

Significance
Pathologic retinal neovascularization commonly causes blindness. Retinoic-acid-receptor–related orphan receptor alpha (RORα), a lipid-sensing nuclear receptor, is genetically associated with the risk of developing neovascular eye disease in humans. We demonstrate that RORα expression was highly increased in a mouse model of oxygen-induced proliferative retinopathy with pathologic neovessels. Both genetic deficiency and pharmacologic inhibition of RORα suppressed pathologic retinal neovascularization in mice with dampened inflammation. RORα transcriptionally regulated suppressors of cytokine signaling 3 (SOCS3), a negative mediator of macrophage function and inflammation. Suppression of SOCS3 attenuated the protective effects of RORα inhibition in retinopathy. Our data demonstrate an important role of RORα in controlling pathologic retinal neovascularization and suggest that RORα may represent a druggable target for treating ocular neovascularization.
Keywords: RORα, neovascularization, retinopathy, inflammation, SOCS3
Abstract
Pathologic ocular angiogenesis is a leading cause of blindness, influenced by both dysregulated lipid metabolism and inflammation. Retinoic-acid-receptor–related orphan receptor alpha (RORα) is a lipid-sensing nuclear receptor with diverse biologic function including regulation of lipid metabolism and inflammation; however, its role in pathologic retinal angiogenesis remains poorly understood. Using a mouse model of oxygen-induced proliferative retinopathy, we showed that RORα expression was significantly increased and genetic deficiency of RORα substantially suppressed pathologic retinal neovascularization. Loss of RORα led to decreased levels of proinflammatory cytokines and increased levels of antiinflammatory cytokines in retinopathy. RORα directly suppressed the gene transcription of suppressors of cytokine signaling 3 (SOCS3), a critical negative regulator of inflammation. Inhibition of SOCS3 abolished the antiinflammatory and vasoprotective effects of RORα deficiency in vitro and in vivo. Moreover, treatment with a RORα inverse agonist SR1001 effectively protected against pathologic neovascularization in both oxygen-induced retinopathy and another angiogenic model of very-low–density lipoprotein receptor (Vldlr)-deficient (Vldlr−/−) mice with spontaneous subretinal neovascularization, whereas a RORα agonist worsened oxygen-induced retinopathy. Our data demonstrate that RORα is a novel regulator of pathologic retinal neovascularization, and RORα inhibition may represent a new way to treat ocular neovascularization.
Pathologic proliferation of blood vessels commonly causes blindness in all age groups, including retinopathy of prematurity in children, proliferative diabetic retinopathy in working-age adults, and neovascular age-related macular degeneration (AMD) in the elderly (1). Development of pathologic ocular angiogenesis is linked with dysregulation of both lipid metabolism (2, 3) and altered inflammation/macrophage function (4). Identification of key controlling mechanisms by which lipids and their metabolites modulate retinal tissue toward (or away from) a proinflammatory, proangiogenic state is critical for developing potential treatments. One potential pivotal regulator of lipid-mediated inflammatory processes is retinoic-acid-receptor–related orphan receptor alpha (RORα), a lipid-sensing nuclear receptor that may modify inflammation (5). Genetic variations in RORα are associated with increased risk of developing neovascular AMD (6–8). However, the functional role of RORα in pathologic retinal angiogenesis is poorly understood.
RORα controls diverse biological processes including immunity, cerebellum development, and circadian rhythm (9). As a ligand-dependent transcription factor, RORα is a suggested receptor for cholesterol, cholesterol sulfate, and other cholesterol-derived oxysterols (10). RORα regulates lipid metabolism including cholesterol and lipoprotein levels, and hence is implicated in atherosclerosis and vascular contractility control (11). RORα is also important for regulating immunity and inflammatory response in allergic inflammation (12) and autoimmune diseases (13), as well as for production of inflammatory cytokines (9, 14–16). RORα controls its target gene expression through binding as a monomer to a core DNA consensus sequence termed RORα responsive element (RORE), comprised of an AGGTCA half-site and a 5′ AT-rich extension (17, 18). Binding of RORα to RORE together with its coactivators and corepressors controls transcription of RORα target genes.
In this study, we investigated whether RORα acted at the crossroad of lipid metabolism and inflammation to control pathologic retinal angiogenesis. RORα deficiency significantly suppressed pathologic retinal angiogenesis in a mouse model of proliferative oxygen-induced retinopathy (OIR) with hypoxia-induced neovascularization (19) that mimics retinopathy of prematurity, and some aspects of proliferative diabetic retinopathy in humans. Loss of RORα resulted in an antiinflammatory retinal environment, which was mediated through its direct transcriptional control of suppressor of cytokine signaling 3 (SOCS3), a critical regulator of tissue inflammation. Suppressing SOCS3 inhibited RORα deficiency-induced inflammatory and vascular effects in vitro and in vivo. Treatment with SR1001, a synthetic small molecular inverse agonist of RORα, effectively inhibited pathologic angiogenesis in OIR and spontaneous subretinal neovascularization in very-low–density lipoprotein receptor (Vldlr)-deficient (Vldlr−/−) mice modeling neovascular AMD. Thus, inhibition of RORα, a novel regulator of pathologic ocular angiogenesis, may be a new approach to potentially treat or prevent pathologic vascular growth in eye diseases.
Results
RORα Deficiency Significantly Attenuated Pathologic Retinal Neovascularization in OIR.
To induce retinopathy, mice were exposed to 75% oxygen from postnatal day (P)7 to P12. Compared with age-matched room air controls, Rora mRNA expression was significantly down-regulated at P8, P10, and P12, during the initial oxygen-induced vessel loss phase (phase I) of OIR, and then up-regulated at P14 and P17 in the second hypoxic and proliferative phase (phase II) (Fig. 1A). Protein levels of RORα were also significantly up-regulated (approximately fivefold) in P17 OIR retinas (Fig. 1B).
Fig. 1.

RORα deficiency significantly attenuated pathologic neovascularization in OIR. (A) Rora mRNA expression in OIR retinas compared with normoxic retinas (n = 6 per group). (B) Quantification of Western blot with RORα and β-ACTIN antibodies in P17 OIR retinas compared with normoxic retinas (Norm). (n = 3). (C) Representative retinal whole mounts from OIR Sg/Sg and WT retinas stained with isolectin IB4 (red) with areas of vasoobliteration (VO) and neovascularization (NV) highlighted (white). Two selected retinal areas (white box) were enlarged to show pathologic neovessels. Quantification of pathologic NV (D) and VO (E) in OIR Sg/Sg and WT retinas was expressed as percentage of total retinal areas. n = 12–20 per group. (Scale bar, 1,000 μm.) *P < 0.05; ***P < 0.001; n.s., no significance.
RORα-deficient Sg/Sg mice have a spontaneous deletion in the Rora gene with loss of RORα activity (20). In OIR, Sg/Sg mice showed markedly decreased levels of pathologic retinal neovascularization at P17 compared with littermate wild-type (WT) controls (WT: 9.22 ± 0.32%; Sg/Sg: 5.61 ± 0.50%; n = 12–20 per group, P < 0.001; Fig. 1 C and D), with comparable vasoobliteration (P = 0.60, Fig. 1 C and E). RORα deficiency did not impact normal retinal vasculature as adult Sg/Sg retinas showed normal vascular structure and morphology as age-matched WT (SI Appendix, Fig. S1). Aortic ring explants isolated from Sg/Sg and WT mice showed similar levels of vascular sprouting (SI Appendix, Fig. S2), suggesting likely marginal effects of endothelium RORα on angiogenesis. This notion is consistent with minimal RORα staining in lectin positive retinal blood vessels (Fig. 2 and SI Appendix, Fig. S3), and strong RORα staining in some lectin positive cells surrounding blood vessels resembling microglia/macrophages morphologically (SI Appendix, Fig. S3).
Fig. 2.

Deficiency of RORα influenced retinal inflammatory cytokine expression and macrophage markers in OIR. (A) Immunohistochemical localization of RORα in macrophages/microglia in P17 WT OIR retina flat mounts, costained with macrophage/microglia markers CD11b, CX3CR1, and F4/80. Isolectin IB4 (red) was used to stain both blood vessels and macrophages/microglia. (Scale bar, 50 μm.) (B) Expression of inflammatory cytokines and macrophage markers in OIR Sg/Sg and WT retinas at P17 (n = 5 per group). Sg/Sg retinas showed decreased expression levels of Tnf, Il6, Il1b, Cxcl10, and Nos2. (C) Expression of Il10, Fizz1, Ccl26, Arg1, and Socs3 were increased in OIR Sg/Sg retinas at P17 compared with WT. **P < 0.01; ***P < 0.001.
RORα Was Localized in Retinal Macrophages/Microglia and Mediated Expression of Inflammatory Cytokines.
Retinal microglia/macrophages are important inflammatory regulators of retinal angiogenesis (21). Increased levels of proinflammatory cytokines were found in vitreous fluid of patients with proliferative retinopathy (22). We next evaluated whether RORα controls pathologic retinal neovascularization through modulation of retinal inflammation. In WT OIR retinas, RORα antibody strongly colocalized with selected population of retinal macrophages/microglia positive for CD11b, CX3CR1, or F4/80 (Fig. 2A). Moreover, RORα deficiency in Sg/Sg retinas significantly suppressed expression of proinflammatory cytokines: interleukin-6 (Il6), interleukin-1 beta (Il1b), and tumor necrosis factor-alpha (Tnf) ranging from 2- to 3-fold (Fig. 2B) and increased expression of antiinflammatory cytokines Il10 (Fig. 2C). In addition, Sg/Sg retinas demonstrated significant decrease of proinflammatory macrophage markers iNOS (encoded by Nos2) and Cxcl10 by 10- to 15-fold compared with WT (Fig. 2B), and reciprocally increased levels of antiinflammatory macrophage markers Fizz1, Ccl26, and Arg1 (∼2- to 10-fold up-regulation) (Fig. 2C), reflecting an overall shift toward antiinflammatory state. Socs3, an inducible negative regulator of inflammation (23, 24), was also significantly increased in Sg/Sg OIR retinas (Fig. 2C), yet vascular endothelial growth factor A (Vegfa) was not significantly changed (SI Appendix, Fig. S4). Together these results indicate that RORα deficiency promotes retinal inflammation state toward an antiinflammatory environment in OIR.
RORα Suppressed Socs3 Transcription Through Binding to Its RORE Site.
RORα controls target gene transcription by binding to RORE sites (9). The proximal promoter sequences of those genes significantly regulated in Sg/Sg OIR retinas all contain at least one potential RORE binding site. Chromatin immunoprecipitation (ChIP) assay was performed with RORα antibody followed by qPCR to quantitate RORα binding to candidate DNA regions in P17 OIR retinas. Among the 10 analyzed genes, binding to Socs3 showed the most significant enrichment compared with IgG control (Fig. 3A). The Socs3 promoter contains four potential RORE sites (SI Appendix, Fig. S5). Direct binding of RORα to the third site, conserved between human and mouse, was the most strongly enhanced in the ChIP-qPCR assay (P = 0.0001, Fig. 3A), with stronger enrichment than the positive control Opn1mw, a known RORα target (25). There was no significant enrichment of other Socs3 RORE sites. In addition, other genes analyzed showed little enrichment except for Arg1 and Il10, with enrichment levels comparable to Opn1mw (Fig. 3A). Together these data indicate that a Socs3 promoter region is directly recognized and bound by RORα.
Fig. 3.

Socs3 was a direct transcriptional target of RORα. (A) ChIP assays were performed with P17 WT OIR retinas. DNA fragments bound to immunoprecipitated RORα were quantified with qPCR with primers flanking potential RORE sites for genes: Il1b, Il6, Tnf, Cxcl10, Nos2, Socs3, Arg1, Il10, Fizz1, and Ccl26. Data were normalized to IgG control. Primers of Opn1mw were used as positive control, and hemoglobin β (Hbb) as a negative control. n = 3. (B) Luciferase reporters with native (WT/Socs3) or mutated RORα (MUT/Socs3) binding sites in Socs3 promoter (covering residues −2,310 bp to −2,314 bp) were cloned and cotransfected with RORα-expressing vector. RORα dose-dependently suppressed WT/Socs3, but not MUT/Socs3 luciferase expression in pGL2 vector, reflecting the transcriptional activity of WT/Socs3 (n = 6). RE, responsive element. (C) SR1001 treatment dose-dependently promoted WT/Socs3 promoter-driven luciferase reporter activity, but not for MUT/Socs3 (n = 6). *P < 0.05; **P < 0.01; ***P < 0.001; n.s., no significance.
Next luciferase reporter assays were performed to assess the direct effect of RORα on Socs3 promoter activity. WT and mutant Socs3:Luc luciferase reporters were constructed around the native or mutated RORE site (from −2,310 bp to −2,314 bp), and cotransfected with RORα-expressing vector into HEK293T cells separately. In the WT Socs3:Luc reporter, expression of RORα significantly reduced the levels of Socs3 promoter activity (>50%) in a dose-dependent manner, as measured by firefly-Renilla luciferase activity (Fig. 3B), with no significant effect in mutant Socs3:Luc reporter activity, suggesting that binding of RORα to this specific RORE region of Socs3 is critical for its transcriptional suppression.
RORα suppression of Socs3 transcription was also validated by treatment with a synthetic inverse agonist of RORα, SR1001, which binds to the ligand-binding domain of RORα to inhibit RORα (13). The transcriptional activity of WT/Socs3:Luc reporter was significantly enhanced with increasing concentrations of SR1001, but not in MUT/Socs3:Luc reporters (Fig. 3C). Together these results indicate that RORα directly binds to this RORE site in the Socs3 promoter to repress its transcription.
RORα Modulated Macrophage Cytokine Expression Through Regulating Socs3 Expression.
To further identify the cellular source of RORα and RORα deficiency-induced Socs3 induction, macrophages/microglia were depleted in WT and Sg/Sg OIR eyes using intraviteal injection of clodronate liposome, which led to significant suppression of Rora expression in WT retinas and abolished Socs3 induction in Sg/Sg retinas (SI Appendix, Fig. S6), suggesting that RORα expression and RORα deficiency-induced SOCS3 expression largely depends on involvement of macrophages/microglia. Next, to determine if SOCS3 mediates the effect of RORα on cytokine expression from macrophages, RAW 264.7 cells were treated with lentivirus expressing Socs3 shRNA (lenti-shSocs3) and/or siRNA targeting RORα (siRora). Treatment with siRora significantly suppressed RORα protein levels and induced SOCS3 protein levels (Fig. 4 A and B). On the other hand, lenti-shSocs3 significantly suppressed SOCS3 protein level as expected without influencing RORα (Fig. 4B). Importantly, siRora treatment resulted in significant suppression of proinflammatory cytokines Tnf, Il1b, Cxcl10, and Il6 (Fig. 4 C–F), and a drastic converse increase of antiinflammatory cytokine and macrophage markers Il10 and Arg1 (Fig. 4 G and H). Knocking down Socs3 in siRora-treated cells markedly reversed the effects of RORα deficiency on inflammatory cytokines and markers (Fig. 4 C–H). These results suggest that RORα deficiency in macrophages promotes SOCS3, which then induces an antiinflammatory tissue environment.
Fig. 4.

RORα regulated inflammatory cytokine expression in macrophages through SOCS3. (A and B) Western blot images and quantification of RORα and SOCS3 protein levels in RAW 264.7 cells treated with siRora and lenti-shSocs3. (C–F) Expression levels of Tnf, Il1b, Cxcl10, and Il6 were suppressed in RAW 264.7 cells treated with siRora and reversed by additional Socs3 knockdown with lenti-shSocs3. (G and H) Arg1 and Il10 were significantly enhanced in siRora-treated RAW 264.7 cells and reversed by lenti-shSocs3 treatment. *P < 0.05; **P < 0.01; ***P < 0.001.
Macrophage RORα Regulated Aortic Ring Vascular Growth Through SOCS3.
WT or RORα-deficient Sg/Sg aortic rings showed no significant difference in sprouting (SI Appendix, Fig. S2). Next, we cocultured WT aortic rings with macrophages to evaluate the effects of modulating macrophage RORα on aortic ring sprouting. RAW 264.7 cells were pretreated with lenti-shSocs3 and siRora with respective controls, then cocultured with WT aortic rings in Transwells (Fig. 5A). RORα-deficient (siRora) RAW 264.7 cells significantly suppressed aortic ring sprouting compared with control (siCon)-treated RAW cells by ∼50% (n = 8, P < 0.05; Fig. 5B). Suppression of Socs3 in RORα-deficient (siRora/shSocs3) macrophages largely reversed the effects of macrophage RORα on aortic ring sprouting (Fig. 5B), indicating that Socs3 mediates the angiogenic effect of macrophage RORα on vascular growth.
Fig. 5.
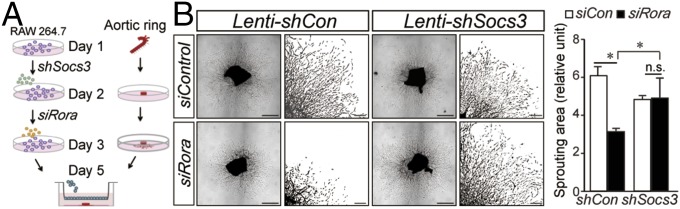
Macrophage RORα-regulated aortic ring vascular growth through modulation of Socs3. (A) Illustration of coculture showing RAW 264.7 cells were pretreated with lenti-shSocs3 and siRora, or respective controls, before being cocultured with normal aortic rings in Transwells. (B) Images of aortic ring sprouts (n = 8 per group) with selected areas enlarged. Sprouting areas were quantified. (Scale bars, 1,000 μm for original images and 150 μm for enlarged images.) *P < 0.05; n.s., no significance.
SOCS3 Mediated the Inflammatory and Vascular Effects of RORα in OIR.
To determine whether SOCS3 is functionally important for RORα-mediated retinal neovascularization, we silenced Socs3 expression with intravitreal injection of lenti-shSocs3 before exposing WT and Sg/Sg mice to OIR. Lenti-shSocs3 effectively suppressed ∼85% of Socs3 expression in both WT and Sg/Sg OIR retinas compared with their respective lenti-controls (Fig. 6A). Lenti-control–treated Sg/Sg retinas exhibited decreased levels of Il6, Il1b, and Tnf, increased levels of Il10 (Fig. 6A), and significantly reduced pathologic neovascularization compared with littermate WT retinas in OIR (P < 0.01, n = 5–9 per group, Fig. 6 B and C), similar to the dampened inflammation and reduced pathologic neovessels observed in nontreated Sg/Sg vs. WT OIR retinas (Fig. 1). Importantly, lenti-shSocs3 injection in Sg/Sg OIR retinas significantly abolished RORα-deficiency–induced dampening of inflammatory cytokines (Fig. 6A) and RORα-deficiency–induced protection from pathologic neovascularization, compared with lenti-control injected Sg/Sg eyes (P < 0.05, n = 5 per group, Fig. 6 B and C), back to the levels comparable to lenti-shSocs3–treated WT retinas (no significance, n = 5–9 per group, Fig. 6 B and C). Together these results support the idea that SOCS3 mechanistically mediates the inflammatory and vascular effects of RORα in OIR.
Fig. 6.

Inhibition of Socs3 abolished the inflammatory and neovascular effects of RORα deficiency in OIR. (A) Expression of Il6, Il1b, Tnf, and Il10 in P17 Sg/Sg and WT OIR retinas intravitreally injected with lenti-shControl or lenti-shSocs3 at P5 (n = 6 per group). (B) Representative retinal whole mounts from P17 OIR WT or Sg/Sg mice intravitreally injected with lenti-shControl or lenti-shSocs3, with retinal vessels stained by isolectin IB4 (red) and selected areas enlarged (white box). (C) Quantification of pathologic neovascular areas normalized to levels in control-treated WT retinas. n = 5–9 per group. (Scale bars, 1,000 μm for original images and 100 μm for enlarged images.) *P < 0.05; **P < 0.01; ***P < 0.001; n.s., no significance.
A Synthetic Inverse Agonist of RORα Suppressed Pathologic Neovascularization in OIR and Vldlr−/− Mice.
SR1001, a synthetic inverse agonist of RORα (13), dose dependently increased Socs3 expression in macrophage RAW 264.7 cells (Fig. 7A). Moreover, SR1001 treatment from P12 to P17 in WT OIR mice significantly reduced pathologic neovascularization at P17 by ∼30% (P < 0.01, n = 14–20 per group) compared with littermate vehicle controls, without affecting vasoobliteration (Fig. 7 B and C), suggesting that pharmacologic inhibition of RORα was effective in suppressing retinopathy. On the other hand, SR1078, a RORα agonist validated in the liver (26), dose-dependently suppressed Socs3 expression level in RAW 264.7 cells and significantly increased the levels of pathologic neovascularization (P < 0.01, n = 14–16 per group) in OIR (SI Appendix, Fig. S7), indicating that activation of RORα worsened retinopathy.
Fig. 7.
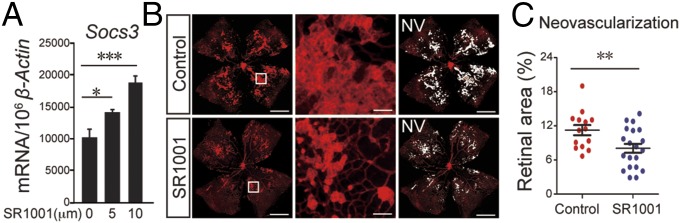
SR1001 suppressed pathologic neovascularization in OIR. (A) Socs3 mRNA expression in SR1001-treated RAW 264.7 cells. (B) Representative retinal whole mounts from WT OIR littermates treated with SR1001 or vehicle control were stained with isolectin IB4 (red). Areas of neovascularization (NV) were highlighted (white) and selected retinal areas (white box) enlarged. (C) Quantification of pathologic NV in SR1001 and vehicle control-treated retinas. n = 14–20 per group. (Scale bars, 1,000 μm for original images and 100 μm for enlarged images.) *P < 0.05; **P < 0.01; ***P < 0.001.
Next SR1001 treatment was evaluated in an additional angiogenic model of Vldlr−/− mice, which develop spontaneous pathologic subretinal neovascularization, modeling neovascular AMD, retinal angiomatous proliferation, and macular telangiectasia (27–29) (Fig. 8A). The spontaneous subretinal neovascularization in Vldlr−/− mice invades the normally avascular photoreceptor layer starting at P12 and reaches retinal pigment epithelium (RPE) at P16 (Fig. 8A). Socs3 expression levels were significantly suppressed in Vldlr−/− retinas compared with WT retinas (Fig. 8B), whereas Rora levels were comparable (SI Appendix, Fig. S8). Daily SR1001 treatment in Vldlr−/− mice from P5 to P15 led to significant induction of retinal Socs3 expression at P16 (Fig. 8C). Importantly, SR1001 treatment significantly inhibited both the number and area of subretinal neovascular lesions in Vldlr−/− mice at P16 by ∼50% (P < 0.001, n = 9–12 per group, Fig. 8 D–F). These data suggest that RORα inhibition by SR1001 suppresses subretinal neovascularization in Vldlr−/− retinas, a relevant model for AMD, corroborating the findings in the OIR model.
Fig. 8.

SR1001 treatment suppressed spontaneous subretinal neovascularization in Vldlr−/− mice. (A) Schematic illustration (Top) and 3D reconstruction (Bottom) of spontaneous subretinal neovascularization in Vldlr−/− mice at P16, stained with isolectin IB4 (red). RGC, retinal ganglion cell; IPL, inner plexiform layer; OPL, outer plexiform layer; RPE, retinal pigment epithelium. (B) Socs3 mRNA expression in WT and Vldlr−/− retinas at P7 and P17, n = 3 per group. (C) Socs3 mRNA expression levels in SR1001 (P5–P15) or vehicle control-treated P16 Vldlr−/− retinas, n = 3 per group. (D) Representative images of a quadrant of Vldlr−/− retinas with SR1001 treatment (P5–P15) or littermate Vldlr−/− retinas with vehicle-control treatment. Lesions were highlighted (white) and enlarged in Inset and 3D. (E and F) Quantification of the number and total area of subretinal vascular lesions in SR1001 or control-treated Vldlr−/− mice. n = 9–12 per group. (Scale bar, 500 μm for flat mount, 250 μm for Inset, and 100 μm for 3D.) *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Our study presents evidence for a critical role of RORα in two models of pathologic ocular neovascularization, an oxygen-induced ischemic retinopathy and the Vldlr−/− mice with spontaneous subretinal neovascularization. Rora expression was significantly regulated in OIR, consistent with a suggested role of Rora as a hypoxia-inducible factor (HIF) target gene (30). RORα may thus be modulated synergistically by tissue ischemia and hypoxia, in addition to lipid-based ligands, to influence the angiogenic response. Our findings support a proangiogenic role of RORα as its deficiency suppressed pathologic retinal neovascularization; yet in a hind limb ischemia model, RORα was reported as a negative regulator of angiogenesis (31), potentially reflecting a functionally plastic role of RORα in regulating angiogenesis in a disease- and organ-dependent manner.
Suppression of pathologic neovascularization in RORα-deficient Sg/Sg retinas was associated with decreased retinal inflammation. Many inflammatory mediators were increased in vitreous fluid of patients with proliferative diabetic retinopathy or retinopathy of prematurity (32) including TNFα, IL-1β (33), IL-6 (34), and chemokines such as CXCL10 (35), suggesting that chemokine-induced recruitment of inflammatory cells and inflammatory cytokines are important for clinical retinopathy. In mice, depletion of TNFα resulted in decreased pathologic retinal neovascularization (36), and inhibition of IL-1β also suppressed diabetic retinopathy (37) and pathologic ocular angiogenesis (38). These studies suggest that proinflammatory cytokines are potentially detrimental to retinopathy, which may underlie the protective effect of RORα deficiency in suppressing inflammation-associated pathologic retinal neovascularization in OIR.
Whereas our study localized RORα mainly in macrophages/microglia, these may not be the sole cellular source of retinal RORα. Clodronate liposome depletion of macrophage largely suppressed but did not completely abolish Rora expression, suggesting other cellular source of RORα. Previous studies have reported the presence of RORα in retinal neurons including retinal ganglion cells (39) and a subset of cone photoreceptors (25), as well as in human aortic vascular cells (40). Therefore, potential contribution from vascular endothelium or neuronal RORα toward the observed vascular effects in the retina is still possible.
Mechanistically RORα-mediated inflammatory and vascular effects may act in part through SOCS3, by direct repression of its transcription. Interestingly RORα is generally considered a constitutively active transcription factor, yet our discovery of transcriptional suppressive activity of RORα on Socs3 may reflect the possibility of a negative response element of the Socs3 RORE site. Negative response elements mediate direct transcriptional suppression by nuclear receptors such as the glucocorticoid receptor (41), and similar mechanisms may also exist for RORα that will require further study. RORα suppression of Socs3 promoted retinal inflammation, consistent with previous reports showing increased inflammation in Socs3-deficient macrophages in myeloid-specific Socs3 knockout mice (28). Depletion of Socs3 in Tie2-expressing cells also promoted pathologic neovascularization in OIR (42), reflecting an endogenous inhibitory role of SOCS3 in blood vessels. Low levels or transient induction of RORα in the endothelium may also potentially mediate endothelial SOCS3-dependent inflammation. Inhibiting SOCS3 effectively reversed the inflammatory and vascular effect of RORα deficiency, yet potential direct RORα transcriptional regulation of other inflammatory mediators, such as Arg1 and Il10, may be additional contributing factors, reflecting a likely multifactorial process regulated by RORα, which will be further investigated.
As a receptor for cholesterol derivatives, RORα is involved in regulation of cholesterol homeostasis and may influence cellular inflammatory and angiogenic responses to lipid metabolites. However, whether RORα-related lipid metabolites may directly influence proliferative retinopathy is still unclear, although dyslipidemia is closely linked with both clinical diabetic retinopathy and AMD. Whereas no data are available regarding the influence of RORα on human proliferative retinopathy, genetic variations of RORα were linked with increased risks of developing neovascular AMD (6–8); however, the functional consequence of the AMD-associated RORα SNPs awaits further investigation.
Our findings indicate that SR1001, a high-affinity synthetic inverse agonist of RORα, effectively suppressed pathologic neovascularization in both OIR and Vldlr−/− mice, without causing the staggering phenotype or obvious gross toxicity, consistent with lack of adverse events observed in previous studies with prolonged SR1001 treatment in adult or diabetic mice (13, 43). However, further evaluation on retinal neuronal function will be needed to fully characterize the safety profile of SR1001. Additional structural optimization of SR1001 may also allow development of more potent and selective RORα inhibitors with even stronger antiangiogenic effect for translational use. Whereas SR1001 binds specifically to the ligand-binding domains of both RORα and RORγ (13), Rorc expression levels were much lower in the retinas compared with Rora, and relatively unchanged in OIR (SI Appendix, Fig. S9), suggesting that potential side-effect contribution from RORγ to the observed effects of SR1001 is likely marginal.
In summary, this study provides the first direct evidence to our knowledge for a critical role of an immunoregulating nuclear receptor RORα in experimental proliferative retinopathy through modulating tissue inflammation via transcriptional regulation of Socs3. Modulation of a nuclear receptor such as RORα may thus serve as a completely new approach to potentially treat vascular eye diseases without directly impacting angiogenic growth factors, such as VEGF, that are essential for vascular homeostasis, and may have broad therapeutic value for potentially other vascular disorders precipitated by inflammation-mediated angiogenesis.
Materials and Methods
Mice.
All animal studies were performed according to protocols reviewed and approved by the Institutional Animal Care and Use Committee at the Boston Children's Hospital. Rora heterozygous staggerer mice (Sg/+) were obtained from The Jackson Laboratory (stock no. 000237) and bred together to generate homozygous and wild-type littermates. C57BL/6J mice (stock no. 000664) and Vldlr−/− mice (stock no. 002529) were both obtained from The Jackson Laboratory.
Oxygen-Induced Retinopathy.
OIR was carried out as described previously (19), with mouse pups exposed to 75% oxygen at postnatal day (P) P7–P12 followed by room air. At P17, mice were anesthetized, and retinas dissected followed by fluoresceinated isolectin IB4 (Invitrogen) staining to visualize vessels on whole-mount retinas. Areas of retinal vasoobliteration and pathologic neovascularization were quantified as a percentage of total retinal areas using Adobe Photoshop and Image J.
Statistics.
Results were presented as mean ± SEM for animal studies and mean ± SD for nonanimal studies. Two-tailed t tests (two groups) or ANOVA (more than two groups) were performed. Differences were considered significant if P ≤ 0.05.
Other expanded materials and methods are available in SI Appendix, SI Methods.
Supplementary Material
Acknowledgments
We thank Drs. Lois Smith, Thomas Burris, Przemyslaw Sapieha, and Zhuo Shao for expert advice and/or critical review of the manuscript; Aimee Juan, Dorothy Pei, Ricky Cui, Christian Hurst, and Roberta Dennison for technical assistance; Drs. Xi He and Xinjun Zhang for providing reagents; and Drs. Martin Friedlander, Edith Aguilar, and Yoshihiko Usui for providing CX3CR1-GFP retinas. This work was supported by NIH/National Eye Institute (R01 EY024963), BrightFocus Foundation, Boston Children's Hospital (BCH) Career Development Award, BCH Ophthalmology Foundation, Massachusetts Lions Eye Research Fund Inc., and Alcon Research Institute (J.C.). A.S. was supported by Deutsche Forschungsgemeinschaft (DFG STA 1102/5-1) and Deutsche Ophthalmologische Gesellschaft.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1504387112/-/DCSupplemental.
References
- 1.Dorrell M, Uusitalo-Jarvinen H, Aguilar E, Friedlander M. Ocular neovascularization: Basic mechanisms and therapeutic advances. Surv Ophthalmol. 2007;52(Suppl 1):S3–S19. doi: 10.1016/j.survophthal.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 2.Busik JV, Esselman WJ, Reid GE. Examining the role of lipid mediators in diabetic retinopathy. Clin Lipidol. 2012;7(6):661–675. doi: 10.2217/clp.12.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curcio CA, Johnson M, Huang JD, Rudolf M. Aging, age-related macular degeneration, and the response-to-retention of apolipoprotein B-containing lipoproteins. Prog Retin Eye Res. 2009;28(6):393–422. doi: 10.1016/j.preteyeres.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen DV, Shaw LC, Grant MB. Inflammation in the pathogenesis of microvascular complications in diabetes. Front Endocrinol (Lausanne) 2012;3:170. doi: 10.3389/fendo.2012.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bitsch F, et al. Identification of natural ligands of retinoic acid receptor-related orphan receptor alpha ligand-binding domain expressed in Sf9 cells—a mass spectrometry approach. Anal Biochem. 2003;323(1):139–149. doi: 10.1016/j.ab.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 6.Silveira AC, et al. Convergence of linkage, gene expression and association data demonstrates the influence of the RAR-related orphan receptor alpha (RORA) gene on neovascular AMD: A systems biology based approach. Vision Res. 2010;50(7):698–715. doi: 10.1016/j.visres.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaumberg DA, et al. Prospective study of common variants in the retinoic acid receptor-related orphan receptor α gene and risk of neovascular age-related macular degeneration. Arch Ophthalmol. 2010;128(11):1462–1471. doi: 10.1001/archophthalmol.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jun G, et al. Influence of ROBO1 and RORA on risk of age-related macular degeneration reveals genetically distinct phenotypes in disease pathophysiology. PLoS One. 2011;6(10):e25775. doi: 10.1371/journal.pone.0025775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jetten AM. Retinoid-related orphan receptors (RORs): Critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal. 2009;7:e003. doi: 10.1621/nrs.07003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kallen J, Schlaeppi JM, Bitsch F, Delhon I, Fournier B. Crystal structure of the human RORalpha ligand binding domain in complex with cholesterol sulfate at 2.2 A. J Biol Chem. 2004;279(14):14033–14038. doi: 10.1074/jbc.M400302200. [DOI] [PubMed] [Google Scholar]
- 11.Saito T, et al. Pivotal role of Rho-associated kinase 2 in generating the intrinsic circadian rhythm of vascular contractility. Circulation. 2013;127(1):104–114. doi: 10.1161/CIRCULATIONAHA.112.135608. [DOI] [PubMed] [Google Scholar]
- 12.Halim TY, et al. Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity. 2012;37(3):463–474. doi: 10.1016/j.immuni.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 13.Solt LA, et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature. 2011;472(7344):491–494. doi: 10.1038/nature10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Journiac N, et al. The nuclear receptor ROR(alpha) exerts a bi-directional regulation of IL-6 in resting and reactive astrocytes. Proc Natl Acad Sci USA. 2009;106(50):21365–21370. doi: 10.1073/pnas.0911782106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delerive P, et al. The orphan nuclear receptor ROR alpha is a negative regulator of the inflammatory response. EMBO Rep. 2001;2(1):42–48. doi: 10.1093/embo-reports/kve007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang XO, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28(1):29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jetten AM, Kurebayashi S, Ueda E. The ROR nuclear orphan receptor subfamily: Critical regulators of multiple biological processes. Prog Nucleic Acid Res Mol Biol. 2001;69:205–247. doi: 10.1016/s0079-6603(01)69048-2. [DOI] [PubMed] [Google Scholar]
- 18.Lechtken A, Zündorf I, Dingermann T, Firla B, Steinhilber D. Overexpression, refolding, and purification of polyhistidine-tagged human retinoic acid related orphan receptor RORalpha4. Protein Expr Purif. 2006;49(1):114–120. doi: 10.1016/j.pep.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 19.Smith LE, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35(1):101–111. [PubMed] [Google Scholar]
- 20.Hamilton BA, et al. Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature. 1996;379(6567):736–739. doi: 10.1038/379736a0. [DOI] [PubMed] [Google Scholar]
- 21.Ritter MR, et al. Myeloid progenitors differentiate into microglia and promote vascular repair in a model of ischemic retinopathy. J Clin Invest. 2006;116(12):3266–3276. doi: 10.1172/JCI29683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaštelan S, Tomić M, Gverović Antunica A, Salopek Rabatić J, Ljubić S. Inflammation and pharmacological treatment in diabetic retinopathy. Mediators Inflamm. 2013;2013:213130. doi: 10.1155/2013/213130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Starr R, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387(6636):917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 24.Qin H, et al. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol. 2012;189(7):3439–3448. doi: 10.4049/jimmunol.1201168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujieda H, Bremner R, Mears AJ, Sasaki H. Retinoic acid receptor-related orphan receptor alpha regulates a subset of cone genes during mouse retinal development. J Neurochem. 2009;108(1):91–101. doi: 10.1111/j.1471-4159.2008.05739.x. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, et al. Identification of SR1078, a synthetic agonist for the orphan nuclear receptors RORα and RORγ. ACS Chem Biol. 2010;5(11):1029–1034. doi: 10.1021/cb100223d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li C, et al. Biochemical alterations in the retinas of very low-density lipoprotein receptor knockout mice: An animal model of retinal angiomatous proliferation. Arch Ophthalmol. 2007;125(6):795–803. doi: 10.1001/archopht.125.6.795. [DOI] [PubMed] [Google Scholar]
- 28.Hua J, et al. Resveratrol inhibits pathologic retinal neovascularization in Vldlr(-/-) mice. Invest Ophthalmol Vis Sci. 2011;52(5):2809–2816. doi: 10.1167/iovs.10-6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dorrell MI, et al. Antioxidant or neurotrophic factor treatment preserves function in a mouse model of neovascularization-associated oxidative stress. J Clin Invest. 2009;119(3):611–623. doi: 10.1172/JCI35977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chauvet C, Bois-Joyeux B, Berra E, Pouyssegur J, Danan JL. The gene encoding human retinoic acid-receptor-related orphan receptor alpha is a target for hypoxia-inducible factor 1. Biochem J. 2004;384(Pt 1):79–85. doi: 10.1042/BJ20040709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Besnard S, et al. Increased ischemia-induced angiogenesis in the staggerer mouse, a mutant of the nuclear receptor Roralpha. Circ Res. 2001;89(12):1209–1215. doi: 10.1161/hh2401.101755. [DOI] [PubMed] [Google Scholar]
- 32.Sato T, Kusaka S, Shimojo H, Fujikado T. Simultaneous analyses of vitreous levels of 27 cytokines in eyes with retinopathy of prematurity. Ophthalmology. 2009;116(11):2165–2169. doi: 10.1016/j.ophtha.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 33.Demircan N, Safran BG, Soylu M, Ozcan AA, Sizmaz S. Determination of vitreous interleukin-1 (IL-1) and tumour necrosis factor (TNF) levels in proliferative diabetic retinopathy. Eye (Lond) 2006;20(12):1366–1369. doi: 10.1038/sj.eye.6702138. [DOI] [PubMed] [Google Scholar]
- 34.Koskela UE, Kuusisto SM, Nissinen AE, Savolainen MJ, Liinamaa MJ. High vitreous concentration of IL-6 and IL-8, but not of adhesion molecules in relation to plasma concentrations in proliferative diabetic retinopathy. Ophthalmic Res. 2013;49(2):108–114. doi: 10.1159/000342977. [DOI] [PubMed] [Google Scholar]
- 35.Nawaz MI, et al. Autocrine CCL2, CXCL4, CXCL9 and CXCL10 signal in retinal endothelial cells and are enhanced in diabetic retinopathy. Exp Eye Res. 2013;109:67–76. doi: 10.1016/j.exer.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 36.Gardiner TA, et al. Inhibition of tumor necrosis factor-alpha improves physiological angiogenesis and reduces pathological neovascularization in ischemic retinopathy. Am J Pathol. 2005;166(2):637–644. doi: 10.1016/s0002-9440(10)62284-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kowluru RA, Odenbach S. Role of interleukin-1beta in the development of retinopathy in rats: Effect of antioxidants. Invest Ophthalmol Vis Sci. 2004;45(11):4161–4166. doi: 10.1167/iovs.04-0633. [DOI] [PubMed] [Google Scholar]
- 38.Lavalette S, et al. Interleukin-1β inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration. Am J Pathol. 2011;178(5):2416–2423. doi: 10.1016/j.ajpath.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steinmayr M, et al. staggerer phenotype in retinoid-related orphan receptor alpha-deficient mice. Proc Natl Acad Sci USA. 1998;95(7):3960–3965. doi: 10.1073/pnas.95.7.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Besnard S, et al. Expression and regulation of the nuclear receptor RORalpha in human vascular cells. FEBS Lett. 2002;511(1-3):36–40. doi: 10.1016/s0014-5793(01)03275-6. [DOI] [PubMed] [Google Scholar]
- 41.Surjit M, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011;145(2):224–241. doi: 10.1016/j.cell.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 42.Stahl A, et al. SOCS3 is an endogenous inhibitor of pathologic angiogenesis. Blood. 2012;120(14):2925–2929. doi: 10.1182/blood-2012-04-422527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Solt LA, Banerjee S, Campbell S, Kamenecka TM, Burris TP. ROR inverse agonist suppresses insulitis and prevents hyperglycemia in a mouse model of type 1 diabetes. Endocrinology. 2015;156(3):869–881. doi: 10.1210/en.2014-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.