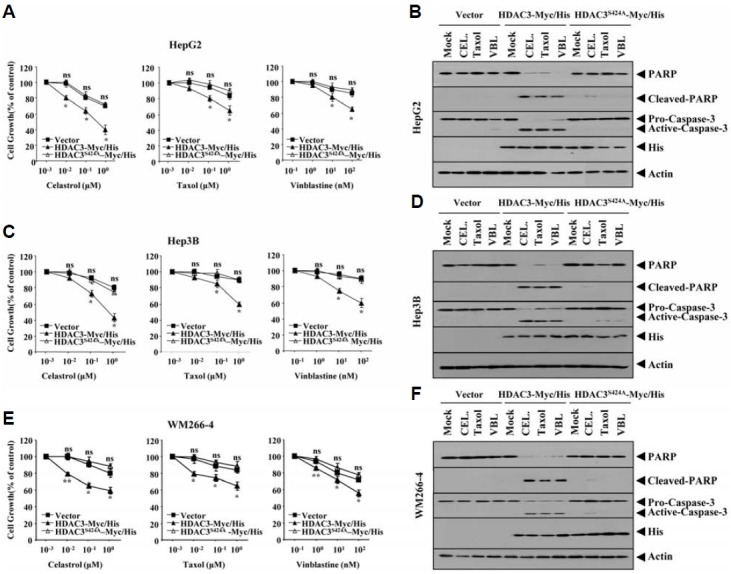
HDAC3 confers sensitivity to celastrol, taxol and vinblastine in cancer cell lines that express low levels of HDAC3. Each indicated drug-resistant cancer cell line was transiently transfected with control vector (1 μg), HDAC3-His (1 μg) or HDAC3 (S424A)-His (1 μg). The next day, cells were treated with or without various concentrations of the indicated drugs for 24 h. MTT assays were performed (A, C, and E). *p < 0.005; **p < 0.0005. P value was determined in comparison with value obtained from HepG2, Hep3B or WM-266-4 cells transfected with control vector. NS denotes not significant. Each indicated drug-resistant cancer cell line was transiently transfected with control vector, HDAC3-Myc/His or HDAC3S424A-Myc/His. The next day, cells were treated with the indicated drugs (1 μM for celastrol and taxol; 100 nM for vinblastine) for 24 h, followed by Western blot analysis (B, D, and F). VBL denotes vinblastine and CEL denotes celastrol.
Table 1.
Drug-sensitivity and relative resistance of SNU387-As-HDAC3 and Malme3M-A-HDAC3 cell lines transiently transfected with wild type or mutant HDAC3. To determine IC50 values, SNU387 or Malme3M cells were treated with or without various concentrations of the indicated drugs for 48 h. To determine the effect of wild type or mutant HDAC3 on IC50 values, SNU387-As-HDAC3 or Malme3M-As-HDAC3 cells were transiently transfected with the indicated construct. At 24 h after transfection, cells were treated with or without the indicated drug at various concentrations for 48 h. MTT assays were performed.
| Drug IC50a (RF) | |||||
|---|---|---|---|---|---|
| Celastrol (μM) | Taxol (μM) | Vinblastine (nM) | |||
| SNU387 | 0.84 ± 0.039 | 0.82 ± 0.184 | 3.47 ± 0.102 | ||
| SNU387-AS-HDAC3 | |||||
| Vector | 2.49 ± 0.020 (2.9c) | 2.75 ± 0.001 (3.3) | 10.21 ± 0.021 (2.9) | ||
| HDAC3-His | 1.08 ± 0.016 (1.2) | 1.03 ± 0.030 (1.3) | 4.27 ± 0.240 (1.2) | ||
| HDAC3S424A-His | 2.11 ± 0.031 (2.5) | 2.38 ± 0.029 (3.0) | 8.89 ± 0.207 (2.6) | ||
| Malme3M | 0.60 ± 0.043b | 0.50 ± 0.070 | 8.00 ±.230 | ||
| Malme3M-AS-HDAC3 | |||||
| Vector | 1.89 ± 0.159 (3.1c) | 1.65 ± 0.205 (3.3) | 17.04 ± 0.121 (2.1) | ||
| HDAC3-His | 0.78 ± 0.019 (1.3) | 0.63 ± 0.003 (1.2) | 8.54 ± 0.020 (1.1) | ||
| HDAC3S424A-His | 1.75 ± 0.250 (2.9) | 1.35 ± 0.107 (2.7) | 16.09 ± 0.153 (2.0) | ||
IC50, the concentration of drug required to inhibit cell growth by 50%.
Mean ± s.d. of at least 3 independent experiments.
RF, resistance factor (IC50 in SNU387-As-HDAC3 or Malme3M-As-HDAC3 transfected with control vector /IC50 in SNU387 or Malme3M cell line).
Fig. 3.
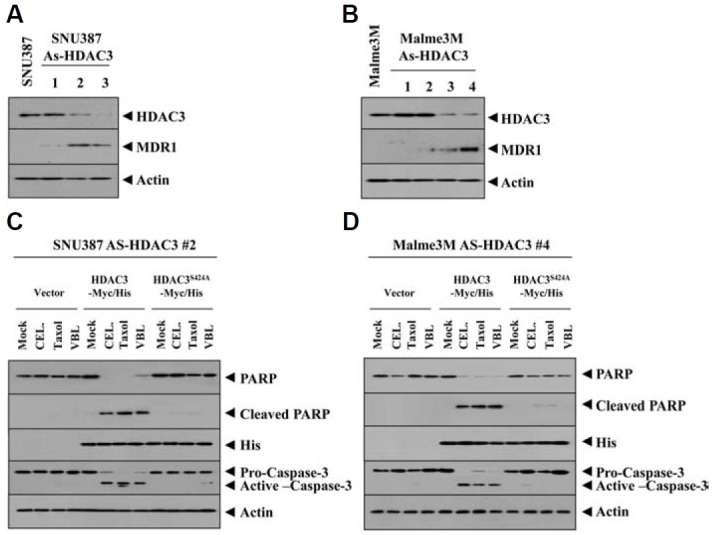
Wild type, but not mutant HDAC3, confers sensitivity to microtubule-disrupting drugs in cancer cell lines that stably express antisense-HDAC3. Cell lysates from the indicated cell line were subjected to Western blot (A, B). (C) SNU387 cells that stably express antsense-HDAC3 (SNU387-As-HDAC3) were transiently transfected with control vector, HDAC3-Myc/His or HDAC3S424A-Myc/His. At 24 h after transfection, cells were treated with or without celastrol (1 μM), taxol (1 μM) or vinblastine (100 nM) for 24 h, followed by Western blot analysis. (D) The same as C except that Malme3M-As-HDAC3 cell line was employed.
HDAC3 directly regulates the expression of tubulin β3
Over-expression of tubulin β3 is involved in resistance to microtubule-targeting drugs (Kavallaris et al., 1997). We hypothesized that HDAC3 would be a negative regulator of tubulin β3. SNU387R and Malme3MR cells showed higher expression levels of tubulin β3 than SNU387 and Malme3M cells (Fig. 4A). HDAC6 interacts with tubulin β3 and causes deacetylation (Zhang et al., 2003). HDAC6 showed an interaction with tubulin β3 in SNU387R and Malme3MR cells (Fig. 4A). SNU387-AS-HDAC3 and Malme3M-AS-HDAC3 cells stably expressing anti sense HDAC3 showed increased expression of HDAC6 and tubulin β3 (Fig. 4B), suggesting that HDAC3 may act as a negative regulator of HDAC6 and tubulin β3. Wild type HDAC3, but not mutant HDAC3 (S424A), decreased the expression of HDAC6 and tubulin β3 and prevented an interaction between HDAC6 and tubulin β3 (Fig. 4C). Celastrol and taxol led to the decreased expression of HDAC3 while increasing the expression of tubulin β3 (Fig. 4D), suggesting that HDAC3 and tubulin β3 are involved in anti-cancer drug-resistance. The decreased expression of HDAC3 preceded the increased expression of tubulin β3 by these anti cancer drugs (Fig. 4D), suggesting that HDAC3 functions upstream of tubulin β3. Tubulin β3 promoter contains putative binding sites for various transcription regulators such as DNMT1, Snail, HDAC2, AP1 and SP1 (Fig. 4E). ChIP assays showed the binding of HDAC3 to the promoter sequences of tubulin β3 (Fig. 4F). The down-regulation of tubulin β3 increased sensitivity to microtubule-targeting drugs via apoptosis (Figs. 5A and 5B). The down-regulation of HDAC6 decreased the expression of tubulin β3 and MDR1, but not HDAC3 in SNU387R and Malme3MR cells (Fig. 5C). The down-regulation of tubulin β3 did not affect the expression of HDAC6 or MDR1 (Fig. 5C). Taken together, these results suggested that tubulin β3 serves as an independent target of HDAC3 and HDAC6.
Fig. 4.
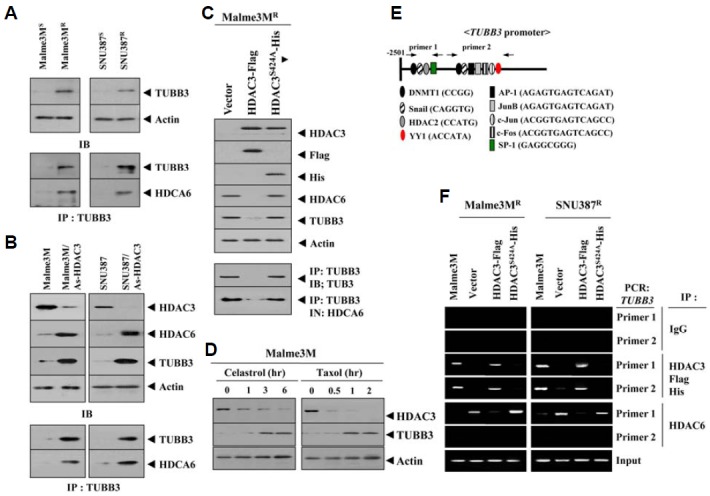
HDAC3 regulates expression of tubulin β3 and the interaction between HDAC6 and tubulin β3. (A) Cell lysates from each cell line were immunoprecipitated with the indicated antibody (2 μg/ml), followed by Western blot (lower panel). Cell lysates were also subjected to Western blot (upper panel). (B) Cell lysates of the indicated cell line were immunoprecipitated with the indicated antibody (2 μg/ml), followed by Western blot (lower panel). Cell lysates were also subjected to Western blot (upper panel). (C) At 48 h after transfection with the indicated construct, cell lysates were immunoprecipitated with the indicated antibody (2 μg/ml), followed by Western blot (lower panel). Cell lysate were also subjected to Western blot (upper panel). (D) Malme3M cells were treated with celastrol (1 μM) or taxol (1 μM) for various time intervals. Cell lysates prepared at each time point were subjected to Western blot analysis. (E) Shows the proximal promoter sequences of tubulin β3. (F) At 48 h after transfection with the indicated construct, ChIP assays were performed. Cell lysates prepared from untransfected Malme3M or SNU387 cells were also subjected to ChIP assays.
Fig. 5.
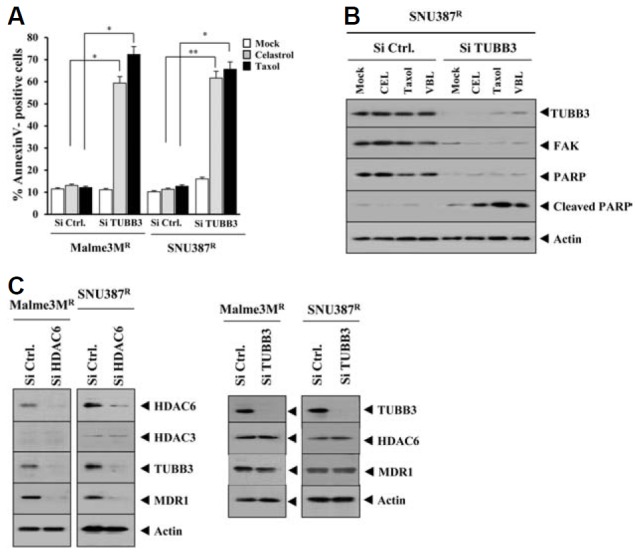
Tubulin β3 acts downstream of HDAC6, and the down-regulation of tubulin β3 enhances sensitivity to microtubule-targeting drugs. (A) The indicated cell line was transfected with the indicated siRNA (10 nM each). The next day, cells were treated with various concentrations of the indicated drug for 24 h, followed by annexin V-FITC staining. *p < 0.05; **p < 0.005. A comparison was made between SNU387R or Malme3MR cells transfected with control siRNA. (B) The same as (A) except that Western blot was performed. (C) At 48 h after transfection with the indicated siRNA (10 nM each), Western blot was performed.
HDAC3 shows binding to the promoter sequences of HDAC6
Because HDAC3 exerted a negative regulation on the expression of HDAC6 (Fig. 4B), we examined the possibility of direct regulation of HDAC6 by HDAC3. HDAC6 promoter contains putative binding sites for HDAC2, DNMT1, YY1, and Snail (Fig.6A). SNU387R and Malme3MR cells showed higher expression levels of HDAC1, -2 and -6 than their respective controls (Fig. 6B). SNU387R and Malme3MR cells also showed higher expression level of YY1 (Fig. 6B). YY1 interacts with histone acetyltransferases p300 and histone deacetylase 1 (HDAC1), HDAC2, and HDAC3 (Yao et al., 2001). The activity of YY1 is regulated through acetylation by p300 and deacetylation by HDACs. In other words, YYI is active in its deacetylated state. ChIP assay showed the binding of HDAC1 and -2 to the promoter sequences of HDAC6 (Fig. 6C). YY1 may recruit HDAC1 and/or HDAC2 to the promoter sequences of HDAC6. HDAC3 did not show binding to the site 1 of HDAC6 promoter (data not shown). We therefore focused on ChIP assays employing sites 2 and 3 of the HDAC6 promoter sequences. Wild type HDAC3, but not mutant HDAC3 (HDAC3 S424A), showed binding to HDAC6 promoter sequences in drug-sensitive Malme3M cells (Fig. 6C). This suggested the direct involvement of HDAC3 in the regulation of HDAC6 expression. HDAC3 exerted negative effects on the binding of HDAC1 and -2 to the promoter sequences of HDAC6 (Fig. 6C). It is probable that HDAC1 and -2 may be involved in resistance to microtubule-targeting drugs. SNU387R cells showed higher expression level of Ac-H3K9/14 and Ac-H4 K16 (data not shown). HDAC3 exerted a negative effect on the binding of Ac-H3K9/14 and Ac-H4 K16 to the promoter sequences of HDAC6 (Fig. 6C). In addition, YY1 showed binding to the promoter sequences of HDAC6 that was prevented by HDAC3 (Fig. 6C). YY1 possibly interacts with HDAC1 and/or HDAC2, and HDAC3 may prevent this interaction by negatively regulating expression of HDAC1 and/or HDAC2. The down- regulation of HDAC6 did not affect the expression of HDAC1, HDAC3 or YY1 (Fig. 6D). This indicated that HDAC1, HDAC3 and YY1 function upstream of HDAC6. The down- regulation of HDAC6 decreased the expression of MDR1 and tubulin β3 (Fig. 6D). The down-regulation of HDAC6 enhanced sensitivity to taxol (Fig. 6E). The down-regulation of HDAC6 enhanced sensitivity to celastrol (data not shown). Taken together, these results showed the regulatory role of HDAC3 in the expression of HDAC6. These results also showed that HDAC6 is a downstream target of HDAC3 and functions upstream of tubulin β3 and MDR1.
Fig. 6.
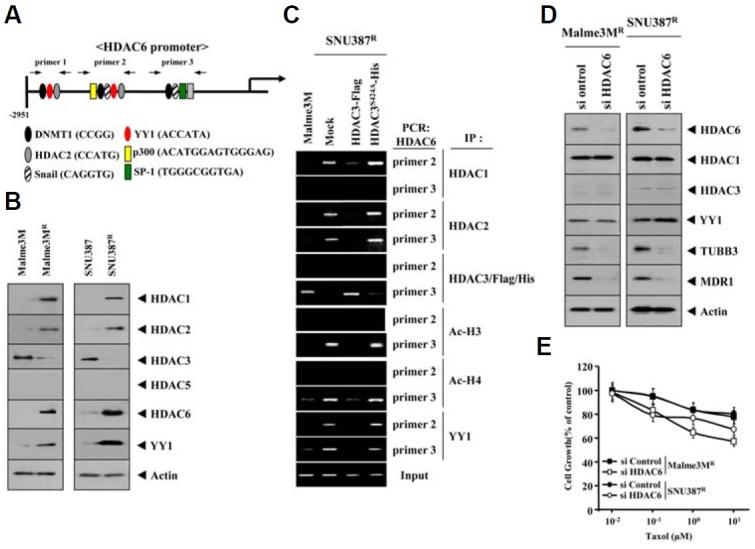
HDAC3 directly regulates the expression of HDAC6 and the down-regulation of HDAC6 enhances sensitivity to taxol. (A) Shows the promoter sequences of the HDAC6. (B) Cell lysates isolated from the indicated cell line were subjected to Western blot analysis. (C) Cell lysates of the indicated cell line were immunoprecipitated with the indicated antibody (2 μg/ml), followed by ChIP assays. (D) SNU387R or Malme3MR cells were transfected with the indicated siRNA (10 nM each). At 48 h after transfection, cell lysates were prepared and subjected to Western blot analysis. (E) SNU387R or Malme3MR cells were transfected with the indicated siRNA (10 nM each). At 24 h after transfection, cells were treated with various concentrations of taxol for 24 h, followed by MTT assays.
MDR1 is necessary for resistance to microtubule-targeting drugs
Because HDAC3 regulated expression of MDR1 (Figs. 3A and 3B), we examined the role of MDR1 in resistance to microtubule-targeting drugs and the relationship between MDR1 and tubulin β3. The down-regulation of MDR1 enhanced sensitivity to microtubule-targeting drugs (Fig. 7A). The down- regulation of MDR1 enhanced the microtubule-targeting drugs induced cleavages of PARP (Fig. 7B). Over-expression of HDAC3 in Malme3MR or SNU387R cells in the presence of MDR1 knock down did not further enhance sensitivity to microtubule-targeting drugs (Fig. 7C) or affect the knock down state of MDR1 (Fig. 7D). This suggested that MDR1 serves as a downstream target of HDAC3. HDAC3 decreased the expression of tubulin β3 (Fig. 7D). Tubulin β3 mediates resistance to microtubule-targeting drugs (Cittelly et al., 2012). The down-regulation of MDR1 did not affect expression of tubulin β3 (Fig. 7D), suggesting that MDR1 and tubulin β3 may serve as independent targets of HDAC3. Taken together, these results suggested that MDR1 serves as a downstream target of HDAC3 that affects response to microtubule-targeting drugs.
Fig. 7.
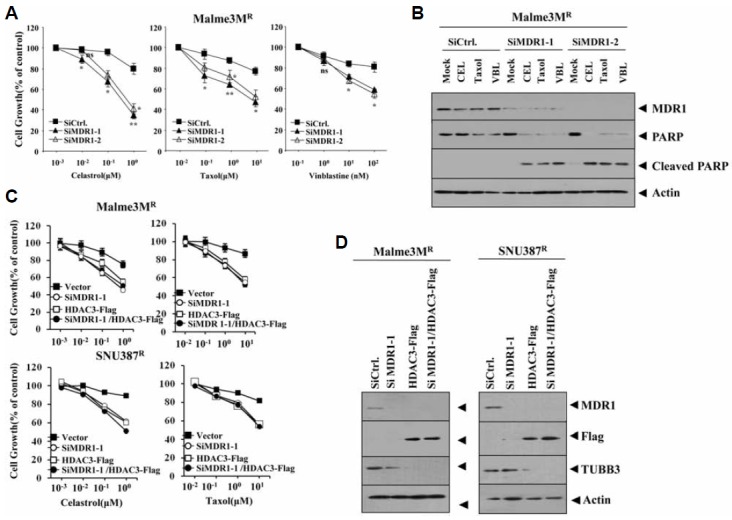
MDR1 is necessary for resistance to microtubule-disrupting drugs. (A) Malme3MR cells were transfected with indicated siRNA. The next day, cells were treated with or without various concentrations of celastrol, vinblastine or taxol for 24 h. *p < 0.05; **p < 0.005. P value was determined in comparison with value obtained from Malme3MR transfected with scrambled siRNA. (B) Malme3MR cells were transfected with indicated siRNA (10 nM each). The next day, cells were treated with celastrol (1 μM), taxol (1 μM) or vinblastine (100 nM) for 24 h, followed by Western blot analysis. (C) The indicated cell line was transfected with the indicated construct alone or in combination. The next day, cells were treated with various concentrations of cealstrol or taxol for 24 h, followed by MTT assays. (D) The indicated cell line was transfected with the indicated construct alone or in combination. Western blot analysis was performed at 48 h after transfection.
DISCUSSION
Microtubule-destabilizer-resistant cancer cell lines show an increased expression of survivin (Hei et al., 2010). Therefore SNU387R and Malme3MR cells may show the increased expression of anti-apoptotic proteins such as survivin. The down-regulation of anti-apoptotic prohibin enhances sensitivity to taxol (Patel et al., 2010). The enhanced sensitivity to microtubule-targeting drugs is related with caspase-3–depedent pathway as evidenced by cleavage of PARP (Figs. 3C and 3D). It will therefore be necessary to examine the effect of HDAC3 on caspase-independent cell death by examining the expression of anti-apoptotic proteins such as surviving and prohibin.
The MDR1 expression level is correlated with resistance to taxol and doxorubicin (Mechetner et al., 1998). It will be interesting to examine whether HDAC3 would also confer sensitivity to doxorubicin and examine the direct regulation of MDR1 by HDAC3. It will be necessary to examine the direct binding of the HDAC3 to the promoter sequences of MDR1. CDX2, a transcription factor, regulates MDR1 expression (Takakura et al., 2010) and the down-regulation of CDX2 enhances sensitivity to cisplatin, doxorubicin and 5-FU (Yan et al., 2013). It will be necessary to examine the effect of HDAC3 on the expression of CDX2.
The down-regulation of HDAC6 leads to the degradation of EGFR (Gao et al., 2010). We found an increased expression of EGFR in Mame3MR cells. In addition, Malme3MR cells show resistance to EGFR inhibitors (data not shown). The inhibition of EGFR by cetuximab enhances sensitivity to taxol (data not shown). It is probable that EGFR signaling may be responsible for the decreased expression of HDAC3 in SNU387R and Malme3MR cells. It is also reasonable that HDAC3 may exert a negative control on the expression and/or activity of EGFR in SNU837 and/or Malme3M cells.
Over-expression of tubulin β3 has been shown in paclitaxel-resistant cells (Kamath et al., 2005; Seve et al., 2010; Verdier-Pinard et al., 2003). We showed the over-expression of tubulin β3 in SNU387R and Malme3MR cells (Fig. 4A). It will be necessary to examine the role of signaling pathways such as MAPK and JNK on the expression of HDAC3 and tubulin β3. Tubulin β3 promoter contains putative binding sites for various transcription factors such as AP1, SP1, Snail and YY1 (Fig. 4E). For example, the expression of SP1 is increased in SNU387R and Malme3MR cells (data not shown). It is lijkely that the down-regulation of HDAC3 induces expression SP1, which in turn binds to the promoter sequences of tubulin β3 and induces the expression of tubulin β3 in SNU387R and Malme3MR cells.
Mutations in tubulin β1 confer resistance to taxol (Yin et al., 2010). It would be interesting to examine possible mutations in tubulin β1 and tubulin β1 expression level in SNU387R and Malme3MR cells. The selective inhibition of HDAC6 by Vorinostat leads to the enhanced sensitivity to taxol (Owonikoko et al., 2010). Furthermore, the down-regulation of HDAC6 enhanced sensitivity to taxol (Fig. 6E). Depletion of HDAC6 enhances cisplatin-induced cytotoxicity by activating the ATR/Chk1 pathway (Wang et al., 2012). These reports indicated the role of HDAC6 in anti-cancer drug-resistance. Because HDAC3 regulates the expression of HDAC6, it will be interesting to examine the effect of HDAC3 on the ATR/Chk1 pathway. Tubacin, a selective inhibitor of HDAC6, enhances DNA damage induced by etoposide or SAHA as indicated by increased accumulation of γH2AX and activation of the checkpoint kinase Chk2 (Namdar et al., 2010). It will also be interesting to examine the effect of HDAC3 in response to DNA damaging drugs.
HDAC6 is necessary for angiogenesis via its interaction with and deacetylation of the actin-remodeling protein cortactin, in endothelial cells (Kaluza et al., 2011). Because HDAC3 regulates the expression of HDAC6, it would be interesting to examine the effect of HDAC3 on the acetylation of cortactin. It will be necessary to examine the effect of HDAC3 on the expression of various angiogenic factor(s). Because HDAC3 confers sensitivity to microtubule-targeting drugs, it possibly exerts a negative effect on tumor-induced angiogenesis. VEGF signaling induces anti cancer drug-resistance by upregulating MDR1 expression (Akiyama et al., 2012). Because HDAC3 negatively regulates MDR1 expression, HDAC3 likely regulates the expression of VEGF.
MicroRNAs (miRNAs) are non-coding RNA molecules that mediate posttranscriptional gene regulation and are strongly implicated in cellular processes such as cell proliferation, carcinogenesis, cell survival and apoptosis. MiRNA-binding factor Lin-28 mediates taxol-resistance in breast cancer cells (Lv et al., 2012). miR-148a attenuates taxol-resistance by regulating MSK1 expression (Fujita et al., 2010). miR-337-3p modulates taxol-sensitivity by targeting STAT3 and RAP1A (Du et al., 2012). These reports suggest the role of miRNAs in taxol-resistance. In this study, we identified miRNAs that were down-regulated in Malme3MR cells. These include miR-138, -189, -211, - 324-3p and -335 (data not shown). It is necessary to further examine the effect of these miRNAs on HDAC3 expression and response to microtubule-targeting drugs. In conclusion, we showed a novel role of HDAC3 in determining response to microtubule-targeting drugs. We showed that HDAC3-HDAC6-tubulin β3 axis determines response to microtubule-targeting drugs.
Acknowledgments
This work was supported by National Research Foundation Grants (2014R1A2A2A01002448, 2015R1A1A3A04001339), a grant from the BK21 Plus Program, and by National R&D Program for Cancer Control, Ministry for Health and Welfare, Republic of Korea Grant 1320160.
REFERENCES
- Akiyama K., Ohga N., Hida Y., Kawamoto T., Sadamoto Y., Ishikawa S., Maishi N., Akino T., Kondoh M., Matsuda A., et al. Tumor endothelial cells acquire drug resistance by MDR1 up-regulation via VEGF signaling in tumor microenvironment. Am. J. Pathol. 2012;180:1283–1293. doi: 10.1016/j.ajpath.2011.11.029. [DOI] [PubMed] [Google Scholar]
- Bonet C., Giuliano S., Ohanna M., Bille K., Allegra M., Lacour J.P., Bahadoran P., Rocchi S., Ballotti R., Bertolotto C. Aurora B is regulated by the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling pathway and is a valuable potential target in melanoma cells. J. Biol. Chem. 2012;287:29887–29898. doi: 10.1074/jbc.M112.371682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae S., Kim Y.B., Lee J.S., Cho H. Resistance to paclitaxel in hepatoma cells is related to static JNK activation and prohibition into entry of mitosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2012;302:1016–1024. doi: 10.1152/ajpgi.00449.2011. [DOI] [PubMed] [Google Scholar]
- Chen L.F., Fischle W., Verdin E., Greene W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- Cittelly D.M., Dimitrova I., Howe E.N., Cochrane D.R., Jean A., Spoelstra N.S., Post M.D., Lu X., Broaddus R.R., Spillman M.A., et al. Restoration of miR-200c to ovarian cancer reduces tumor burden and increases sensitivity to paclitaxel. Mol. Cancer Ther. 2012;11:2556–2565. doi: 10.1158/1535-7163.MCT-12-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L., Subauste M.C., DeSevo C., Zhao Z., Baker M., Borkowski R., Schageman J.J., Greer R., Yang C.R., Suraokar M., et al. miR-337-3p and its targets STAT3 and RAP1A modulate taxane sensitivity in non-small cell lung cancers. PLoS One. 2012;7:e39167. doi: 10.1371/journal.pone.0039167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadri-Moskwik M., Weiderhold K.N., Deeraksa A., Chuang C., Pan J., Lin S.H., Yu-Lee L.Y. Aurora B is regulated by acetylation/deacetylation during mitosis in prostate cancer cells. FASEB J. 2012;26:4057–4067. doi: 10.1096/fj.12-206656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y., Kojima K., Ohhashi R., Hamada N., Nozawa Y., Kitamoto A., Sato A., Kondo S., Kojima T., Deguchi T., et al. MiR-148a attenuates paclitaxel resistance of hormone-refractory, drug-resistant prostate cancer PC3 cells by regulating MSK1 expression. J. Biol. Chem. 2010;285:19076–19084. doi: 10.1074/jbc.M109.079525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.S., Hubbert C.C., Yao T.P. The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J. Biol. Chem. 2010;285:11219–11226. doi: 10.1074/jbc.M109.042754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hei C., Cheung A., Wu S.Y., Lee T.R., Chang C.Y., Wu J.S., Hsieh H.P., Chang J.Y. Cancer cells acquire mitotic drug resistance properties through beta I-tubulin mutations and alterations in the expression of beta-tubulin isotypes. PLoS One. 2010;5:e12564. doi: 10.1371/journal.pone.0012564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X.F., Yao T.P. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Ishii S., Kurasawa Y., Wong J., Yu-Lee L.Y. Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore-microtubule attachment. Proc. Natl. Acad. Sci. USA. 2008;105:4179–4184. doi: 10.1073/pnas.0710140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaluza D., Kroll J., Gesierich S., Yao T.P., Boon R.A., Hergenreider E., Tjwa M., Rössig L., Seto E., Augustin H.G., et al. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 2011;30:4142–4156. doi: 10.1038/emboj.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath K., Wilson L., Cabral F., Jordan M.A. BetaIII-tubulin induces paclitaxel resistance in association with reduced effects on microtubule dynamic instability. J. Biol. Chem. 2005;280:12902–12907. doi: 10.1074/jbc.M414477200. [DOI] [PubMed] [Google Scholar]
- Kavallaris M., Kuo D.Y., Burkhart C.A., Regl D.L., Norris M.D., Haber M., Horwitz S.B. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J. Clin. Invest. 1997;100:1282–1293. doi: 10.1172/JCI119642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.C., Choi K.C., Choi H.K., Kang H.B., Kim M.J., Lee Y.H., Lee O.H., Lee J., Kim Y.J., Jun W., et al. HDAC3 selectively represses CREB3-mediated transcription and migration of metastatic breast cancer cells. Cell. Mol. Life Sci. 2010a;67:3499–3510. doi: 10.1007/s00018-010-0388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Park H., Park D., Lee Y.S., Choe J., Hahn J.H., Lee H., Kim Y.M., Jeoung D. Cancer/testis antigen CAGE exerts negative regulation on p53 expression through HDAC2 and confers resistance to anti-cancer drugs. J. Biol. Chem. 2010b;285:25957–25968. doi: 10.1074/jbc.M109.095950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levallet G., Bergot E., Antoine M., Creveuil C., Santos A.O., Beau-Faller M., de Fraipont F., Brambilla E., Levallet J., Morin F., et al. High TUBB3 expression, an independent prognostic marker in patients with early non-small cell lung cancer treated by preoperative chemotherapy, is regulated by K-Ras signaling pathway. Mol. Cancer Ther. 2012;11:1203–1213. doi: 10.1158/1535-7163.MCT-11-0899. [DOI] [PubMed] [Google Scholar]
- Li J., Wang J., Wang J., Nawaz Z., Liu J.M., Qin J., Wong J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000;19:4342–4350. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv K., Liu L., Wang L., Yu J., Liu X., Cheng Y., Dong M., Teng R., Wu L., Fu P., et al. Lin28 mediates paclitaxel resistance by modulating p21, Rb and Let-7a miRNA in breast cancer cells. PLoS One. 2012;7:e40008. doi: 10.1371/journal.pone.0040008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlknecht U., Emiliani S., Najfeld V., Young S, Verdin E. Genomic organization and chromosomal localization of the human histone deacetylase 3 gene. Genomics. 1999;56:197–202. doi: 10.1006/geno.1998.5645. [DOI] [PubMed] [Google Scholar]
- Mahlknecht U., Will J., Varin A., Hoelzer D., Herbein G. Histone deacetylase 3, a class I histone deacetylase, suppresses MAPK11-mediated activating transcription factor-2 activation and represses TNF gene expression. J. Immunol. 2004;173:3979–3990. doi: 10.4049/jimmunol.173.6.3979. [DOI] [PubMed] [Google Scholar]
- Mechetner E., Kyshtoobayeva A., Zonis S., Kim H., Stroup R., Garcia R., Parker R.J., Fruehauf J.P. Levels of multidrug resistance (MDR1) P-glycoprotein expression by human breast cancer correlate with in vitro resistance to taxol and doxorubicin. Clin. Cancer Res. 1998;4:389–398. [PubMed] [Google Scholar]
- Namdar M., Perez G., Ngo L., Paul A., Marks P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA. 2010;107:20003–20008. doi: 10.1073/pnas.1013754107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott P.A., Chang J., Madden K., Kannan R., Muren C., Escano C., Cheng X., Shao Y., Mendoza S., Gandhi A., et al. Oblimersen in combination with temozolomide and albumin-bound paclitaxel in patients with advanced melanoma: a phase I trial. Cancer Chemother. Pharmacol. 2013;71:183–191. doi: 10.1007/s00280-012-1995-7. [DOI] [PubMed] [Google Scholar]
- Owonikoko T.K., Ramalingam S.S., Kanterewicz B., Balius T., Belani C.P., Hershberger P.A. Vorinostat increases carboplatin and paclitaxel activity in non-small cell lung cancer cells. Int. J. Cancer. 2010;126:743–755. doi: 10.1002/ijc.24759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N., Chatterjee S.K., Vrbanac V., Chung I., Mu C.J., Olsen R.R., Waghorne C., Zetter B.R. Rescue of paclitaxel sensitivity by repression of Prohibitin1 in drug-resistant cancer cells. Proc. Natl. Acad. Sci. USA. 2010;107:2503–2508. doi: 10.1073/pnas.0910649107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seve P., Reiman T., Dumontet C. The role of betaIII tubulin in predicting chemoresistance in non-small cell lung cancer. Lung Cancer. 2010;67:136–143. doi: 10.1016/j.lungcan.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Takakura Y., Hinoi T., Oue N., Sasada T., Kawaguchi Y., Okajima M., Akyol A., Fearon E.R., Yasui W., Ohdan H. CDX2 regulates Multidrug Resistance 1 gene expression in malignant intestinal epithelium. Cancer Res. 2010;70:6767–6778. doi: 10.1158/0008-5472.CAN-09-4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdier-Pinard P., Wang F., Martello L., Burd B., Orr G.A., Horwitz S.B. Analysis of tubulin isotypes and mutations from taxol-resistant cells by combined isoelectrofocusing and mass spectrometry. Biochemistry. 2003;42:5349–5357. doi: 10.1021/bi027293o. [DOI] [PubMed] [Google Scholar]
- Wang L., Xiang S., Williams K.A., Dong H., Bai W., Nicosia S.V., Khochbin S., Bepler G., Zhang X. Depletion of HDAC6 enhances cisplatin-induced DNA damage and apoptosis in non-small cell lung cancer cells. PLoS One. 2012;7:e44265. doi: 10.1371/journal.pone.0044265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H., Lee M.H., Zhu F., Reddy K., Peng C., Li Y., Lim do Y., Kim D.J., Li X., Kang S., et al. Identification of an Aurora kinase inhibitor specific for the Aurora B isoform. Cancer Res. 2013;73:716–724. doi: 10.1158/0008-5472.CAN-12-2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R., Sato N., Yanai K., Akiyoshi T., Nagai S., Wada J., Koga K., Mibu R., Nakamura M., Katano M. Enhancement of paclitaxel-induced apoptosis by inhibition of mitogen-activated protein kinase pathway in colon cancer cells. Anti-cancer Res. 2009;29:261–270. [PubMed] [Google Scholar]
- Xu R., Nakano K., Iwasaki H., Kumagai M., Wakabayashi R., Yamasaki A., Suzuki H., Mibu R., Onishi H., Katano M. Dual blockade of phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways overcomes paclitaxel-resistance in colorectal cancer. Cancer Lett. 2011;306:151–160. doi: 10.1016/j.canlet.2011.02.042. [DOI] [PubMed] [Google Scholar]
- Yan L.H., Wang X.T., Yang J., Lian C., Kong F.B., Wei W.Y., Luo W., Xiao Q., Xie Y.B. Reversal of multidrug resistance in gastric cancer cells by CDX2 downregulation. World J. Gastroenterol. 2013;19:4155–4165. doi: 10.3748/wjg.v19.i26.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y.L., Yang W.M., Seto E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol. Cell Biol. 2001;21:5979–5991. doi: 10.1128/MCB.21.17.5979-5991.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S., Bhattacharya R., Cabral F. Human mutations that confer paclitaxel resistance. Mol. Cancer Ther. 2010;9:327. doi: 10.1158/1535-7163.MCT-09-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Kalkum M., Chait B.T., Roeder R.G. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol. Cell. 2002;9:611–623. doi: 10.1016/s1097-2765(02)00468-9. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Li N., Caron C., Matthias G., Hess D., Khochbin S., Matthias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Ozawa Y., Lee H., Wen Y.D., Tan T.H., Wadzinski B.E., Seto E. Histone deacetylase 3 (HDAC3) activity is regulated by interaction with protein serine/threonine phosphatase 4. Genes Dev. 2005;19:827–839. doi: 10.1101/gad.1286005. [DOI] [PMC free article] [PubMed] [Google Scholar]