

Significance
We demonstrate that glutathione biosynthesis is controlled by hypoxia-inducible factor 1 and is critical for chemotherapy-induced enrichment of breast cancer stem cells, making it an attractive therapeutic target in triple-negative breast cancer, which is the only subset of breast cancers for which there is no available targeted therapy. We also delineate a molecular mechanism in which glutathione functions as a signaling molecule to activate the breast cancer stem cell phenotype, establishing cross-talk between cancer metabolism and signal transduction. We also demonstrate that mitogen-activated protein kinase kinase (MEK)-ERK inhibitors and copper chelators have the countertherapeutic effect of inducing breast cancer stem cell enrichment.
Keywords: hypoxia-inducible factor, paclitaxel, pluripotency factors, chemotherapy resistance, tumor-initiating cells
Abstract
Triple negative breast cancer (TNBC) accounts for 10–15% of all breast cancer but is responsible for a disproportionate share of morbidity and mortality because of its aggressive characteristics and lack of targeted therapies. Chemotherapy induces enrichment of breast cancer stem cells (BCSCs), which are responsible for tumor recurrence and metastasis. Here, we demonstrate that chemotherapy induces the expression of the cystine transporter xCT and the regulatory subunit of glutamate-cysteine ligase (GCLM) in a hypoxia-inducible factor (HIF)-1–dependent manner, leading to increased intracellular glutathione levels, which inhibit mitogen-activated protein kinase kinase (MEK) activity through copper chelation. Loss of MEK-ERK signaling causes FoxO3 nuclear translocation and transcriptional activation of the gene encoding the pluripotency factor Nanog, which is required for enrichment of BCSCs. Inhibition of xCT, GCLM, FoxO3, or Nanog blocks chemotherapy-induced enrichment of BCSCs and impairs tumor initiation. These results suggest that, in combination with chemotherapy, targeting BCSCs by inhibiting HIF-1–regulated glutathione synthesis may improve outcome in TNBC.
Triple negative breast cancer (TNBC), which is defined as breast cancer that lacks expression of the estrogen receptor (ER), progesterone receptor, and human epidermal growth factor receptor 2 (HER2), accounts for 10–15% of all breast carcinomas (1). TNBCs are more aggressive than other subtypes of breast cancer, as exemplified by the increased risk of recurrence, metastasis, and patient mortality (2). Unlike ER+ breast cancer, which can be treated with an ER antagonist such as tamoxifen, or HER2+ breast cancer, which can be treated with the anti-HER2 antibody trastuzumab, patients with TNBC do not benefit from targeted therapy. Current systemic treatment options for TNBC are limited to cytotoxic chemotherapy, which may reduce tumor bulk initially, but the majority of patients have residual disease or early relapse with a median survival of only 13 mo (3). Therefore, better understanding of chemotherapy resistance mechanisms and better treatment options are urgently needed.
Tumor heterogeneity is a key characteristic of breast cancer (4). Breast cancer stem cells (BCSCs) are a small population of cancer cells that possess an infinite proliferative potential and the capacity to initiate tumors clonally (5). BCSCs play a critical role in the metastatic process, which is the major cause of death for breast cancer patients, because only BCSCs are capable of forming clinically relevant metastases at a secondary site (6). In addition, BCSCs have increased resistance to chemotherapy through expression of drug transporter proteins, expression of survival factors, and other mechanisms (7). BCSCs are enriched in response to chemotherapy, further potentiating the risk of tumor recurrence and metastasis (8–10). The enrichment of BCSCs can be measured by the Aldefluor assay, which is based on the high aldehyde dehydrogenase activity (ALDH) in BCSCs (11), or by the mammosphere assay, which is based on the ability of BCSCs to generate multicellular spheroids in suspension culture (12).
Similar to other solid tumors, most breast cancers develop regions of intratumoral hypoxia because of the imbalance between the delivery and consumption of O2 (13). Hypoxia-inducible factors (HIFs) are transcription factors that serve as master regulators of cellular responses to hypoxia (14) and are associated with chemotherapy resistance in breast cancer (10, 15). Chemotherapy induces HIF-dependent expression of interleukin (IL)-6 and IL-8, which promote the BCSC phenotype, and of multidrug resistance protein 1 (MDR-1), which mediates chemotherapy efflux from BCSCs, such that the percentage of BCSCs is increased following treatment with paclitaxel or gemcitabine (10). Coadministration of digoxin, which inhibits HIF-1α protein accumulation, blocked chemotherapy-induced expression of IL-6, IL-8, and MDR-1, and blocked BCSC enrichment (10).
Cancer stem cells require an increased antioxidant capacity to prevent oxidative stress and to maintain stemness and the capacity for tumor initiation (16, 17). The major antioxidant within cells is glutathione, a tripeptide that is synthesized from cysteine, glutamate, and glycine (18). Cystine is taken up by cells primarily through the xC(-) system, which is a heterodimeric protein consisting of a transporter subunit (xCT, encoded by the SLC7A11 gene) and a regulatory subunit (4F2hc, also known as CD98, encoded by the SLC3A2 gene) (19). In cells, cystine is reduced to cysteine and reacts with glutamate to form γ-glutamylcysteine in a reaction catalyzed by glutamate cysteine ligase (GCL), which consists of a catalytic subunit, GCLC, and a modifier subunit, GCLM. Glycine is added to γ-glutamylcysteine by the enzyme glutathione synthetase (GSS) to form glutathione. The glutathione synthesis pathway has been shown to promote cancer initiation and progression, and targeting this pathway by inhibiting xCT or GCL has shown some promise in inhibiting tumor growth in combination with chemotherapy in mouse models of breast cancer (20, 21), although the underlying molecular mechanisms have not been fully delineated. Because chemotherapy induces oxidative stress, it has been assumed that the glutathione synthesis pathway promotes chemotherapy resistance through its antioxidant effects (17). Here, we demonstrate that in TNBC, glutathione synthesis is induced by chemotherapy in a HIF-1–dependent manner, resulting in increased intracellular glutathione levels, which activate expression of pluripotency factors that directly specify the BCSC phenotype. Moreover, rather than solely functioning in its traditional role as an antioxidant, glutathione induces the BCSC phenotype by chelating copper and, thereby, inhibiting mitogen-activated protein kinase kinase (MEK)-ERK signaling.
Results
Chemotherapy Induces HIF-1–Dependent Glutathione Biosynthesis.
We hypothesized that chemotherapy induces glutathione synthesis in breast cancer cells to protect against oxidative stress. Paclitaxel, gemcitabine, and carboplatin are all Food and Drug Administration-approved chemotherapy drugs that are used for the treatment of TNBC. We treated two TNBC cell lines, MDA-MB-231 and SUM-149, with paclitaxel, gemcitabine, or carboplatin for 72 h at the concentration of drug that inhibited growth by 50% (IC50). Each of these chemotherapeutic agents increased xCT and GCLM mRNA levels in both cell lines as determined by reverse transcription (RT) and quantitative real-time PCR (qPCR) (Fig. 1A and Fig. S1A). In MDA-MB-231 cells transfected with a nontargeting control (NTC) short hairpin RNA (shRNA), paclitaxel treatment increased xCT and GCLM protein levels (Fig. 1B), and intracellular total glutathione levels (Fig. 1C). Knockdown of xCT or GCLM expression by specific shRNAs (Fig. 1B) blocked glutathione induction in response to paclitaxel treatment (Fig. 1C), indicating that both xCT and GCLM are required for paclitaxel-induced glutathione synthesis.
Fig. 1.

Chemotherapy induces glutathione synthesis in a HIF-1–dependent manner. (A) MDA-MB-231 cells were treated with vehicle (V), 10 nM paclitaxel (P), 10 nM gemcitabine (G), or 100 μM carboplatin (C) for 72 h. Aliquots of total cellular RNA were subjected to RT and qPCR to analyze xCT (SLC7A11) and GCLM mRNA expression. The results were normalized to cells treated with vehicle (mean ± SEM; n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle. (B) MDA-MB-231 subclones, which were stably transfected with an expression vector encoding nontargeting control (NTC) short hairpin RNA (shRNA), or vector encoding shRNA against xCT or GCLM, were treated without (-) or with (+) 10 nM paclitaxel (Pac) for 72 h, and immunoblot assays were performed to analyze xCT and GCLM protein expression. (C) MDA-MB-231 subclones were treated without or with 10 nM Pac for 72 h, glutathione levels were measured, and the results were normalized to NTC Pac (-) (mean ± SEM; n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. NTC Pac (-); ###P < 0.001 vs. NTC Pac (+). (D) MDA-MB-231 subclones, which were stably transfected with NTC vector or vector encoding shRNA against HIF-1α or HIF-2α, were treated without or with Pac for 72 h. RT-qPCR was performed to assay xCT and GCLM mRNA levels (mean ± SEM; n = 3). **P < 0.01, ***P < 0.001 vs. NTC Pac (-); #P < 0.05, ###P < 0.001 vs. NTC Pac (+); ns, not significant. (E) MDA-MB-231 cells were implanted into the mammary fat pad of female SCID mice. When tumor volume reached 200 mm3, mice were randomly assigned to treatment with saline (vehicle, V), Pac (P; 10 mg/kg on days 5 and 10), digoxin (D; 2 mg/kg on days 1–12), or Pac and digoxin (P + D). Tumors were harvested on day 12 for RT-qPCR analysis of xCT and GCLM mRNA. *P < 0.05, **P < 0.01 vs. vehicle; ##P < 0.01, ###P < 0.001 vs. Pac. (F) MDA-MB-231 cells were incubated at 20% or 1% O2 for 16 h, and ChIP assays were performed by using IgG or antibodies against HIF-1α or HIF-1β. Primers flanking candidate binding sites in the SLC7A11 and GCLM genes were used for qPCR, and the results were normalized to cells exposed to 20% O2 and immunoprecipitated with anti-HIF-1α (mean ± SEM; n = 3). **P < 0.01, ***P < 0.001 vs. 20% O2. The nucleotide sequences surrounding the HIF-1–binding sites (colored fonts) within intron 3 of SLC7A11 and the 5′-flanking region of GCLM are shown.
Fig. S1.
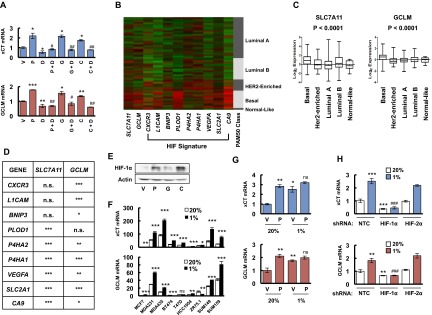
Chemotherapy and hypoxia induce xCT and GCLM expression in a HIF-1–dependent manner. (A) SUM-149 cells were treated with vehicle (V), 5 nM paclitaxel (P), 10 nM gemcitabine (G), 100 μM carboplatin (C), either alone or in combination with 100 nM digoxin (P + D, G + D, C + D) for 72 h, and RT-qPCR were performed to analyze the expression of xCT and GCLM mRNA. The results were normalized to cells treated with vehicle (mean ± SEM; n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle; #P < 0.05, ##P < 0.01 vs. chemotherapy alone. (B) The relative expression levels of nine mRNAs encoded by HIF target genes, as well as SLC7A11 (xCT) and GCLM mRNA, are shown for breast cancers accessed from the Cancer Genome Atlas (TCGA) database, which were classified based on the expression of a 50-mRNA (PAM50) signature that defines five molecular subtypes (Luminal A, Luminal B, HER2-enriched, Basal, and Normal-like) (22). Color code: green, less than the median; red, greater than the median. (C) Log2 expression of SLC7A11 (xCT) and GCLM mRNA from 1,215 human breast cancer specimens from TCGA database were compared among five molecular subgroups. Data were analyzed by using one-way ANOVA. (D) SLC7A11 (xCT) and GCLM mRNA levels in each of 1,215 human breast cancers from the TCGA database were compared with the levels of nine HIF-regulated mRNAs by using Pearson’s correlation test. *P < 0.05, **P < 0.01, ***P < 0.001; n.s., not significant. (E) MDA-MB-231 cells were treated with vehicle (V), 10 nM paclitaxel (P), 10 nM gemcitabine (G), or 100 μM carboplatin (C) for 72 h, and immunoblot assays were performed. (F) Nine breast cancer cell lines were exposed to 20% or 1% O2 for 24 h, total cellular RNA was isolated, and RT-qPCR was performed to analyze the expression of xCT and GCLM mRNA. The results were normalized to MCF-7 cells that were exposed to 20% O2 (mean ± SEM; n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. 20% O2. (G) MDA-MB-231 cells were treated with vehicle (V) or 10 nM paclitaxel (P) in 20% or 1% O2 for 72 h, total RNA was isolated, and RT-qPCR was performed to analyze the expression of xCT and GCLM mRNA (mean ± SEM; n = 3). *P < 0.05, **P < 0.01 vs. vehicle in 20% O2; ns, not significant vs. paclitaxel in 20% O2. (H) MDA-MB-231 subclones, which were stably transfected with nontargeting control shRNA (NTC) vector or expression vector encoding shRNA against HIF-1α or HIF-2α, were exposed to 20% or 1% O2 for 24 h, total RNA was isolated, and RT-qPCR was performed to analyze the expression of xCT and GCLM mRNA (mean ± SEM; n = 3). **P < 0.01, ***P < 0.001 vs. NTC in 20% O2; ###P < 0.001 vs. NTC in 1% O2.
Gene expression data from 1,215 human breast cancers in the Cancer Genome Atlas (TCGA) database were analyzed to compare the expression patterns of xCT and GCLM mRNA in different molecular subtypes of breast cancer (Basal, HER2-enriched, Luminal A, Luminal B, and Normal-like) that are based on a 50-mRNA (PAM50) signature (22) (Fig. S1B). Compared with other breast cancer subtypes, xCT and GCLM mRNA levels were significantly increased in basal breast cancers (Fig. S1C), in which hypoxia-inducible factor (HIF)-target gene expression is also increased (10). A detailed comparison of xCT and GCLM mRNA levels with those of CXCR3, L1CAM, BNIP3, PLOD1, P4HA1, P4HA2, VEGFA, SLC2A1, and CA9 mRNA, which are all HIF-regulated gene products in TNBC cells, showed a significant correlation of xCT and GCLM expression with six of nine and eight of nine HIF-target genes, respectively (Fig. S1D). Exposure of MDA-MB-231 cells to paclitaxel, gemcitabine, or carboplatin at IC50 induced HIF-1α protein expression (Fig. S1E). In addition, xCT and GCLM mRNA levels were induced in most breast cancer cell lines when they were exposed to 1% O2 for 24 h (Fig. S1F). In MDA-MB-231 cells, xCT and GCLM mRNA levels were significantly induced by either paclitaxel or hypoxia alone, although no additive effect was observed by the combined treatment (Fig. S1G). These data suggest that SLC7A11 and GCLM gene expression are regulated by HIFs.
To test this hypothesis, we analyzed MDA-MB-231 subclones that were stably transfected with an expression vector encoding shRNA targeting HIF-1α or HIF-2α, and found that knockdown of HIF-1α, but not HIF-2α, decreased xCT and GCLM mRNA basal levels and blocked their induction in response to paclitaxel treatment (Fig. 1D). Knockdown of HIF-1α, but not HIF-2α, also abrogated xCT and GCLM induction in response to hypoxia (Fig. S1H). Pharmacological inhibition of HIF-1α expression by treatment of cells with the HIF-1 inhibitor digoxin (10, 23) also blocked expression of xCT and GCLM mRNA in SUM-149 cells exposed to paclitaxel, gemcitabine, or carboplatin (Fig. S1A). Taken together, these data indicate that expression of the SLC7A11 and GCLM genes is regulated by HIF-1, but not HIF-2. We also implanted MDA-MB-231 cells into the mammary fat pad of female severe combined immunodeficiency (SCID) mice and treated the mice with paclitaxel, either alone or in combination with digoxin. Digoxin treatment decreased xCT and GCLM mRNA levels, and blocked their induction by paclitaxel (Fig. 1E), demonstrating that paclitaxel treatment induces xCT and GCLM mRNA expression in a HIF-1–dependent manner in vivo.
To investigate whether HIF-1 directly regulates SLC7A11 and GCLM expression, genomic DNA sequences were searched for matches to the consensus HIF-1 binding-site sequence 5′-(A/G)CGTG-3′ and candidate sites were evaluated by chromatin immunoprecipitation (ChIP) assays performed in MDA-MB-231 cells. Hypoxia induced the binding of HIF-1α and HIF-1β to sites located in the third intron of SLC7A11 and in the 5′-flanking region of GCLM (Fig. 1F). Taken together, these data indicate that chemotherapy drugs induce HIF-1–dependent activation of SLC7A11 and GCLM transcription.
Inhibition of Glutathione Synthesis Blocks Paclitaxel-Induced BCSC Enrichment.
We recently demonstrated that paclitaxel treatment increases the percentage of BCSCs in a HIF-dependent manner (10). xCT and GCLM expression in breast cancer cell lines is correlated with expression of CD44, an important BCSC marker (20, 24). To test the role of the glutathione synthesis pathway in paclitaxel-induced BCSC enrichment, MDA-MB-231 cells were sorted into an ALDH+ population, which is highly enriched for BCSCs, and an ALDH− population, which is depleted of BCSCs (11). xCT and GCLM mRNA levels were increased four- to fivefold in ALDH+ cells compared with ALDH− cells (Fig. 2A). We also enriched BCSCs from MDA-MB-231 and SUM-149 cells through mammosphere formation (12) and found 3- to 10-fold increased xCT and GCLM mRNA levels in primary and secondary mammospheres compared with cells cultured in monolayer (Fig. 2B).
Fig. 2.
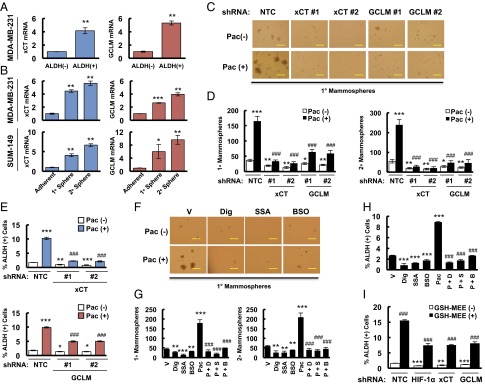
Paclitaxel-induced BCSC enrichment is blocked by inhibition of glutathione synthesis. (A) MDA-MB-231 cells were sorted into aldehyde dehydrogenase negative [ALDH (-)] and positive [ALDH (+)] populations by flow cytometry. RT-qPCR was performed to analyze the expression of xCT (SLC7A11) and GCLM mRNA, and the results were normalized to ALDH (-) cells (mean ± SEM; n = 3). **P < 0.01 vs. ALDH (-). (B) MDA-MB-231 (Upper) and SUM-149 (Lower) cells were cultured on ultra-low adherence plates for 7 d to generate primary mammospheres (1° sphere). Half of the mammospheres were harvested for RT-qPCR analyses, and the other half of the mammospheres were dissociated and cultured for 7 d to generate secondary mammospheres (2° sphere), which were harvested for RT-qPCR analyses. Results were normalized to MDA-MB-231 and SUM-149 adherent cells (mean ± SEM; n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. adherent cells. (C and D) MDA-MB-231 subclones were treated without or with 10 nM paclitaxel (Pac) for 72 h. The number of 1° and 2° mammospheres per 1,000 cells initially seeded was calculated (mean ± SEM; n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. NTC Pac (-); ###P < 0.001 vs. NTC Pac (+). (E) MDA-MB-231 subclones were treated without or with 10 nM Pac for 4 d, and the percentage of ALDH (+) cells was determined (mean ± SEM; n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. NTC Pac (-); ###P < 0.001 vs. NTC Pac (+). (F and G) SUM-149 cells were treated without or with 5 nM Pac for 72 h, in combination with vehicle (V), 100 nM digoxin (Dig), 100 μM sulfasalazine (SSA), or 100 μM buthionine sulphoximine (BSO). Primary and secondary mammospheres were counted (mean ± SEM; n = 3). **P < 0.01, ***P < 0.001 vs. vehicle; ###P < 0.001 vs. Pac. (H) SUM-149 cells were treated with V, Dig, SSA, BSO, or Pac alone, or Pac in combination with Dig (P + D), SSA (P + S), or BSO (P + B) for 4 d, and the percentage of ALDH (+) cells was determined (mean ± SEM; n = 3). ***P < 0.001 vs. V; ###P < 0.001 vs. Pac. (I) MDA-MB-231 subclones were treated without or with 2 mM glutathione monoethyl ester (GSH-MEE) for 4 d and the percentage of ALDH (+) cells was determined (mean ± SEM; n = 3). **P < 0.01, ***P < 0.001 vs. GSH-MEE (-) in NTC group; ###P < 0.001 vs. GSH-MEE (-) in the same subclone. (Scale bars: 200 μm.)
To test the role of xCT and GCLM in chemotherapy-induced BCSC enrichment, MDA-MB-231 subclones with shRNA-mediated knockdown of xCT or GCLM expression were exposed to paclitaxel for 72 h and mammosphere assays were performed as a measure of BCSCs. Paclitaxel treatment of the NTC subclone significantly increased the number of mammosphere-forming cells, whereas knockdown of xCT or GCLM expression decreased the number of mammosphere-forming cells and abrogated the induction by paclitaxel in both primary and secondary mammosphere assays (Fig. 2 C and D). Aldefluor assays revealed that paclitaxel treatment of the NTC subclone significantly increased the percentage of ALDH+ cells, and this effect was attenuated by knockdown of xCT or GCLM expression (Fig. 2E and Fig. S2).
Fig. S2.

Knockdown of xCT or GCLM abrogates paclitaxel-induced BCSC enrichment. MDA-MB-231 subclones were treated without or with 10 nM paclitaxel (Pac) for 4 d, and the percentage of ALDH (+) cells was determined. Representative flow cytometry results are shown.
Pharmacologic inhibition was also used to investigate the role of the glutathione synthesis pathway in BCSCs. SUM-149 cells were treated with paclitaxel, either alone or in combination with the HIF-1 inhibitor digoxin, xCT inhibitor sulfasalazine (SSA) (21), or GCL inhibitor buthionine sulphoximine (BSO) (25) for 72 h. Paclitaxel increased the number of mammosphere-forming cells, whereas administration of digoxin, SSA, or BSO decreased the number of mammosphere-forming cells and abolished the effect of paclitaxel treatment (Fig. 2 F and G). Coadministration of digoxin, SSA, or BSO also abrogated the increased percentage of ALDH+ cells induced by paclitaxel treatment (Fig. 2H). To further confirm the role of glutathione synthesis in BCSCs, we exposed MDA-MB-231 cells to 2 mM glutathione monoethyl ester (GSH-MEE), a cell membrane-permeable form of glutathione, for 4 d. GSH-MEE treatment alone was sufficient to increase the percentage of ALDH+ cells, even in HIF-1α, xCT, or GCLM knockdown subclones (Fig. 2I). Taken together, these data demonstrate that increased glutathione levels are both necessary and sufficient for induction of the BCSC phenotype.
Paclitaxel Induces the Expression of Pluripotency Factors.
To delineate the mechanism through which paclitaxel-induced glutathione synthesis regulates BCSCs, we analyzed mRNA samples from ALDH− and ALDH+ populations sorted from MDA-MB-231 cells and found that the pluripotency factors Nanog and Sox2 were markedly overexpressed in ALDH+ compared with ALDH− cells (Fig. 3A). Increased expression of Nanog and Sox2 was observed previously in MCF-7 cells that were selected for resistance to tamoxifen (26). Nanog and Sox2 were also overexpressed in BCSC-enriched mammosphere cultures of MDA-MB-231 cells (Fig. 3B, Upper). Sox2 mRNA was not expressed in SUM-149 cells; however, Nanog and another pluripotency factor, Oct4, were overexpressed in SUM-149 mammosphere cultures (Fig. 3B, Lower). Paclitaxel treatment of MDA-MB-231 cells induced the expression of Nanog and Sox2, which was completely abrogated by knockdown of HIF-1α, xCT, or GCLM; in contrast, Oct4 expression was not affected by either paclitaxel or knockdown of HIF-1α, xCT, or GCLM (Fig. 3C). In SUM-149 cells, paclitaxel treatment increased the expression of Nanog and Oct4 (Fig. 3D), whereas Sox2 was not detectable. The induction of Nanog and Oct4 by paclitaxel was completely abolished by cotreatment with the HIF-1 inhibitor digoxin, xCT inhibitor SSA, or GCL inhibitor BSO (Fig. 3D).
Fig. 3.
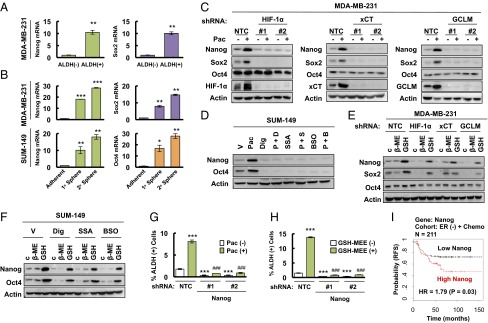
Paclitaxel-induced expression of pluripotency factors mediates BCSC enrichment. (A) MDA-MB-231 cells were sorted into ALDH (-) and ALDH (+) populations by flow cytometry. RT-qPCR was performed to analyze the expression of Nanog and Sox2 mRNA, and the results were normalized to ALDH (-) cells (mean ± SEM; n = 3). **P < 0.01 vs. ALDH (-). (B) Primary and secondary mammospheres from MDA-MB-231 (Upper) and SUM-149 (Lower) cells were harvested for RT-qPCR analyses. The results were normalized to adherent cells (mean ± SEM; n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. adherent. (C) MDA-MB-231 subclones were treated without or with 10 nM paclitaxel (Pac) for 72 h, and immunoblot assays were performed. (D) SUM-149 cells were treated with vehicle (V), 5 nM paclitaxel (Pac), 100 nM digoxin (Dig), 100 μM sulfasalazine (SSA), 100 μM buthionine sulphoximine (BSO), or Pac combined with Dig (P + D), SSA (P + S), or BSO (P + B) for 72 h, and immunoblot assays were performed. (E) MDA-MB-231 subclones were treated with 2 μM β-mercaptoethanol (β-ME) or 2 mM glutathione monoethyl ester (GSH) for 72 h and immunoblot assays were performed. (F) SUM-149 cells were treated with Dig, SSA, or BSO, in combination with vehicle, β-ME, or GSH for 72 h, and immunoblot assays were performed. (G and H) MDA-MB-231 subclones stably transfected with NTC or Nanog shRNA vector were treated without or with Pac (G), and without or with glutathione monoethyl ester (GSH-MEE) (H), and the percentage of ALDH (+) cells was determined (mean ± SEM; n = 3). ***P < 0.001 vs. NTC Pac (-) or GSH-MEE (-); ###P < 0.001 vs. NTC Pac (+) or GSH-MEE (+). (I) Kaplan–Meier analysis of relapse-free survival (RFS) was performed based on clinical and molecular data from 211 ER (-) breast cancer patients who received chemotherapy. The patients were stratified according to Nanog mRNA levels in the primary tumor. High Nanog, Nanog mRNA levels greater than the median; low Nanog, Nanog mRNA levels less than the median. The P value (log-rank test) and hazard ratio (HR) are shown.
To directly demonstrate the effect of glutathione on pluripotency factor expression, we treated MDA-MB-231 subclones with GSH-MEE and found that addition of glutathione was sufficient to induce expression of Nanog and Sox2, even in subclones with knockdown of HIF-1α, xCT, or GCLM (Fig. 3E). Similarly, treatment of SUM-149 cells with GSH-MEE induced the expression of Nanog and Oct4, even in the presence of digoxin, SSA, or BSO (Fig. 3F). Thus, GSH-MEE treatment phenocopied the cell type-specific pluripotency factor expression that was induced by chemotherapy. β-mercaptoethanol reduces extracellular cystine to cysteine, which is taken up by alanine-serine-cysteine transporters, thereby bypassing genetic or pharmacologic inhibition of xCT. Addition of β-mercaptoethanol to cell culture media partially rescued the inhibition of Nanog and Sox2 caused by xCT knockdown, but not HIF-1α or GCLM knockdown, in MDA-MB-231 cells (Fig. 3E), and partially rescued the inhibition of Nanog and Oct4 caused by SSA, but not digoxin or BSO, in SUM-149 cells (Fig. 3F).
Although paclitaxel treatment induced the expression of a different combination of pluripotency factors in different TNBC cell lines, Nanog expression was induced in both MDA-MB-231 and SUM-149. To test whether Nanog is required for BCSC maintenance, we generated stable Nanog-knockdown subclones of MDA-MB-231 cells. Knockdown of Nanog dramatically decreased the percentage of ALDH+ cells and blocked the enrichment of ALDH+ cells induced by paclitaxel (Fig. 3G). Unlike HIF-1α, xCT, or GCLM knockdown (Fig. 2I), the effect of Nanog knockdown on BCSCs could not be rescued by addition of GSH-MEE (Fig. 3H), confirming that Nanog is downstream of glutathione in the regulation of the BCSC phenotype. In addition, knockdown of Nanog in MDA-MB-231 cells dramatically decreased expression of Sox2 and Oct4 mRNA (Fig. S3A), which suggested that Nanog functions as the master pluripotency factor that specifies the BCSC phenotype in these cells. We also analyzed survival data from breast cancer patients that received chemotherapy by stratifying them according to Nanog mRNA levels in the primary tumor. Nanog mRNA levels above the median were associated with decreased patient survival in the ER− (Fig. 3I), but not in the ER+ (Fig. S3B), cohort. These results are consistent with the overexpression of SLC7A11/xCT and GCLM mRNA in basal breast cancers (Fig. S1C), the majority of which are ER− (27).
Fig. S3.

Role of Nanog expression in the regulation of other pluripotency factors and clinical outcome for ER (+) breast cancer patients treated with chemotherapy. (A) Total cellular RNA from MDA-MB-231 subclones stably transfected with NTC or Nanog shRNA vector were subjected to RT-qPCR to analyze Sox2 and Oct4 mRNA expression. The results were normalized to NTC (mean ± SEM; n = 3). ***P < 0.001 vs. NTC. (B) Kaplan–Meier analysis of relapse-free survival was performed based on clinical and molecular data from 173 ER (+) breast cancer patients who received chemotherapy. The patients were stratified according to Nanog mRNA levels in the primary tumor. High, Nanog mRNA greater than the median; Low, Nanog mRNA less than the median. The P value (log-rank test) and hazard ratio (HR) are shown.
Paclitaxel-Induced Glutathione Synthesis Activates NANOG Transcription by FoxO3.
Next, we investigated how glutathione regulates pluripotency factor expression in TNBC. The FoxO family of transcription factors has been implicated in the maintenance of somatic stem cells and several types of cancer stem cells (28–30). We first analyzed the correlation between survival of breast cancer patients who received chemotherapy and the expression of FoxO1 and FoxO3, two FoxO family members that are expressed in human breast cancers. FoxO3 mRNA levels above the median were significantly associated with decreased patient survival, with an even larger survival difference when only patients with ER− breast cancer were analyzed (Fig. S4A, Upper). In contrast, FoxO1 mRNA levels were not significantly associated with patient survival (Fig. S4A, Lower). Neither FoxO1 nor FoxO3 mRNA or protein levels were affected by changes in glutathione levels resulting from paclitaxel treatment or knockdown of HIF-1α, xCT, or GCLM (Fig. S4 B and C). However, GSH-MEE treatment induced nuclear translocation of FoxO3 (Fig. 4A), which is required for its transcriptional activity. Paclitaxel treatment also induced nuclear translocation of FoxO3 (but not FoxO1) in the MDA-MB-231 NTC subclone, whereas knockdown of HIF-1α, xCT, or GCLM was associated with loss of FoxO3 nuclear localization (Fig. 4B).
Fig. S4.
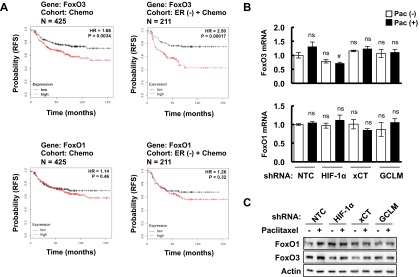
Analysis of FoxO1 and FoxO3 expression in patients and breast cancer cell lines treated with chemotherapy. (A) Kaplan–Meier analysis of relapse-free survival was performed based on clinical and molecular data from 425 breast cancer patients who received chemotherapy (Left) or from a subgroup of 211 ER (-) breast cancer patients who received chemotherapy (Right). The specimens were stratified according to levels of FoxO3 (Upper) or FoxO1 (Lower) mRNA in the primary tumor. High, FoxO mRNA levels greater than the median; Low, FoxO mRNA levels less than the median. The P value (log-rank test) and hazard ratio (HR) for each comparison are shown. (B) MDA-MB-231 subclones expressing the indicated shRNA were treated without or with paclitaxel for 72 h. RT-qPCR was performed to assay FoxO1 (Bottom) and FoxO3 (Top) mRNA levels (mean ± SEM; n = 3). #P < 0.05 vs. NTC Pac (+); ns, not significant. (C) MDA-MB-231 subclones were treated without or with paclitaxel for 72 h, and immunoblot assays were performed to analyze FoxO1 and FoxO3 protein expression.
Fig. 4.
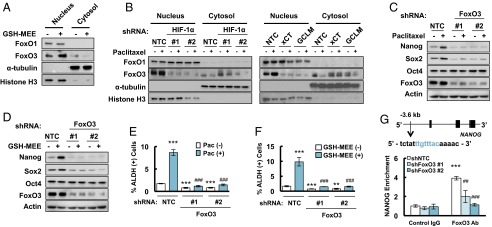
Paclitaxel promotes nuclear translocation of FoxO3 to activate NANOG transcription. (A and B) MDA-MB-231 cells were treated without or with 2 mM GSH-MEE (A), or 10 nM paclitaxel (B), for 72 h. Cytosolic and nuclear lysates were prepared, and immunoblot assays were performed. (C and D) MDA-MB-231 subclones expressing NTC or a FoxO3 shRNA vector were treated without or with paclitaxel (C), or GSH-MEE (D) for 72 h, and immunoblot assays were performed. (E and F) MDA-MB-231 NTC and FoxO3 shRNA subclones were treated without or with paclitaxel (Pac, E), or GSH-MEE (F), and the percentage of ALDH (+) cells was determined (mean ± SEM; n = 3). **P < 0.01, ***P < 0.001 vs. NTC Pac (-) or GSH-MEE (-); ###P < 0.001 vs. NTC Pac (+) or GSH-MEE (+). (G) Chromatin immunoprecipitation (ChIP) was performed by using IgG or FoxO3 antibodies in NTC and FoxO3 shRNA subclones. qPCR was performed using primers that flanked the candidate binding site, and the results were normalized to NTC ChIP with IgG (mean ± SEM; n = 3). The nucleotide sequence of the FoxO3 binding site, which is located 3.6 kb 5′ to the NANOG transcription start site, is shown. ***P < 0.001 vs. NTC with IgG; ##P < 0.01, ###P < 0.001 vs. NTC with FoxO3 antibody.
To test the hypothesis that glutathione regulates pluripotency factor expression and the BCSC phenotype through activation of FoxO3, we generated FoxO3 knockdown subclones of MDA-MB-231 cells and found that knockdown of FoxO3 completely abrogated the induction of Nanog and Sox2 expression that was induced by treatment with paclitaxel (Fig. 4C) or GSH-MEE (Fig. 4D). FoxO3 knockdown also blocked the enrichment of ALDH+ cells in response to paclitaxel (Fig. 4E) or GSH-MEE (Fig. 4F). Taken together, these results establish that FoxO3 is required for induction of the BCSC phenotype.
To investigate whether FoxO3 directly regulates NANOG gene expression, we searched the NANOG genomic DNA sequence for matches to the consensus FoxO binding-site sequence (31) and performed ChIP assays in MDA-MB-231 cells, which revealed that FoxO3 bound to a site located 3.6 kb 5′ to the transcription start site and immunoprecipitation of this DNA sequence by anti-FoxO3 antibody was significantly reduced in FoxO3 knockdown subclones compared with the NTC subclone (Fig. 4G), validating the specificity of the FoxO3 antibody. Taken together, these data indicate that paclitaxel-induced synthesis of glutathione promotes nuclear translocation of FoxO3, which activates expression of the pluripotency factor Nanog.
Glutathione Activates FoxO3 by Chelating Copper To Inhibit MEK Activity.
Glutathione is the most abundant antioxidant within mammalian cells, and its role as an antioxidant is well understood. We therefore investigated whether glutathione promotes FoxO3 nuclear translocation by decreasing intracellular oxidative stress. We exposed MDA-MB-231 cells to different concentrations of the ROS scavenger manganese (III) tetrakis(1-methyl-4-pyridyl)porphyrin (MnTMPyP) and found that at 100 μM, MnTMPyP had an effect on mitochondrial superoxide levels that was comparable to 2 mM GSH-MEE (Fig. 5A), without changing intracellular glutathione levels (Fig. 5B). Surprisingly, 100 μM MnTMPyP failed to induce nuclear translocation of FoxO3, or expression of Nanog and Sox2 in MDA-MB-231 cells, which were observed when cells were treated with 2 mM GSH-MEE (Fig. 5 C and D). These data raised the intriguing possibility that glutathione was acting as a signaling molecule, rather than as an antioxidant, to regulate FoxO3 activity.
Fig. 5.
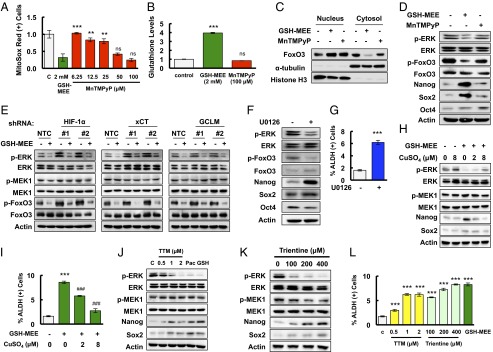
Glutathione activates FoxO3 and induces pluripotency factor expression through inhibition of MEK1 activity by chelating copper. (A) MDA-MB-231 cells were treated with GSH-MEE or MnTMPyP for 72 h. The percentage of cells positive for MitoSox Red staining was determined by flow cytometry, and the results were normalized to vehicle control (C) (mean ± SEM; n = 3). **P < 0.01, ***P < 0.001 vs. GSH-MEE; ns, not significant. (B) MDA-MB-231 cells were treated with GSH-MEE or MnTMPyP for 72 h. Glutathione levels were measured, and the results were normalized to control (mean ± SEM; n = 3). ***P < 0.001 vs. control; ns, not significant. (C) Cytosolic and nuclear lysates were prepared from MDA-MB-231 cells treated with GSH-MEE or MnTMPyP, and immunoblot assays were performed. (D) MDA-MB-231 cells were treated with GSH-MEE or MnTMPyP, and immunoblot assays were performed. (E) MDA-MB-231 subclones were treated without or with GSH-MEE, and immunoblot assays were performed. (F and G) MDA-MB-231 cells were treated with 0.625 μM U0126 (+) or vehicle (-) for 72 h and immunoblot assays were performed (F), or the percentage of ALDH (+) cells was determined by flow cytometry (G; mean ± SEM; n = 3). ***P < 0.001 vs. control. (H) MDA-MB-231 cells were treated with 2 mM GSH-MEE, either alone or in combination with CuSO4 for 72 h, and immunoblot assays were performed. (I) MDA-MB-231 cells were treated with GSH-MEE, either alone or in combination with CuSO4, and the percentage of ALDH (+) cells was determined (mean ± SEM; n = 3). ***P < 0.001 vs. control; ###P < 0.001 vs. GSH-MEE alone. (J) MDA-MB-231 cells were treated with tetrathiomolybdate (TTM), paclitaxel (Pac), or GSH-MEE (GSH) for 72 h, and immunoblot assays were performed. (K) MDA-MB-231 cells were treated with trientine for 72 h, and immunoblot assays were performed. (L) MDA-MB-231 cells were treated with TTM, trientine, or GSH-MEE, and the percentage of ALDH (+) cells was determined (mean ± SEM; n = 3). ***P < 0.001 vs. control.
ERK phosphorylates FoxO3 on serine residues S294, S344, and S425 and induces its nuclear exclusion, leading to loss of FoxO3 transcriptional activity (32). We found that glutathione treatment inhibited phosphorylation of ERK at T202/Y204 and FoxO3 at S294 (Fig. 5D), suggesting that glutathione may activate FoxO3 by inhibition of ERK signaling. To test this hypothesis, MDA-MB-231 subclones were treated with 2 mM GSH-MEE for 72 h. Knockdown of HIF-1α, xCT, or GCLM, which inhibited glutathione synthesis and decreased intracellular glutathione levels, also promoted phosphorylation of ERK and FoxO3 (Fig. 5E). GSH-MEE treatment not only inhibited ERK and FoxO3 phosphorylation in the NTC subclone, but also reversed the activation of ERK signaling in the HIF-1α, xCT, and GCLM knockdown subclones (Fig. 5E). In contrast, paclitaxel treatment inhibited ERK and FoxO3 phosphorylation in the NTC subclone, but not in the HIF-1α, xCT, or GCLM knockdown subclone (Fig. S5). These results were consistent with the failure of paclitaxel to increase intracellular glutathione levels when the HIF-1–regulated glutathione synthesis pathway was blocked (Fig. 1C). The phosphorylation of MEK1 was not affected by changes in glutathione levels (Fig. 5E). Pharmacological inhibition of ERK signaling by the MEK1 inhibitor U0126 precisely phenocopied the effect of glutathione on inhibition of FoxO3 phosphorylation and induction of pluripotency factors in MDA-MB-231 and SUM-149 cells (Fig. 5F and Fig. S6), and enrichment of the ALDH+ population in MDA-MB-231 cells (Fig. 5G). Taken together, these data indicate that glutathione promotes FoxO3 activity through inhibition of MEK1-ERK signaling.
Fig. S5.

Paclitaxel inhibits phosphorylation of ERK and FoxO3, which is reversed by inhibition of glutathione synthesis. MDA-MB-231 subclones (NTC, HIF-1α, xCT, or GCLM knockdown) were treated without or with 10 nM paclitaxel for 72 h, and immunoblot assays were performed.
Fig. S6.

MEK1 inhibitor U0126 induces expression of pluripotency factors in SUM-149 cells. SUM-149 cells were treated with 0.625 μM U0126 for 72 h, and immunoblot assays were performed.
Glutathione is a known chelator of metal ions (33). Recently, it has been reported that copper is a required cofactor for the kinase activity of MEK1 (34, 35). We hypothesized that glutathione inhibits MEK1 activity through chelating copper. MDA-MB-231 cells were treated with GSH-MEE for 72 h, in the absence or presence of CuSO4. GSH-MEE treatment inhibited ERK phosphorylation and increased expression of Nanog and Sox2, which were partially reversed by copper supplementation in a dose-dependent manner (Fig. 5H). Coadministration of CuSO4 with GSH-MEE also decreased the percentage of glutathione-induced ALDH+ cells dose-dependently (Fig. 5I). Treatment of MDA-MB-231 cells with the copper chelator tetrathiomolybdate (TTM) or trientine for 72 h phenocopied the inhibition of ERK phosphorylation and activation of Nanog and Sox2 by glutathione (Fig. 5 J and K). In addition, both TTM and trientine increased the percentage of ALDH+ cells (Fig. 5L). Taken together, these results indicate that glutathione chelates copper and, thereby, inhibits MEK1 activity, leading to dephosphorylation and nuclear translocation of FoxO3.
Paclitaxel Promotes BCSC Enrichment by Induction of Glutathione Synthesis in Vivo.
To investigate whether paclitaxel induces BCSC enrichment in vivo through the molecular mechanisms that we had delineated in cell culture, MDA-MB-231 NTC, xCT, or GCLM knockdown cells were implanted in the mammary fat pad of SCID mice. When the tumors reached a volume of 200 mm3, the mice were treated with 10 mg/kg paclitaxel by i.p. injection every 5 d for three doses. Tumors were harvested 3 d after the last dose for analysis of glutathione levels (Fig. 6A), ALDH+ cells (Fig. 6B), and expression of mRNA (Fig. S7A) and protein (Fig. 6C and Fig. S7B). Paclitaxel induced xCT and GCLM mRNA and protein expression, and increased intracellular glutathione levels, which were blocked by knockdown of xCT or GCLM. Paclitaxel also inhibited phosphorylation of ERK and FoxO3, whereas knockdown of xCT or GCLM, which decreased glutathione levels, increased the phosphorylation of ERK and FoxO3. In addition, paclitaxel treatment induced expression of Nanog and Sox2 mRNA and protein, and increased the percentage of ALDH+ cells, which were abrogated by knockdown of xCT or GCLM (Fig. 6 B and C and Fig. S7).
Fig. 6.

Paclitaxel induces synthesis of glutathione, which inhibits MEK1 activity, increases FoxO3 activity and pluripotency factor expression and promotes tumor initiation. (A–C), MDA-MB-231 subclones were implanted into SCID mice. When tumor volume reached 200 mm3 (day 0), mice were randomly assigned to treatment with i.p. injections of saline [Pac (-)] or 10 mg/kg paclitaxel [Pac (+)] on days 0, 5, and 10. Tumors were harvested on day 13, and samples were analyzed for intracellular total glutathione levels (A), percentage of ALDH (+) cells (B), and protein expression as indicated (C) (mean ± SEM; n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. NTC Pac (-); #P < 0.05, ##P < 0.01, ###P < 0.001 vs. NTC Pac (+); ns, not significant. (D) One thousand cells of each MDA-MB-231 subclone were implanted into the mammary fat pad of female SCID mice. The number of mice that developed palpable tumors after 65 d in each group was reported, and Fisher’s exact test was performed to determine statistical significance vs. NTC group.
Fig. S7.
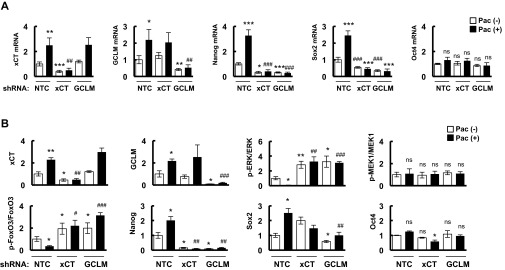
Paclitaxel increases expression of xCT, GCLM, and pluripotency factors in vivo. (A and B) MDA-MB-231 subclones (NTC, xCT, or GCLM knockdown) were implanted into the mammary fat pad of female severe combined immunodeficiency (SCID) mice. When tumor volume reached 200 mm3 (day 0), mice were grouped randomly and treated with saline or 10 mg/kg paclitaxel (Pac) on days 0, 5, and 10. Tumors were harvested on day 13. Total RNA was isolated, and RT-qPCR was performed (A). Tissue protein lysates were prepared from tumors, and immunoblot assays were performed and quantified (B) (mean ± SEM; n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. NTC Pac (-); #P < 0.05, ##P < 0.01, ###P < 0.001 vs. NTC Pac (+).
To test the role of xCT, GCLM, and FoxO3 in determining the tumor initiation potential of breast cancer cells in vivo, we injected 1,000 MDA-MB-231 NTC, xCT, GCLM, or FoxO3 knockdown cells into the mammary fat pad of female SCID mice. NTC subclone cells formed tumors in all nine mice injected after 65 d, whereas xCT, GCLM, and FoxO3 knockdown subclone cells all showed significantly decreased tumor initiating capacity (Fig. 6D), supporting their role in the maintenance of BCSCs. Taken together, these data support our model that chemotherapy induced glutathione synthesis through HIF-1–mediated SLC7A11 and GCLM gene expression. Increased glutathione levels inhibited MEK1-ERK signaling and FoxO3 phosphorylation, leading to nuclear translocation of FoxO3, which transcriptionally activated NANOG gene expression to promote the BCSC phenotype (Fig. 7).
Fig. 7.

Pluripotency factor expression and the BCSC phenotype are induced through HIF-1–dependent glutathione synthesis in response to chemotherapy. Chemotherapy treatment induces HIF-1–dependent SLC7A11 and GCLM transcriptional activation, leading to increased glutathione synthesis. Glutathione chelates copper, which is a required cofactor for MEK1, leading to inactivation of MEK1-ERK signaling, FoxO3 dephosphorylation, and nuclear translocation, leading to transcriptional activation of NANOG, which encodes a pluripotency factor that specifies the BCSC phenotype.
Discussion
BCSCs are resistant to cytotoxic chemotherapy and play a major role in breast cancer relapse and metastasis. Recent studies have suggested that chemotherapy increases the percentage of BCSCs (8–10) and that HIFs are required for this process (10). However, the molecular mechanisms through which HIFs contribute to BCSC enrichment in response to chemotherapy are only beginning to be elucidated. In the present study, we have demonstrated that HIF-1 regulates BCSCs by increasing the synthesis of glutathione for use as a signaling molecule. Our previous study implicated chemotherapy-induced and HIF-1–dependent expression of IL-6, IL-8, and MDR1 as a mechanism underlying the differential survival, relative to bulk cancer cells, of BCSCs (10). In contrast, the present study has delineated a mechanism that results in an active induction of the BCSC phenotype mediated by increased expression of pluripotency factors.
Cancer cells are metabolically active, under increased oxidative stress, and require robust antioxidant capacity to maintain viability (36). Glutathione is the major endogenous antioxidant produced by mammalian cells to detoxify free radicals. However, the role of glutathione in tumor initiation, cancer progression, and chemotherapy resistance has been controversial for many years, perhaps because glutathione may play both protective and pathogenic roles with respect to cancer (18, 37). Recently it has been shown that inhibition of xCT or GCL by treatment with SSA or BSO, respectively, potentiates the anticancer effect of chemotherapy on breast cancer cell lines (38, 39). Our results demonstrate that targeting the glutathione synthesis pathway also decreases the number of BCSCs, which are required for breast cancer recurrence and metastasis. Thus, inhibiting the glutathione synthesis pathway in combination with chemotherapy may improve patient outcome in TNBC. In addition, we demonstrate that administration of glutathione is sufficient to enrich BCSCs, which may serve as a caveat to the use of glutathione as a dietary supplement (21).
Although novel roles for glutathione in cancer metabolism have been described recently (40), they have been related to its role as an antioxidant. Our studies revealed that glutathione induces the BCSC phenotype through a molecular mechanism that is independent of its antioxidant role. Glutathione also functions as a chelator to protect cells from damage caused by heavy metal ions (33). Although excess copper is toxic to cells, copper is indispensable for cells under physiological conditions and controls many important biochemical processes by regulating protein structure, catalytic activity, and protein–protein interactions (41). It has been reported recently that copper is an obligate cofactor for MEK1 activity (35). In this study, we demonstrate that glutathione inhibits ERK phosphorylation through copper chelation, which is phenocopied by two other copper chelators, tetrathiomolybdate and trientine. Copper has been considered as a target for cancer therapy (42), but our results suggest it may not be a good target because copper chelators may enrich for cancer stem cells, whereas copper ionophores will increase cancer cell proliferation (43), neither of which is desired. The inhibition of ERK phosphorylation by glutathione is consistent with the effect of ATN-224, a specific copper chelator that is under clinical trials, which also inhibits ERK phosphorylation (44). The regulatory role of glutathione in cancer cell signaling highlights the importance of glutathione in balancing proliferation, stem cell maintenance, and redox homeostasis.
The Ras-Raf-MEK-ERK signal transduction pathway plays a critical role in mediating fundamental biological processes, including cell survival, proliferation, and migration (35). This pathway is commonly activated in many types of cancer, making it a rational target for cancer therapeutics. Several MEK inhibitors have been tested in preclinical and clinical studies in combination with chemotherapy. However, recent studies showed that ERK signaling is important for promoting stem cell differentiation and antagonizing self-renewal (45, 46), findings that are consistent with our results. In the current study, we demonstrate that inhibition of MEK1 activity by U0126 at a low dose (0.625 μM) but for a relatively long time (72 h), promotes FoxO3 nuclear translocation and activates FoxO3 transcriptional activity, leading to the expression of pluripotency factors and enrichment of BCSCs in TNBC. The negative correlation between ERK phosphorylation and pluripotency factor expression in our study is also consistent with the inhibitory effect of ERK on pluripotency factors that was observed in embryonic stem cells (47, 48). These observations raise the concern that although inhibition of MEK1-ERK signaling pathway may decrease primary breast tumor growth, it might enrich for BCSCs, thereby increasing the risk of cancer recurrence and/or metastasis. More detailed studies are required to evaluate MEK-ERK inhibitors for their potential countertherapeutic effects on BCSCs.
The reported roles of FoxO3 are also contradictory in different types of cancer and in different phases of cancer progression, which is likely due to the fact that FoxO3 regulates diverse gene expression programs involved in cell division, survival, metabolism, and redox homeostasis (49). Our analysis revealed that the survival of breast cancer patients who received chemotherapy was negatively correlated with expression of FoxO3 in the primary tumor: Higher FoxO3 expression predicted decreased survival, supporting the role of FoxO3 in promoting chemotherapy resistance and tumor recurrence in breast cancer.
In summary, we have delineated a signaling pathway through which HIF-1–mediated glutathione synthesis contributes to chemotherapy-induced cancer stem cell enrichment in TNBC, which can be targeted by inhibiting HIF-1α, xCT, or GCLM activity or expression. Glutathione plays an important role in regulating the expression of Nanog and other pluripotency factors that determine the BCSC phenotype, through its activity as a signaling molecule rather than exclusively as an antioxidant. In addition, we have demonstrated that glutathione, MEK1-ERK inhibitors, and copper chelators promote the BCSC phenotype, indicating that caution should be exercised regarding their use as cancer therapeutics. Coadministration of a HIF-1 inhibitor, such as digoxin (10, 23, 50) or ganetespib (51), with cytotoxic chemotherapy may be particularly effective because of the multiple mechanisms by which HIF-1 promotes BCSCs and other aspects of cancer progression (52).
Methods
Cell Culture and Reagents.
MDA-MB-231 and SUM-149 cells were maintained as described (10) at 37 °C in a 5% CO2, 95% air incubator (20% O2). Hypoxic cells were placed in a modular incubator chamber (Billups-Rothenberg) flushed with a gas mixture containing 1% O2, 5% CO2, and 94% N2. Paclitaxel, gemcitabine, carboplatin, digoxin, SSA, BSO, β-mercaptoethanol, TTM, trientine, and CuSO4 were purchased from Sigma-Aldrich. MnTMPyP and U0126 were purchased from EMD Millipore and Life Technologies.
Lentivirus Transduction.
Lentiviral vectors encoding shRNA targeting HIF-1α and HIF-2α were described (50). pLKO.1-puro lentiviral vectors encoding shRNA targeting xCT, GCLM, Nanog, and FoxO3 were purchased from Sigma-Aldrich, and shRNA sequences are shown in Table S1. Lentiviruses were packaged, and MDA-MB-231 cells were transduced and subjected to puromycin selection as described (50).
Table S1.
Oligonucleotide sequence of shRNAs
| shRNA | Clone ID | Sequence |
| SLC7A11 #1 | NM_014331.2–1384s1c1 | CCGGCCTGCGTATTATCTCTTTATTCTCGAGAATAAAGAGATAATACGCAGGTTTTTG |
| SLC7A11 #2 | NM_014331.3–1995s21c1 | CCGGTATTCTATGAGTCGCACAATTCTCGAGAATTGTGCGACTCATAGAATATTTTTG |
| GCLM #1 | NM_002061.2–840s21c1 | CCGGCCAAATAGTAACCAAGTTAATCTCGAGATTAACTTGGTTACTATTTGGTTTTTG |
| GCLM #2 | NM_002061.1–842s1c1 | CCGGCCACCAGATTTGACTGCATTTCTCGAGAAATGCAGTCAAATCTGGTGGTTTTTG |
| Nanog #1 | NM_024865.2–1135s21c1 | CCGGAGATGAGTGAAACTGATATTACTCGAGTAATATCAGTTTCACTCATCTTTTTTTG |
| Nanog #2 | NM_024865.2–1047s21c1 | CCGGACAGCAGACCACTAGGTATTTCTCGAGAAATACCTAGTGGTCTGCTGTTTTTTTG |
| FoxO3 #1 | NM_001455.x-2766s1c1 | CCGGCATGTTCAATGGGAGCTTGGACTCGAGTCCAAGCTCCCATTGAACATGTTTTTG |
| FoxO3 #2 | NM_001455.3–1105s21c1 | CCGGGGACAATAGCAACAAGTATACCTCGAGGTATACTTGTTGCTATTGTCCTTTTTG |
RT-qPCR.
Total RNA was extracted, cDNA was synthesized, and qPCR analysis was performed as described (50). PCR primer sequences are shown in Table S2.
Table S2.
Oligonucleotide sequence of RT-qPCR primers
| mRNA | Nucleotide sequence (5′ to 3′) |
| SLC7A11 | Forward: AGGGTCACCTTCCAGAAATC |
| Reverse: GAAGATAAATCAGCCCAGCA | |
| GCLM | Forward: CATTTACAGCCTTACTGGGAGG |
| Reverse: ATGCAGTCAAATCTGGTGGCA | |
| FOXO1 | Forward: TCGTCATAATCTGTCCCTACACA |
| Reverse: CGGCTTCGGCTCTTAGCAAA | |
| FOXO3 | Forward: CGGACAAACGGCTCACTCT |
| Reverse: GGACCCGCATGAATCGACTAT | |
| NANOG | Forward: TTTGTGGGCCTGAAGAAAACT |
| Reverse: AGGGCTGTCCTGAATAAGCAG | |
| SOX2 | Forward: GCCGAGTGGAAACTTTTGTCG |
| Reverse: GGCAGCGTGTACTTATCCTTCT | |
| OCT4 | Forward: TTCAGCCAAACGACCATCTG |
| Reverse: CACGAGGGTTTCTGCTTTGC | |
| 18S rRNA | Forward: CGGCGACGACCCATTCGAAC |
| Reverse: GAATCGAACCCTGATTCCCCGTC |
ChIP.
MDA-MB-231 cells were cross-linked in 3.7% formaldehyde for 15 min, quenched in 0.125 M glycine for 5 min, and lysed with SDS lysis buffer. Chromatin was sheared by sonication, and lysates were precleared with salmon sperm DNA/protein A agarose slurry (Millipore) for 1 h and incubated with IgG or antibodies against HIF-1α (Santa Cruz), HIF-1β, or FoxO3 (Novus Biologicals) in the presence of protein A agarose beads overnight. After sequential washes of the agarose beads, DNA was eluted in 1% SDS with 0.1 M NaHCO3, and cross-links were reversed by addition of 0.2 M NaCl. DNA was purified by phenol-chloroform extraction and ethanol precipitation, and analyzed by qPCR (Table S3).
Table S3.
Oligonucleotide sequence of ChIP primers
| Binding site | Nucleotide sequence (5′ to 3′) |
| SLC7A11 HIF-1 binding site | Forward: ATCTTGGCTCACTGCAAGGT |
| Reverse: GATCGAGACCATCCTGGCTA | |
| GCLM HIF-1 binding site | Forward: AGCTGCTTCTGAGAACGAAAA |
| Reverse: CGCCTGGTGAGGTAGACAC | |
| NANOG FoxO3 binding site | Forward: TGAGGCAAAGAGCATTGTTG |
| Reverse: TGATGAAGGAATGGGCATAA |
Immunoblot Assay.
Whole-cell lysates were prepared in modified RIPA buffer, and proteins were separated by SDS/PAGE, blotted onto nitrocellulose membranes, and probed with primary antibodies (Table S4) followed by HRP-conjugated secondary antibodies (GE Healthcare). The chemiluminescent signal was detected by using ECL Plus (GE Healthcare).
Table S4.
Primary antibody information for immunoblot assays
| Antibody | Manufacturer | Catalog no. |
| HIF-1α | BD Biosciences | 610959 |
| xCT | Novus Biologicals | NB300-318 |
| GCLM | Novus Biologicals | NBP1-33405 |
| Nanog | Novus Biologicals | NB100-588 |
| Sox2 | Novus Biologicals | NB100-2379 |
| Oct4 | Novus Biologicals | NB110-37235 |
| FoxO3 | Novus Biologicals | NBP2-16521 |
| p-ERK (T202/Y204) | Novus Biologicals | NBP1-19924 |
| ERK | Novus Biologicals | NBP1-61866 |
| p-MEK1 (S221) | Novus Biologicals | NB100-82033 |
| MEK1 | Novus Biologicals | NBP1-47833 |
| p-FoxO3 (S294) | Cell Signaling | #5538 |
| Actin | Santa Cruz | sc-1616 |
Breast Cancer Stem Cell Assays.
Aldefluor assays were performed according to manufacturer’s instructions (Stem Cell Technologies) as described (10). For cell sorting, the top and bottom 5% of Aldefluor-expressing cells were collected by flow cytometry and considered as ALDH+ and ALDH−, respectively. Mammosphere assays were performed as described (10).
Subcellular Fractionation.
Cells were resuspended in 10 mM Hepes, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.05% Igepal, and protease inhibitor mixture (Roche), incubated on ice for 15 min, and centrifuged for 10 min at 1,000 × g. The supernatant was reserved as the cytosolic fraction. The pellet was resuspended in 5 mM Hepes, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 26% (vol/vol) glycerol and protease inhibitor mixture, homogenized with a Dounce homogenizer (30 strokes), incubated on ice for 30 min, and centrifuged to collect supernatant as the nuclear fraction.
Glutathione Assay.
Cultured cells were trypsinized, collected by centrifugation, resuspended in 5% (wt/vol) 5-sulfosalicylic acid, subjected to three freeze-and-thaw cycles, and centrifuged to remove debris. Tumor tissues were frozen in liquid nitrogen, ground with a mortar and pestle, resuspended in 5% 5-sulfosalicylic acid, homogenized in a Dounce homogenizer, and centrifuged to remove debris. Supernatants were analyzed by using a glutathione assay kit (Sigma-Aldrich) based on the enzymatic reaction of glutathione with 5,5′-dithiobis-2-nitrobenzoic acid. Absorbance was quantified with a spectrophotometer at the wavelength of 405 nm.
Orthotopic Transplantation.
Protocols were approved by the Johns Hopkins University Animal Care and Use Committee and were in accordance with the NIH Guide for the Care and Use of Laboratory Animals. MDA-MB-231 subclones were injected into the mammary fat pad of 5- to 7-wk-old female SCID mice in a 1:1 suspension of Matrigel (BD Biosciences) in PBS.
Acknowledgments
We thank Karen Padgett of Novus Biologicals for providing antibodies against xCT, GCLM, HIF-1β, Nanog, Sox2, Oct4, FoxO3, ERK, phospho-ERK, MEK1, and phospho-MEK1. This work was supported by Impact Award W81XWH-12-1-0464 from the Breast Cancer Research Program of the Department of Defense. G.L.S. is an American Cancer Society Research Professor and the C. Michael Armstrong Professor at the Johns Hopkins University School of Medicine. L.X. was supported by a China Scholarship Council grant and National Natural Science Foundation of China Grant 81402526.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1513433112/-/DCSupplemental.
References
- 1.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 2.Dent R, et al. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 3.André F, Zielinski CC. Optimal strategies for the treatment of metastatic triple-negative breast cancer with currently approved agents. Ann Oncol. 2012;23(Suppl 6):vi46–vi51. doi: 10.1093/annonc/mds195. [DOI] [PubMed] [Google Scholar]
- 4.Polyak K. Heterogeneity in breast cancer. J Clin Invest. 2011;121(10):3786–3788. doi: 10.1172/JCI60534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pece S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140(1):62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Oskarsson T, Batlle E, Massagué J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem Cell. 2014;14(3):306–321. doi: 10.1016/j.stem.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 8.Li X, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100(9):672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 9.Bhola NE, et al. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123(3):1348–1358. doi: 10.1172/JCI65416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci USA. 2014;111(50):E5429–E5438. doi: 10.1073/pnas.1421438111. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Ginestier C, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dontu G, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17(10):1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaupel P, Mayer A, Briest S, Höckel M. Hypoxia in breast cancer: Role of blood flow, oxygen diffusion distances, and anemia in the development of oxygen depletion. Adv Exp Med Biol. 2005;566:333–342. doi: 10.1007/0-387-26206-7_44. [DOI] [PubMed] [Google Scholar]
- 14.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flamant L, Notte A, Ninane N, Raes M, Michiels C. Anti-apoptotic role of HIF-1 and AP-1 in paclitaxel exposed breast cancer cells under hypoxia. Mol Cancer. 2010;9:191–205. doi: 10.1186/1476-4598-9-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9(4):298–310. doi: 10.1016/j.stem.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 17.Perales-Clemente E, Folmes CD, Terzic A. Metabolic regulation of redox status in stem cells. Antioxid Redox Signal. 2014;21(11):1648–1659. doi: 10.1089/ars.2014.6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Traverso N, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. 2013;2013:972913. doi: 10.1155/2013/972913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okuno S, et al. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br J Cancer. 2003;88(6):951–956. doi: 10.1038/sj.bjc.6600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Timmerman LA, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24(4):450–465. doi: 10.1016/j.ccr.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris IS, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27(2):211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 22.Parker JS, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci USA. 2008;105(50):19579–19586. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheridan C, et al. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: An early step necessary for metastasis. Breast Cancer Res. 2006;8(5):R59. doi: 10.1186/bcr1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qu Y, et al. Thioredoxin-like 2 regulates human cancer cell growth and metastasis via redox homeostasis and NF-κB signaling. J Clin Invest. 2011;121(1):212–225. doi: 10.1172/JCI43144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piva M, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. 2014;6(1):66–79. doi: 10.1002/emmm.201303411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyamoto K, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1(1):101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Naka K, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463(7281):676–680. doi: 10.1038/nature08734. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, et al. FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat Cell Biol. 2011;13(9):1092–1099. doi: 10.1038/ncb2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J. 2000;349(Pt 2):629–634. doi: 10.1042/0264-6021:3490629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang JY, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10(2):138–148. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Freedman JH, Ciriolo MR, Peisach J. The role of glutathione in copper metabolism and toxicity. J Biol Chem. 1989;264(10):5598–5605. [PubMed] [Google Scholar]
- 34.Turski ML, et al. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol Cell Biol. 2012;32(7):1284–1295. doi: 10.1128/MCB.05722-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brady DC, et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature. 2014;509(7501):492–496. doi: 10.1038/nature13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trachootham D, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10(3):241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 37.Balendiran GK, Dabur R, Fraser D. The role of glutathione in cancer. Cell Biochem Funct. 2004;22(6):343–352. doi: 10.1002/cbf.1149. [DOI] [PubMed] [Google Scholar]
- 38.Lo M, Ling V, Wang YZ, Gout PW. The xc- cystine/glutamate antiporter: A mediator of pancreatic cancer growth with a role in drug resistance. Br J Cancer. 2008;99(3):464–472. doi: 10.1038/sj.bjc.6604485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Narang VS, Pauletti GM, Gout PW, Buckley DJ, Buckley AR. Sulfasalazine-induced reduction of glutathione levels in breast cancer cells: Enhancement of growth-inhibitory activity of Doxorubicin. Chemotherapy. 2007;53(3):210–217. doi: 10.1159/000100812. [DOI] [PubMed] [Google Scholar]
- 40.Sullivan LB, et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013;51(2):236–248. doi: 10.1016/j.molcel.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4(3):176–185. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- 42.Gupte A, Mumper RJ. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat Rev. 2009;35(1):32–46. doi: 10.1016/j.ctrv.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 43.Ishida S, Andreux P, Poitry-Yamate C, Auwerx J, Hanahan D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc Natl Acad Sci USA. 2013;110(48):19507–19512. doi: 10.1073/pnas.1318431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Juarez JC, et al. Copper binding by tetrathiomolybdate attenuates angiogenesis and tumor cell proliferation through the inhibition of superoxide dismutase 1. Clin Cancer Res. 2006;12(16):4974–4982. doi: 10.1158/1078-0432.CCR-06-0171. [DOI] [PubMed] [Google Scholar]
- 45.Booth A, Trudeau T, Gomez C, Lucia MS, Gutierrez-Hartmann A. Persistent ERK/MAPK activation promotes lactotrope differentiation and diminishes tumorigenic phenotype. Mol Endocrinol. 2014;28(12):1999–2011. doi: 10.1210/me.2014-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burdon T, Smith A, Savatier P. Signalling, cell cycle and pluripotency in embryonic stem cells. Trends Cell Biol. 2002;12(9):432–438. doi: 10.1016/s0962-8924(02)02352-8. [DOI] [PubMed] [Google Scholar]
- 47.Hamilton WB, Brickman JM. Erk signaling suppresses embryonic stem cell self-renewal to specify endoderm. Cell Reports. 2014;9(6):2056–2070. doi: 10.1016/j.celrep.2014.11.032. [DOI] [PubMed] [Google Scholar]
- 48.Leitch HG, et al. Embryonic germ cells from mice and rats exhibit properties consistent with a generic pluripotent ground state. Development. 2010;137(14):2279–2287. doi: 10.1242/dev.050427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eijkelenboom A, Burgering BM. FOXOs: Signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14(2):83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- 50.Zhang H, et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene. 2012;31(14):1757–1770. doi: 10.1038/onc.2011.365. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Xiang L, et al. Ganetespib blocks HIF-1 activity and inhibits tumor growth, vascularization, stem cell maintenance, invasion, and metastasis in orthotopic mouse models of triple-negative breast cancer. J Mol Med (Berl) 2014;92(2):151–164. doi: 10.1007/s00109-013-1102-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Semenza GL. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim Biophys Acta. 2015;pii:S0167-4889(15):00192–00195. doi: 10.1016/j.bbamcr.2015.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]