

Significance
Binding of nucleotides to proteins is arguably the most important regulation process in our body. How the signal of nucleotide binding gets transmitted through the protein eventually changing its conformation and function is, however, still a poorly understood process. In this study, we set out to investigate the energy balance within the nucleotide binding domain (NBD) of the heat shock protein of 70 kDa (Hsp70) chaperone DnaK. Using single molecule force spectroscopy and theoretical simulations, we were able to dissect the contribution of the individual subdomains of the NBD to the mechanical stability of the protein and identified how the mechanical hierarchy is modulated by nucleotide binding. Our results have immediate consequences for the signal transduction within the whole actin/sugar kinase superfamily.
Keywords: ATPase, laser trapping, elasticity, force, protein extension
Abstract
The regulation of protein function through ligand-induced conformational changes is crucial for many signal transduction processes. The binding of a ligand alters the delicate energy balance within the protein structure, eventually leading to such conformational changes. In this study, we elucidate the energetic and mechanical changes within the subdomains of the nucleotide binding domain (NBD) of the heat shock protein of 70 kDa (Hsp70) chaperone DnaK upon nucleotide binding. In an integrated approach using single molecule optical tweezer experiments, loop insertions, and steered coarse-grained molecular simulations, we find that the C-terminal helix of the NBD is the major determinant of mechanical stability, acting as a glue between the two lobes. After helix unraveling, the relative stability of the two separated lobes is regulated by ATP/ADP binding. We find that the nucleotide stays strongly bound to lobe II, thus reversing the mechanical hierarchy between the two lobes. Our results offer general insights into the nucleotide-induced signal transduction within members of the actin/sugar kinase superfamily.
Proteins are complex biomolecular nanomachines that fulfill many vital tasks in the cell. One of the remarkable features of protein machines is their ability to undergo conformational changes that can be regulated selectively by dedicated ligands. Because the interaction network within a protein structure is highly dynamic and fine-tuned, it is extremely difficult to predict to what extent a ligand will affect conformational states and, hence, the remodeling of substructures within a folded protein (1). Many large conformational transitions in proteins can be viewed as a change in a delicate energy balance between alternative folded conformations; the signal transduction is triggered by the ligand-induced disruption of the weak interface between stable substructures, which further propagates and results in a coordinated domain movement. Single molecule mechanical methods have the potential to supply a valuable tool to study mechanical hierarchies within folded protein molecules, for they can probe nanometer-sized collective protein fluctuations as a response to ligand binding (2–4). In this study, we use an integrated approach of single molecule measurements together with steered molecular simulations to investigate the influence of ligand binding (5–8) on the mechanical stability of the subdomains of an ATPase domain of DnaK, a member of the heat shock protein of 70 kDa (Hsp70) family of chaperones.
The Hsp70 family is a ubiquitous group of proteins displaying ATP-regulated chaperone function (9, 10). The Hsp70 proteins are conserved in all kingdoms of life—from bacteria to human. Hsp70 from Escherichia coli, DnaK—the most prominent member of the Hsp70 family (10)—comprises two domains reflecting their distinct functions: a nucleotide binding domain (NBD; Fig. 1A), and a substrate binding domain (SBD). The domains are connected by a short, flexible linker (for structure, see refs. 11, 12). The NBD binds MgADP and MgATP at nanomolar affinity (13); the SBD binds a large repertoire of protein clients and confers chaperone function (14, 15). The binding of the substrates is strongly coupled to the nucleotide state of the NBD, which is triggered by allosteric communication between the domains. The nucleotide binding and regulation of ATPase activity play a central role in the biological function of DnaK, and disruption of the ATPase activity or interdomain communication impairs biological function in vivo (16, 17). The chaperone activity of DnaK is further enhanced by the cochaperones DnaJ and GrpE (reviewed in ref. 18); both proteins regulate the nucleotide turnover of the NBD.
Fig. 1.

Structure of the NBD and the nucleotide binding pocket. (A) The 3D structure of the NBD from DnaK (PDB ID code 2KHO) (11). The position for the insertions (D45, K183, A290, and D364) are shown in black as a stick representation. (B) The protein comprises two lobes—lobe I (blue surface) and lobe II (red surface)—which are further subdivided into “a” and “b” subdomains. The lobes are connected by the C-terminal helix (gray surface). The ATP molecule (yellow spheres) binds between the lobes (PDB ID code 4B9Q) (39). The figures were prepared using DS Viewer (Accelrys Software, Inc.). (C) The analysis and mapping of the contacts between the protein’s residues and Mg2+ATP4- ligand (ball-and-stick representation, Mg2+ ion in green) using LigPlot+ software (42). The residue names have background-shading based on their correspondence to the lobes. Black broken lines indicate the hydrogen bonding between atoms; gray broken lines indicate hydrophobic/van der Waals contact.
Here, we focus on one specific question: How does nucleotide binding regulate the mechanical stability of the NBD and its substructures? Owing to its characteristic 3D butterfly-like structure, the NBD belongs to the actin/sugar kinase superfamily; it comprises four subdomains—Ia, Ib and IIa, IIb—which assemble into two lobes—I and II (Fig. 1). The binding site for MgATP/MgADP is located in a deep cleft at the bottom of the lobes, and it comprises residues from both lobes (Fig. 1C). The two lobes are held tightly by the C-terminal helix. A remarkable symmetric arrangement of the lobes and conservation of the phosphate binding residues might be an indication for divergent evolution after gene duplication of the single-lobe precursor protein (19). In other words, one speculates that lobe I and lobe II had a common ancestor protein and their specialization occurred later. Even though many biophysical, mutational, and biochemical analyses have provided us with structural/thermodynamic insights on protein stability and nucleotide binding (20, 21), the energetic coupling to the mechanics of the individual structures within NBD has remained elusive. We show that nucleotide binding is strongly coupled to the mechanical stability of lobe II, and we identify the residues of lobe II as the major contributors to the binding-free energy of the nucleotide.
Results
Single Molecule Force Experiments on the NBD of the DnaK Chaperone.
To study the unfolding pathways of the NBD by optical tweezers, we prepared a construct for trapping experiments carrying two cysteine residues at the termini of the protein (Fig. 2A) (22). The cysteine residues were introduced for modification by a single-stranded oligonucleotide containing a maleimide group. The DNA–protein–DNA construct was then hybridized to a single-stranded overhang of a long DNA handle containing biotin or a digoxygenin group (Fig. 2A and Methods). These groups bind to functionalized streptavidin or α-digoxygenin decorated glass beads, so that mechanical forces can be applied to the DNA–protein tether. In a first set of experiments, we probed the mechanical NBD unfolding pathway in the absence of nucleotides (the “apo-form”). A typical force–extension curve of the NBD–DNA construct obtained at a pulling speed of 20 nm/s is depicted in Fig. 2B (black curve). At extensions below ca. 350 nm, the force response reflects the elastic stretching of the linking DNA tether. At a force of 34 pN, a sudden rip can be observed corresponding to the unfolding of the protein. After the unfolding event, the contour length of the construct has increased by 134 nm as measured using a worm-like chain (WLC) analysis (23) (SI Appendix, Fig. S1). The length increase agrees very well with the contour-length increase (∼136 nm) expected for complete unfolding of the structured part of the NBD (aa 4–380) (11). For statistics on unfolding forces and total contour-length increase, see SI Appendix. In a subsequent relaxing cycle at the same pulling velocity, the force on the NBD molecule is reduced to allow for refolding (Fig. 2B, purple curve). Refolding intermediates can be observed (purple curve), and full refolding of the NBD occurs at zero force. Successful unfolding and refolding cycles of one single molecule can be observed multiple times (SI Appendix, Figs. S2 and S3).
Fig. 2.

Single molecule force experiments of the NBD by optical tweezers. (A) Optical trap assay. The protein is tethered to the beads by two DNA handles containing a corresponding epitope (biotin, green hexagon; digoxygenin, brown hexagon). The connection between the DNA and protein is realized by the modification of the two cysteine residues of the protein by the single-stranded DNA–maleimide oligonucleotide complementary to the DNA handle overhang. The 1 μm-sized functionalized (α-digoxygenin, purple square; streptavidin, light blue square) glass beads are trapped in the highly focused laser beam. One of the beams is reflected by the steerable mirror, which enables pulling and stretching of a single protein. (B) Force–extension curves of a single NBD domain (the apo-form). The protein–DNA construct was stretched (black curve, 1 kHz filtered) and relaxed (purple curve, 1 kHz filtered) at a constant velocity of 20 nm/s. The trace depicts two parts corresponding to the stretching of the DNA handles and a sudden rip at 34 pN corresponding to the unfolding of the protein. The dashed lines correspond to a WLC fit to the data yielding the contour-length increase of 134 nm. (C) A magnification of the force–extension trace. The unfolding phase of the protein reveals transient populations of the intermediates (black squares; for details, see Single Molecule Force Experiments on the NBD of the DnaK Chaperone). (D) Contour-length transformation of the force–extension data. Shown are multiple unfolding phases. (E) Contour-length increase versus time for the wild-type (black) and insert variants (purple for K183-Insert, red for A290-Insert, and gray for D364-Insert). The insert variants have an additional increase in the contour length (∼7 nm) due to insertion of a highly flexible 20 aa large loop. The plus sign (+) indicates an increase in the length at a particular position. (F) Comparison of the unfolding phases between different protein forms: apo, MgATP, and MgADP. The conditions were 50 mM Tris⋅HCl, 150 mM KCl, 5 mM MgCl2, and 1 mM ATP/ADP, pH 7.8. (G) Contour-length increase versus time for the wild-type (black) and insert variants (purple for K183-Insert, red for A290-Insert, and gray for D364-Insert) under holo-conditions (1 mM ATP, 5 mM MgCl2).
A closer examination of the unfolding phase of the stretching cycle revealed the population of two short-lived unfolding intermediates: Iapo1 and Iapo2 (Fig. 2C). A contour-length transformation using a WLC model (for details, see ref. 24) shows contour lengths of the unfolded portions of the intermediates of 23.5 ± 1 nm for Iapo1 (n = 64) and 79 ± 1.2 nm (n = 73) for Iapo2. These lengths are related to the number of residues that sequentially detach in each step (ca. 60 for Iapo1 and 160 for Iapo2). To gain lifetime information about the intermediate states from the force versus extension plots, we display the unfolding phase as a function of time (exemplified for seven sample traces in Fig. 2D). Note that instead of force we use contour length of the unfolded chain as the vertical axis. The average lifetime of Iapo1 is 0.8 ± 0.2 ms (n = 64), whereas Iapo2 lives for 11 ± 3 ms (n = 73), on average.
The sequence of unfolding events appeared strictly obligatory. It is important to note that identification of intermediates always depends on the temporal resolution of the technique used. At the measurement bandwidth of 30 kHz, we can confidently detect intermediate lifetimes of around 200 µs and longer. In addition to the regular unfolding intermediates, we sometimes observed transient substructures in the unfolding phase (e.g., additional dots in the first trace of Fig. 2D between Iapo1 and Iapo2). We focused on the well-populated intermediates.
Even though these experiments already give us coarse information about the number of amino acids unfolded in each intermediate, unequivocal assignment to the regions that unfold within the protein is not possible based on these data alone.
Loop Insertions in Subdomains of the NBD Reveal the Structure of Intermediates.
To further assess the structural nature of the observed intermediates and the associated unfolding pathway, we designed four different constructs to probe different subdomains within the NBD. Because length is the natural coordinate in single molecule force experiments, we inserted additional amino acid residues into various loop regions of the protein. The additional length gain upon unfolding of those loop inserts identifies the region of the protein that has unfolded (25, 26). We inserted a flexible Gly–Ser-rich loop of 20 residues (for the L20 sequence and activity, see SI Appendix) at the positions shown in Fig. 1A. Three (K183, A290, and D364) out of four constructs showed robust folding and unfolding forces as well as intermediate lifetimes very similar to the wild-type protein and were hence used for structural mapping. The D45-Insertion mutant did not refold completely under apo conditions and hence was not used in the analysis under these conditions.
In the K183-Insert mutant, we probed the interface between lobe I (blue in Fig. 1B) and lobe II (red). The purple trace in Fig. 2E represents a sample unfolding trace for this mutant. The contour lengths of both unfolding intermediates as well as the unfolded state are shifted by 7 ± 0.5 nm (n = 59) compared with the wild type (black trace). The contour-length shift reflects the 20 residues inserted. Apparently, for the K183-Insert, the insert had already unfolded in the transition from the native state to Iapo1, indicating that the two lobes separated early on (see + in the schematics of Fig. 2E). In the A290-Insert variant, the L20 loop was inserted at position A290, thus probing lobe II. In this construct, the Iapo1 species occurs at the same length as in WT, but now Iapo2 is shifted by 7 ± 0.7 nm (n = 42; Fig. 2E, red), indicating that lobe II unfolds during the transition from Iapo1 to Iapo2. In the D364-Insert, the very C-terminal region of the NBD is probed. As it was with the K183-Insert, the 7 ± 0.4 nm (n = 43) contour-length shift occurred in the transition from the native state to Iapo1, proving that the C-terminal part also unfolds in the first unfolding transition. In conclusion, the insert mutants provide the following sequence of unfolding events for the apo NBD. First, the C-terminal region unfolds including residue 364 together with a separation of lobe I and lobe II through unfolding of loop residues including K183 (N→Iapo1). In the next step, lobe II unfolds (Iapo1→Iapo2). Finally, the stretching of lobe I (Iapo2→U) finishes the long journey of the protein to the fully unfolded state U.
Nucleotides Switch the Unfolding Pathway and Stabilize the Intermediates.
The NBD binds MgADP or MgATP at nanomolar affinities (the “holo-form”). In a next set of experiments, we aimed to elucidate the role of nucleotide binding for the mechanical stabilization and hierarchies within the subdomains of the NBD. Surprisingly, at first sight, bound nucleotides did not affect the unfolding force of the native state of the protein (SI Appendix, Fig. S3). However, inspection of the unfolding phase of the holo-form NBD curve reveals a very different pattern of lengths and lifetimes of the intermediates compared with apo NBD (Fig. 2F). Three different intermediates could be clearly identified, none of them observed under apo conditions: two shorter intermediates with Lc = 11.7 ± 1 nm for Iholo1 (n = 77) and Lc = 22.3 ± 0.9 nm for Iholo2 (n = 51) as well as a third intermediate with Iholo3 Lc = 74.7 ± 2 nm (n = 77). This shows the ligand affects the relative stability of NBD’s substructures as well as the unfolding pathway. Moreover, we do not observe any difference in the overall mechanical stability of the apo-form (Funf, avg. = 33.1 ± 4.2 pN, n = 73), MgADP-bound form (Funf, avg. = 32.1 ± 4.0 pN, n = 53), and MgATP-bound form (Funf, avg. = 32.7 ± 3.5 pN, n = 77).
NBD of the Mitochondrial Hsp70 Chaperone.
We were curious whether such a strict order of unfolding events is limited to the NBD of DnaK or might be a more general attribute of the NBDs of different Hsp70s. Single molecule force experiments on the mitochondrial NBD from yeast Hsp70 (n = 31; SI Appendix) show the mean unfolding force of 32.5 ± 4.2 pN and contour length of 134.3 ± 1.9 nm compares well with the mean unfolding force 32.7 ± 3.5 pN (n = 77) and contour length of 133.9 ± 2.6 nm for the DnaK chaperone (n = 77; SI Appendix, Fig. S3). The positions of unfolding intermediates for the mtNBD (11.2 ± 1 nm, n = 31 for Iholo1; 22.0 ± 1.5 nm, n = 14 for Iholo2; and 75.0 ± 2.2 nm, n = 31 for Iholo3) match almost exactly those of the NBD of DnaK. Thus, these two proteins display the same mechanical stability and hierarchy of the subdomains.
Nucleotide Binding Regulates the Mechanical Stability of the Subdomains.
Similar to the apo case, we also attempted to unravel the structural nature of the unfolding intermediates for the holo NBD using insertion mutants. In the presence of nucleotides, all four insertion mutants were able to refold completely to the native form, with native unfolding forces making them amenable to a length analysis (Fig. 2D). In the D45-Insert construct (blue trace), the positions of Iholo1 and Iholo2 remained identical to the wild-type protein (black). However, the position of Iholo3 is shifted relative to the wild-type protein by 7 ± 1 nm (n = 29), implying that domain Ib unfolds in the transition from Iholo2 to Iholo3. In the K183-Insert construct (Fig. 2G, purple), a 7 ± 1 nm (n = 41) nm shift in the transition from Iholo1 to Iholo2 can be detected. In the case of the A290-Insert (Fig. 2G, red), the positions of all intermediates are identical to the wild-type, and the loop insert (7 ± 1.1 nm, n = 61) is released only in the last unfolding step from Iholo3 to the unfolded state. The D364-Insert variant exhibits a change in the position of the first intermediate, Iholo1 (the shift 7 ± 1.6 nm, n = 18; Fig. 2D, gray). In conclusion, the insert mutants provide the following sequence of unfolding events for the holo NBD. First, the C-terminal region unfolds including residue 364 (Iholo1). Second, lobe I and lobe II separate through the unfolding of the lobe-connecting residues including the K183 residue (Iholo2). In the third step, lobe I unfolds including residue D45 (Iholo3), and finally unfolding of lobe II including residue A290 leads to the fully unfolded state U.
The detailed experimental characterization described earlier already provides a coarse picture of the unfolding pathway and how a ligand stabilizes the individual subdomains. To refine this picture, we conducted molecular dynamics simulations to get even more detailed structural insight.
Molecular Simulations by Forced Unfolding of the NBD Provide Detailed Structural Models for Intermediates.
We previously used the self-organized polymer (SOP) model (27) to identify the origin of kinetic partitioning and the details of each pathway during the unfolding of the green fluorescent protein. The resulting computational predictions have been confirmed by our joint studies combining simulation and atomic force microscopy experiments (28). Importantly, this model, which enables one to follow the dynamics of proteins on experimental timescales up to seconds (29), has been successfully applied to study the mechanical response of systems from kinesin (30) to fibrinogen (31) to viral capsids (32) to full microtubules (33) as well as to provide insights into protein refolding following chemical denaturation (34, 35).
We performed molecular dynamics pulling simulations of the NBD from DnaK using the SOP model (27) (Methods) to delineate the nature of the intermediates seen in experiments and to determine the mechanism of stabilization of lobe II in the presence of the nucleotide. Unfolding simulations were performed for both the apo- and the holo-forms of the NBD at a computationally accessible pulling speed of 2,680 nm/s (compare Movies S1 and S2) by fixing the N-terminal end and pulling at the C-terminal end of the chain. The force–extension trace for the apo-NBD simulations (Fig. 3A) exhibits eight peaks. Detectable force peaks were determined using our usual ad hoc criterion (27, 28), based on the general behavior of globular proteins in water: We assume that structural fragments with more than 50% of hydrophobic residues exposed to solvent [hydrophobic surface area (HSA) > 0.5] are not stable, and hence the respective peaks are not experimentally detectable (28) (SI Appendix, Fig. S8). Using this HSA criterion, only the two peaks marked with red dots, as well as the first and final ones, are detectable (Fig. 3A). We assigned these peaks to the N, Iapo1, Iapo2, and U states. To determine the precise boundaries of the folded portions of the intermediates, we monitored the tension propagation along each bond in the chain and the time evolution of the structural overlap for each position in the chain (36). Fig. 3B depicts the tension along the backbone residues of the apo-NBD (see SI Appendix, Fig. S5 A and C for additional details). The top graph in Fig. 3B shows that high tension (blue regions) is found only in the C-terminal part of the chain and across the spacer connecting the two lobes. This indicates that these two disjointed regions of the chain are the only force-bearing elements. More precisely, unfolding of helices 15 and 16 (positions 339–383) and stretching of the lobe-connecting residues (positions 168–187) occur simultaneously when Iapo1 is formed. In the next unfolding step (lower graph in Fig. 3B), the load-bearing region extends to lobe II (blue region), whereas lobe I shows no sizeable increase in tension (red region). Hence, we conclude that in the transition from Iapo1 to Iapo2, lobe II unravels, as seen also in the evolution of native contacts from SI Appendix, Fig. S9. In the last step, lobe I unfolds.
Fig. 3.

Forced unfolding simulations of the NBD. Simulation results for apo- and holo-NBD. (A) Force extension curve (FEC) for the unfolding of the apo-form. Intermediates Iapo1 and Iapo2 are labeled on the respective force peaks. (B) Tension propagation in the chain and conformational snapshots for the two intermediates evaluated from the red points in A. (Top) For Iapo1, tension is greatest in the connecting loop and in helices 15 and 16 (residues 168–187 and 339–383). The snapshot shows that this corresponds to the separation of the lobes and the unfolding of those helices. (Bottom) For Iapo2, before lobe I unravels, the connecting loop and the entire lobe II experiences increased tension (residues from Iapo1 plus 188–338). (C) FEC for the unfolding of the holo-form. Intermediates Iholo1, Iholo2, and Iholo3 are labeled. (D) Tension propagation in the chain and conformational snapshots for the three intermediates evaluated from the red points in C. (Top) For Iholo1, helices 15 and 16 are extracted from lobe II due to increased tension (residues 346–383). (Middle) For Iholo2, the two lobes separate due to the increased tension in the connecting loop (residues from Iholo1 plus 169–187). (Bottom) In Iholo3, only lobe II remains folded (residues from Iholo2 and positions 1–168 unfold).
Simulations of the ADP-bound NBD (the holo-form) showed that the unfolding occurs according to two pathways with 89% (32 of 36 trajectories) and 11% (4 of 36 trajectories) probability. We find the partitioning between the two pathways is driven by the affinity of the two lobes for the nucleotide. In the first pathway (pathway I), the nucleotide remains bound to lobe I (SI Appendix, Figs. S6 and S9), whereas in pathway II the nucleotide is bound to lobe II (SI Appendix, Figs. S5 and S9). We also performed simulations of ATP-bound NBD, which all followed pathway I described before. In the caption to SI Appendix, Fig. S9, we provide details about the trigger for kinetic partitioning in simulations. These results, together with the fact that the position of the transition state likely changes with force in the holo-NBD case, strongly suggest that the discrepancy between experiments and computations arises from differences in the respective loading rates. Here, we focus on pathway II, which we relate to the experimentally observed pathway because, as discussed earlier, in experiments the nucleotide remains bound to lobe II. The discussion regarding pathway I is in SI Appendix, Figs. S6, S8, and S9. The HSA criterion indicates that only three peaks (with red dots) as well as the first and final ones are experimentally detectable (Fig. 3 and SI Appendix, Fig. S8). We assigned the peaks to the N, Iholo1, Iholo2, Iholo3, and U states. Fig. 3D depicts the tension along holo-NBD. The top plot in Fig. 3D shows that only the C-terminal part of the chain is under substantial tension. Thus, in the simulations, the unfolding of helices 15 and 16 (positions 347–383) leads to the formation of Iholo1. The middle graph in Fig. 3D indicates that the spacer connecting the two lobes (positions 168–185) unfolds during the transition from Iholo1 to Iholo2. Finally, the bottom graph in Fig. 3D shows that from Iholo2 to Iholo3, lobe I unravels. In the last step, lobe II unravels and the nucleotide dissociates.
Discussion
Deciphering the Unfolding Pathway of NBD.
Single molecule force spectroscopy by optical tweezers is a superb method for studying protein folding/unfolding at very high temporal (μs) and spatial resolution (nm). Combining this method with mutational approaches as well as with computational MD simulations even allows deriving structural interpretation of the complex processes observed during mechanical unfolding (28, 37, 38). Using our insertion variants in combination with pulling molecular dynamics simulations, we decipher the structure of the intermediates along the unfolding pathway of the complex multidomain subunit of DnaK NBD. Furthermore, the question of how nucleotide binding couples to the domain mechanics can be addressed.
A combined picture of the experimental and simulation results is presented in Fig. 4 (for quantitative comparison, see SI Appendix). The very first event in mechanical unfolding of the NBD both in apo- and holo-form is pulling out the C-terminal helix. In the apo case, stretching of the C-terminal helix occurs concomitantly with separation of lobe I and lobe II (Iapo1), which indicates that these two events are highly cooperative. Apparently, the lobe–lobe interactions in the absence of nucleotide are not strong enough to resist force even transiently.
Fig. 4.

Summary of the unfolding pathway of the NBD of DnaK. The structures of intermediates are those from the pulling simulations. The red surface indicates lobe II; the blue surface indicates lobe I and the yellow sphere is an ADP molecule.
In the holo-form, stretching of the last helix and lobe separation proceeds in two distinct steps. First, the helix unfolds (N→Iholo1), followed by the splitting up of lobe I and lobe II by stretching of the connecting spacer (Iholo1→Iholo2). Apparently, the interface between the two lobes is now stabilized by the nucleotide, in addition to the already existing lobe–lobe interactions (compare Fig. 1 B and C). After lobe separation, the nucleotide stays bound to lobe II, indicating stronger interactions with this subdomain (see also discussion below). Analysis of the time evolution of native contacts along the holo-case pathways (SI Appendix, Fig. S9 C–F) indicates that the release of the connecting loop between the two lobes can only occur following the partial disruption of the nucleotide binding pocket involving amino acids in lobe II. Importantly, simulations show also that, upon extension of the loop, the binding pocket is able to reform and this leads to the stabilization of lobe II against unraveling. In the Protein Data Bank (PDB) structure (4B9Q), the 186–195 region makes contacts with the local (along the sequence) region 196–210, as well as with the remote (along the sequence) 330–345 region. Both the local and the remote regions contain five nucleotide binding residues each (shown in Fig. 1C): 195–199 in the local region and 341–345 in the remote one. We found that during the Iholo1→Iholo2 event in pathway II (SI Appendix, Fig. S9D), 186–195 loses contacts with its local partner, while retaining the contacts with its remote partner. This indicates that in vitro stabilization of lobe II requires the preservation of the 341–345 region of the nucleotide binding site. At the same time, transient loss of the 195–199 region of the binding site does not destabilize lobe II. Using simulation and experimental results, we can estimate the time required to detect Iholo2, once Iholo1 occurs. This is as short as 2 ms (from simulations) and as long as 9 ± 2 ms (from experiments).
In the next step, further unfolding continues now from the domain that is less force resistant. In the apo-case (Fig. 4), lobe II is apparently less stable than lobe I and, hence, unfolds first. The unfolding of the apo-form is then completed by unfolding of lobe I. In the holo-case, binding of the nucleotide stabilizes lobe II strongly over lobe I so that now lobe I is mechanically less stable and unfolds first (Iholo3). The last step is the unfolding of lobe II and only then is the ligand released from the polypeptide chain. Remarkably, the unfolding intermediates of the holo-form and pathways we find for the E. coli Hsp70 are identical to the NBD from yeast mitochondria (SI Appendix, Fig. S3), and thus the observed nucleotide-dependent domain hierarchy is likely a more general property.
Nucleotide Binding Regulates the Mechanical Coupling Between Subdomains.
The nucleotides bind to the intact NBD with about 100 nM affinity corresponding to 16 kBT of free energy of binding under standard conditions. How does this binding energy stabilize the different subdomains of the NBD? The binding free energy results from the multiple interactions between ligand and the contacting residues of the NBD (Fig. 1C). Simple counting of the number of interactions between protein and nucleotide reveals that most of the interacting residues (13 out of 18) come from lobe II (highlighted in red, Fig. 1C) and very few (5 out of 18) from lobe I (highlighted in blue, Fig. 1C). This consideration already highlights the very different roles of the subdomains in binding. One could speculate that the majority of residues specializing in the binding of the ligand are located in lobe II and the signal transducing residues are located in lobe I. We provide, to our knowledge, the first evidence for nucleotide binding to the isolated lobe II in the absence of a folded lobe I.
Exclusive Role of the C-Terminal Helix as Molecular Glue.
For the last C-terminal helix, a very special role emerges from our single molecule force experiments. The C-terminal helix is sandwiched between both lobes and is nearly completely buried within the structure. The extensive network of interactions with both lobes might already imply a mediating role in coupling between the two lobes. The function of this helix as molecular glue is further supported by the observation that removal of this helix results in incomplete folding of the NBD (SI Appendix, Fig. S4) and unfolding forces dropped dramatically (10–15 pN). Our atomistic simulations of the apo-form of the NBD (PDB ID code 2KHO) in the absence of this last helix (SI Appendix, Fig. S7) support the experimental conclusions that the last helix is a major folding element of the NBD: Its loss leads to the transition between closed and open cleft conformations, even on the relatively short (∼400 ns) timescale of our simulations. Interestingly, the C-terminal helix is the main determinant for the mechanical stability of the whole NBD. We now can reinterpret the observed major unfolding force peak of ∼34 pN as the force necessary to pull out the helix from the folded structure. After unfolding of the C-terminal helix, the remaining domains are more mechanically labile. Starting from this step, the unfolding pathway is modulated by the presence of the nucleotide. The strong interaction with both lobes also explains why the major unfolding force peak is independent of the nucleotide status. Clearly, there is no energetic coupling between ligand binding and C-terminal helix unfolding, and hence, the presence of nucleotide does not matter for the initial unfolding event. This has an interesting consequence for the communication between the NBD and the SBD of the two domain DnaK chaperones. The C-terminal helix continues into the SBD domain through a short linker sequence. After binding of ATP to the NBD, the SBD undergoes a large structural rearrangement (12, 39). How does the allosteric signal propagate? Our experiments exclude a simple scenario according to which the information about the nucleotide state is transmitted along the backbone through the C-terminal helix from the NBD to the SBD as the C-terminal helix is firmly glued between the two lobes independent of the nucleotide status of the NBD. Thus, the signal must be transmitted by contacts of noncontiguous residues through interactions over space.
Methods
Experimental Procedures.
For cloning, protein purification, and design, see SI Appendix. The experimental setup used for optical trapping is a custom-built high-resolution dual trap optical tweezers with back-focal plane detection as described previously (40, 41).
SOP Model.
We simulated the mechanical response of the NBD of Hsp70 using the topology-based SOP model (27) starting from the Cα positions of each amino acid residue in the respective PDB entry (ID codes 2KHO, for apo, and 4B9Q, for holo). Brownian dynamics simulations followed the behavior of the chain under force applied using constant loading rate conditions. The details are provided in SI Appendix.
Supplementary Material
{kind=link}
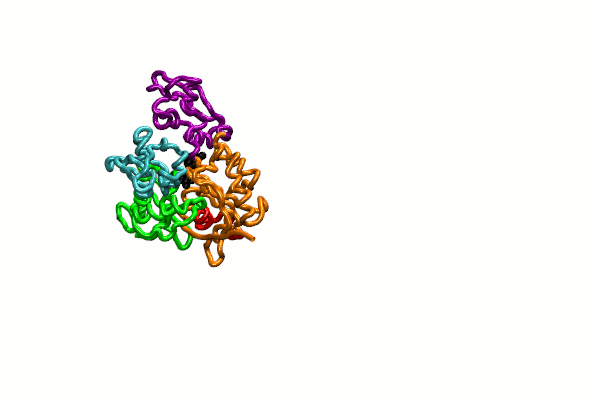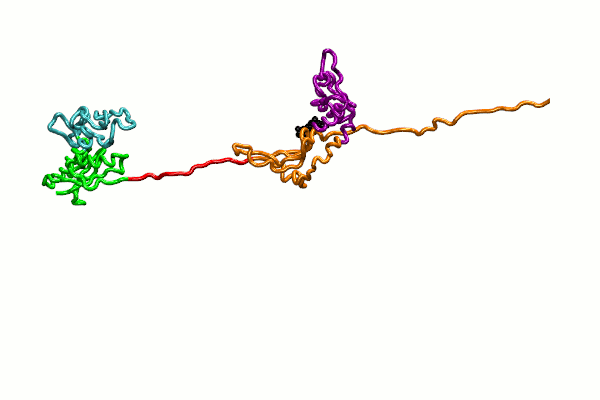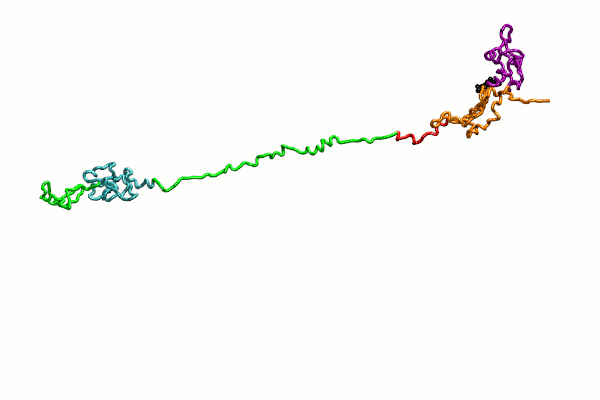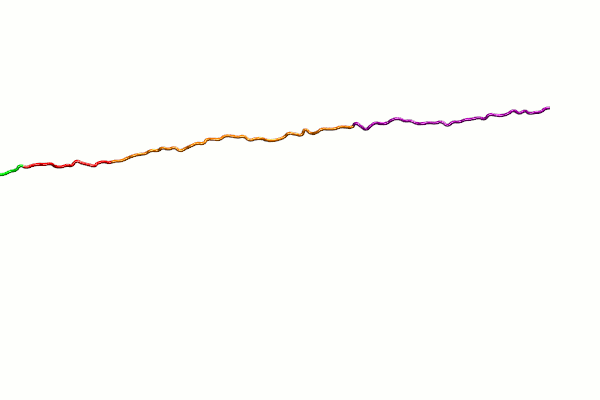{kind=link}
Acknowledgments
G.Z. would like to dedicate this paper to Prof. Franz X. Schmid on the occasion of his 65th birthday. We thank the members of our group for suggestions and comments. This work was supported by Projects SFB 1035/A5, ZO 291/1-1, and MO1944/1-1 of Deutsche Forschungsgemeinschaft (to M.R., G.Z., and D.M.). This work was also supported in part by National Science Foundation CAREER Award MCB-0845002 (to R.I.D.). M.R. received support from the Nanosystems Initiative Munich.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1504625112/-/DCSupplemental.
References
- 1.Smock RG, Gierasch LM. Sending signals dynamically. Science. 2009;324(5924):198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janovjak H, Sapra KT, Kedrov A, Muller DJ. From valleys to ridges: Exploring the dynamic energy landscape of single membrane proteins. Chemphyschem. 2008;9(7):954–966. doi: 10.1002/cphc.200700662. [DOI] [PubMed] [Google Scholar]
- 3.Kedrov A, Ziegler C, Muller DJ. Differentiating ligand and inhibitor interactions of a single antiporter. J Mol Biol. 2006;362(5):925–932. doi: 10.1016/j.jmb.2006.07.049. [DOI] [PubMed] [Google Scholar]
- 4.Kedrov A, Krieg M, Ziegler C, Kuhlbrandt W, Muller DJ. Locating ligand binding and activation of a single antiporter. EMBO Rep. 2005;6(7):668–674. doi: 10.1038/sj.embor.7400455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manibog K, Li H, Rakshit S, Sivasankar S. Resolving the molecular mechanism of cadherin catch bond formation. Nat Commun. 2014;5:3941. doi: 10.1038/ncomms4941. [DOI] [PubMed] [Google Scholar]
- 6.Junker JP, Hell K, Schlierf M, Neupert W, Rief M. Influence of substrate binding on the mechanical stability of mouse dihydrofolate reductase. Biophys J. 2005;89(5):L46–L48. doi: 10.1529/biophysj.105.072066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puchner EM, et al. Mechanoenzymatics of titin kinase. Proc Natl Acad Sci USA. 2008;105(36):13385–13390. doi: 10.1073/pnas.0805034105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao Y, Li H. Dynamics of protein folding and cofactor binding monitored by single-molecule force spectroscopy. Biophys J. 2011;101(8):2009–2017. doi: 10.1016/j.bpj.2011.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daugaard M, Rohde M, Jäättelä M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581(19):3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 10.Genevaux P, Georgopoulos C, Kelley WL. The Hsp70 chaperone machines of Escherichia coli: A paradigm for the repartition of chaperone functions. Mol Microbiol. 2007;66(4):840–857. doi: 10.1111/j.1365-2958.2007.05961.x. [DOI] [PubMed] [Google Scholar]
- 11.Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ER. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci USA. 2009;106(21):8471–8476. doi: 10.1073/pnas.0903503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qi R, et al. Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat Struct Mol Biol. 2013;20(7):900–907. doi: 10.1038/nsmb.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhuravleva A, Gierasch LM. Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc Natl Acad Sci USA. 2011;108(17):6987–6992. doi: 10.1073/pnas.1014448108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calloni G, et al. DnaK functions as a central hub in the E. coli chaperone network. Cell Reports. 2012;1(3):251–264. doi: 10.1016/j.celrep.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Tomoyasu T, Mogk A, Langen H, Goloubinoff P, Bukau B. Genetic dissection of the roles of chaperones and proteases in protein folding and degradation in the Escherichia coli cytosol. Mol Microbiol. 2001;40(2):397–413. doi: 10.1046/j.1365-2958.2001.02383.x. [DOI] [PubMed] [Google Scholar]
- 16.Barthel TK, Zhang J, Walker GC. ATPase-defective derivatives of Escherichia coli DnaK that behave differently with respect to ATP-induced conformational change and peptide release. J Bacteriol. 2001;183(19):5482–5490. doi: 10.1128/JB.183.19.5482-5490.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vogel M, Mayer MP, Bukau B. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J Biol Chem. 2006;281(50):38705–38711. doi: 10.1074/jbc.M609020200. [DOI] [PubMed] [Google Scholar]
- 18.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475(7356):324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 19.Bork P, Sander C, Valencia A. An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins. Proc Natl Acad Sci USA. 1992;89(16):7290–7294. doi: 10.1073/pnas.89.16.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palleros DR, Shi L, Reid KL, Fink AL. Three-state denaturation of DnaK induced by guanidine hydrochloride. Evidence for an expandable intermediate. Biochemistry. 1993;32(16):4314–4321. doi: 10.1021/bi00067a021. [DOI] [PubMed] [Google Scholar]
- 21.Montgomery D, Jordan R, McMacken R, Freire E. Thermodynamic and structural analysis of the folding/unfolding transitions of the Escherichia coli molecular chaperone DnaK. J Mol Biol. 1993;232(2):680–692. doi: 10.1006/jmbi.1993.1418. [DOI] [PubMed] [Google Scholar]
- 22.Cecconi C, Shank EA, Bustamante C, Marqusee S. Direct observation of the three-state folding of a single protein molecule. Science. 2005;309(5743):2057–2060. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- 23.Bustamante C, Marko JF, Siggia ED, Smith S. Entropic elasticity of lambda-phage DNA. Science. 1994;265(5178):1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 24.Puchner EM, Franzen G, Gautel M, Gaub HE. Comparing proteins by their unfolding pattern. Biophys J. 2008;95(1):426–434. doi: 10.1529/biophysj.108.129999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carrion-Vazquez M, Marszalek PE, Oberhauser AF, Fernandez JM. Atomic force microscopy captures length phenotypes in single proteins. Proc Natl Acad Sci USA. 1999;96(20):11288–11292. doi: 10.1073/pnas.96.20.11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwaiger I, Kardinal A, Schleicher M, Noegel AA, Rief M. A mechanical unfolding intermediate in an actin-crosslinking protein. Nat Struct Mol Biol. 2004;11(1):81–85. doi: 10.1038/nsmb705. [DOI] [PubMed] [Google Scholar]
- 27.Hyeon C, Dima RI, Thirumalai D. Pathways and kinetic barriers in mechanical unfolding and refolding of RNA and proteins. Structure. 2006;14(11):1633–1645. doi: 10.1016/j.str.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 28.Mickler M, et al. Revealing the bifurcation in the unfolding pathways of GFP by using single-molecule experiments and simulations. Proc Natl Acad Sci USA. 2007;104(51):20268–20273. doi: 10.1073/pnas.0705458104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhmurov A, Dima RI, Kholodov Y, Barsegov V. Sop-GPU: Accelerating biomolecular simulations in the centisecond timescale using graphics processors. Proteins. 2010;78(14):2984–2999. doi: 10.1002/prot.22824. [DOI] [PubMed] [Google Scholar]
- 30.Hyeon C, Onuchic JN. Mechanical control of the directional stepping dynamics of the kinesin motor. Proc Natl Acad Sci USA. 2007;104(44):17382–17387. doi: 10.1073/pnas.0708828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhmurov A, et al. Mechanism of fibrin(ogen) forced unfolding. Structure. 2011;19(11):1615–1624. doi: 10.1016/j.str.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kononova O, et al. Structural transitions and energy landscape for Cowpea Chlorotic Mottle Virus capsid mechanics from nanomanipulation in vitro and in silico. Biophys J. 2013;105(8):1893–1903. doi: 10.1016/j.bpj.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kononova O, et al. Tubulin bond energies and microtubule biomechanics determined from nanoindentation in silico. J Am Chem Soc. 2014;136(49):17036–17045. doi: 10.1021/ja506385p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reddy G, Liu Z, Thirumalai D. Denaturant-dependent folding of GFP. Proc Natl Acad Sci USA. 2012;109(44):17832–17838. doi: 10.1073/pnas.1201808109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z, Reddy G, O’Brien EP, Thirumalai D. Collapse kinetics and chevron plots from simulations of denaturant-dependent folding of globular proteins. Proc Natl Acad Sci USA. 2011;108(19):7787–7792. doi: 10.1073/pnas.1019500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JY, Iverson TM, Dima RI. Molecular investigations into the mechanics of actin in different nucleotide states. J Phys Chem B. 2011;115(1):186–195. doi: 10.1021/jp108249g. [DOI] [PubMed] [Google Scholar]
- 37.Marszalek PE, et al. Mechanical unfolding intermediates in titin modules. Nature. 1999;402(6757):100–103. doi: 10.1038/47083. [DOI] [PubMed] [Google Scholar]
- 38.Li YD, Lamour G, Gsponer J, Zheng P, Li H. The molecular mechanism underlying mechanical anisotropy of the protein GB1. Biophys J. 2012;103(11):2361–2368. doi: 10.1016/j.bpj.2012.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kityk R, Kopp J, Sinning I, Mayer MP. Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol Cell. 2012;48(6):863–874. doi: 10.1016/j.molcel.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 40.von Hansen Y, Mehlich A, Pelz B, Rief M, Netz RR. Auto- and cross-power spectral analysis of dual trap optical tweezer experiments using Bayesian inference. Rev Sci Instrum. 2012;83(9):095116. doi: 10.1063/1.4753917. [DOI] [PubMed] [Google Scholar]
- 41.Žoldák G, Stigler J, Pelz B, Li H, Rief M. Ultrafast folding kinetics and cooperativity of villin headpiece in single-molecule force spectroscopy. Proc Natl Acad Sci USA. 2013;110(45):18156–18161. doi: 10.1073/pnas.1311495110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laskowski RA, Swindells MB. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.